
Abstract
Amyotrophic lateral sclerosis (ALS) is a worldwide problem with no effective treatment. Patients usually die of respiratory failure. The basic pathological process of ALS is the degeneration and necrosis of motor neurons. Neuroglial cell dysfunction is considered closely related to the development of ALS. Sleep plays an important role in repairing the nervous system, and sleep disorders can worsen ALS. Herein, we review the pathogenesis of ALS and the neuroprotective mechanism of sleep‐based therapy. Sleep‐based therapy could be a potential strategy to treat ALS.
Keywords
1 Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and irreversible neuromuscular disease characterized by the degeneration of upper and lower motor neurons. It causes gradual weakness and atrophy of the limb and trunk muscles. The clinical manifestations of ALS are progressive muscle atrophy, muscle bundle tremor, medulla oblongata paralysis, and pyramidal tract damage, which eventually lead to dysphagia, respiratory failure, and death. Approximately 10% of ALS cases are familial (familial ALS, fALS), with a mean patient age of 58–63 years. The remaining 90% of ALS cases are sporadic (sporadic ALS, sALS), with a mean patient age of 43–52 years [1]. The incidence of ALS is 1/100,000 to 2/100,000, and the patient survival time is 2 to 3 years after the onset of symptoms [2]. The mean patient age of ALS onset in China is younger, while the survival time is generally longer. The etiology and pathogenesis of ALS are unclear, but many hypotheses exist regarding excitotoxicity, neuroinflammation, autoimmunity, and oxidative stress. Many environmental factors (such as smoking, heavy metals or pesticides, radiation, and several sports activities) are considered related to the neuropathological changes of ALS [3]. More than 120 genetic variants, including SOD1, TARDBP, C9orf72, and TBK1, have been associated with the risk of ALS [4]. The same mutation gene can be detected in approximately 60%–80% of fALS cases [5]. Currently, only riluzole and edaravone have been approved by the United States Food and Drug Administration for ALS treatment, but they only delay the progression of certain cases [6]. The 5‐year survival of ALS cases is 10%. ALS is painful for the patients and their families, so it is urgent to develop new treatment strategies to delay or treat ALS.
The basic pathological process of ALS is the degeneration and necrosis of motor neurons. Neuroinflammation characterized by the activation of microglia and astrocytes, oxidative stress, and elevated inflammatory cytokines levels is the hallmark of ALS [7]. Sleep disorders are observed in 70% of patients with ALS [8]. Sleep disorders impair the cell clearance of misfolded neurotoxin proteins and affect the immunological and redox system resulting in neuroinflammation and oxidative stress [9]. Prolonging sleep time and improving sleep structure may be helpful in the treatment of ALS.
Since neurons cannot regenerate after necrosis, ALS becomes an incurable disease. However, understanding the pathogenesis of ALS and the importance of sleep may shed light on its future treatment.
2 Neuroglial cells and ALS
The human brain consists of two types of cells, neurons and neuroglial cells. Neuroglial cells include astrocytes, oligodendrocytes, and microglia. 100 billion neurons are found in the human brain, making it an extremely complex neural network. These cells process all kinds of neural information and are responsible for several brain functions such as feeling, learning, memory, movement, and creation. There are around 10–15 times more neuroglial cells than neurons, and the total volume is almost the same [10]. Neuroglial cells are widely distributed in the central nervous system (CNS). Although they do not transmit nerve impulses (action potentials), they demonstrate multiple physiological functions and play an important role in ALS pathogenesis.
Astrocytes participate in the glutamate cycle in the brain. Glutamic acid is the main excitatory amino acid in the brain. It is generated from the intermediate product α‐ketoglutarate (α‐KG) of the tricarboxylic acid cycle. Neurons cannot synthesize glutamic acid from glucose, and due to the lack of pyruvate carboxylase in neurons, the tricarboxylic acid cycle cannot be completed. Astrocytes mainly provide glutamate in the brain. Astrocytes are rich in two types of glutamate transporter (GLT): glutamate transporter‐1 (GLT‐1) and glutamate aspartate transporter. During neuronal activity, neurons release glutamate in the form of vesicles. These vesicles are absorbed by astrocytes through GLT and converted into glutamine. Subsequently, glutamine is released into the synaptic cleft and absorbed by neurons. Astrocytes convert L‐serine into D‐serine, and D‐serine activates the glycine regulatory N‐methyl‐D‐aspartate receptor and promotes glutamate uptake by astrocytes [11]. In addition, astrocytes release glutamate through GLT and cystine–glutamate exchanger (xCT). When glutamate is excessively released, astrocytes negatively regulate the release of glutamate in neurons [12]. Glutamate excitotoxicity is one of the contributors to ALS pathogenesis [13]. High‐level glutamate aggregation in the local environment leads to excessive ion channel activity, calcium inflow, damaged endoplasmic reticulum, and neuronal death.
Astrocytes exhibit an anti‐inflammatory effect to protect CNS from injury. Neuroinflammation induces the formation of an astrocyte scar and a blood–brain barrier (BBB) tight junction, thus preventing inflammatory cells from entering the CNS. During the early stage of ALS, the damaged BBB can increase vascular permeability, shorten capillary length, and reduce the expression of barrier structural components [14, 15]. Therefore, BBB damage may be initiating factor of ALS. Neuroinflammation induces differentiation of mature astrocytes into neural progenitor cells, exhibiting an anti‐inflammatory effect and promoting the regeneration of the injured CNS [16]. Astrocytes can also release lipoxin A4 (LxA4) and lipoxin B4 (LxB4) to inhibit neuroinflammation; LxB4 provides more effective neuroprotection [17].
Microglial cells are the main cells involved in neuroinflammation. These cells are further divided into M1 and M2. M1 cells promote T cell neurotoxicity and release inflammatory factors, such as reactive oxygen species and tumor necrosis factor‐α (TNF‐α). M2 cells produce anti‐inflammatory factors such as IL‐4, IL‐10, and insulin‐like growth factor‐1 (IGF‐1). During the early stages of ALS, M2 microglia and auxiliary T cells play an anti‐inflammatory role. When they enter the stage of rapid progression, microglial cells transform into M1 cells and co‐accelerate motor neuron loss with auxiliary T cells [18]. Astrocytes regulate microglia activation by releasing orosomucoid‐2 (ORM2), lipocalin‐2 (LCN2), transforming growth factor‐β (TGF‐β), and prostaglandin E2 (PGE2), thereby inhibiting neuroinflammation and preventing neurodegeneration [19].
Oxidative stress is also thought to be a cause of ALS. The imbalance between the production and scavenging of reactive oxygen species (ROS) causes oxidative stress damage in the body. In ALS, superoxide dismutase (SOD) and 8‐hydroxy‐2‐deoxyguanosine (8‐OHDG) levels decrease, while the ratio of oxidized glutathione to reduced glutathione increases. Neurons exhibit a lower antioxidant capacity than astrocytes and are more vulnerable to injuries. Therefore, neurons usually require astrocyte coupling, the main executor of glutathione (GSH) reduction in the brain, to resist oxidative stress. The reduced GSH is oxidized into oxidized GSH (GSSG), and ROS is eliminated [20]. Astrocytes produce IGF‐1, which is anti‐oxidative by activating protein kinase B (PKB) [21].
Considering the tight relationship between abnormal neuroglial cell function and the occurrence of ALS, restoring normal neuroglial cell function could be critical to ALS treatment.
3 Sleep and ALS
Neurodegenerative diseases present various sleep disorders, such as insomnia, somnolence, circadian rhythm disorder, parasomnia, and sleep‐disordered breathing (SDB) [22]. Degenerative motor neurons in ALS cases are mainly distributed in the motor cortex, low brain stem, spinal pyramidal tract, and anterior horn cells of the spinal cord. They are not in the sleep regulation center (thalamus, hypothalamus, and pontine nucleus). Sleep structural disorders are mainly caused by exogenous factors, such as cough, excessive oropharyngeal secretions, a sleep breathing event, restless legs syndrome, nocturia, muscle stiffness or pain, difficulty turning over, and anxiety or depression [23]. Previous studies found that ALS patients exhibit a decreased sleep time length, prolonged sleep latency, increased number of awakenings, an increased proportion of non‐rapid eye movement (NREM) sleep phases 1 and 2, and a decreased proportion of the rapid eye movement (REM) sleep phase [24, 25]. Sleep disorders can aggravate ALS progression. A decrease in REM sleep can significantly shorten the median survival time of patients with ALS [26], forming a vicious circle of insomnia-aggravation-insomnia.
Adequate sleep is crucial to ensure the brain can detoxify metabolic wastes and toxins, avoid neuronal degeneration and necrosis, and promote nerve regeneration [27]. During NREM sleep, the activity of neurons in the cerebral cortex decreases, arteries constrict, and blood entering brain tissue decreases. Cerebrospinal fluid flows into the brain through arteries and helps to regularly “flush” the brain [28]. Sleep disorders and sleep deprivation can cause a structural sleep disorder and change the peak time of inflammatory cytokines, thus aggravating the inflammatory response [29]. Sleep deprivation can promote astrocyte phagocytosis and microglial activation in mice brains [30], and further stimulate peripheral inflammatory cytokines, such as TNF‐α, IL‐1, and IL‐6 [31]. Short‐term sleep (≤ 6 h) or sleep deprivation can reduce the secretion of growth hormone (GH), IGF‐1, and testosterone [32]. The decrease of GH, IGF‐1, or testosterone can reduce the synthesis and increase the decomposition of muscle proteins. It can also enhance the expression of myostatin by reducing the activity of IGF/P13K/Akt/mTOR [33].
Therefore, improving sleep in patients with ALS might be a potential strategy to treat ALS.
4 Sleep‐based therapy
Dexmedetomidine (DEX) is a selective α2‐adrenergic receptor (α2‐AR) agonist with sedative and analgesic effects; however, it exhibits little effect on respiration. DEX has been widely used for perioperative sedation and analgesia, but it induces a common side effect of bradycardia. As an intravenous anesthetic, DEX demonstrates a strong neuroprotective function. It activates α2‐AR coupled with G0 and Gi proteins in the presynaptic membrane, decreases intracellular calcium concentration, and inhibits glutamate release. In addition, DEX increases brain‐derived neurotrophic factor (BDNF) expression in astrocytes and prevents neuronal cell death induced by glutamate receptor agonists [34]. It reduces neuroinflammation by inhibiting the Toll‐like receptor 4/ nuclear factor kappa (TLR4/BNF‐κB) pathway and reducing the expression of TNF‐α [35]. DEX can also counteract oxidative stress, which could be attributed to increasing the ratio of GSH to GSSG, increasing the activity of SOD, and reducing the levels of malondialdehyde and nitric oxide [34]. Conversely, continuous infusion of DEX can significantly improve the sleep quality of patients without interfering with the natural sleep pattern [36]. After discontinuation of dexmedetomidine, patients achieve continuously improved sleep quality [37]. DEX improves sleep quality by increasing NREM sleep time, reducing the latency of the NREM sleep phase, reducing sleep segments, and partially restoring the normal circadian rhythm [38, 39] From the mechanisms mentioned above, DEX seems to be a potential drug for ALS treatment.
Anticholinergic drugs, such as atropine and scopolamine, might also be beneficial for ALS. Around half of the patients with ALS exhibit salivation symptoms, and excessive salivation can cause aspiration pneumonia [40]. Anticholinergic drugs reduce salivary gland secretion and salivation. They also increase the heart rate to relieve the bradycardia caused by DEX. They excite the respiratory center to decrease SDB events during sleep. Several patients with ALS are prone to anxiety and depression symptoms, which usually lead to low quality of life and accelerate ALS progression [41]. By coincidence, researchers found that intravenous administration of scopolamine results in a rapid and lasting antidepressant effect [42]. Moreover, anticholinergic drugs can also improve microcirculation and provide a stable internal environment for neuronal repair.
Therefore, sleep‐based therapy might be a potential strategy to treat ALS in the following mechanisms: (1) sleep‐based therapy uses hypnotic drugs to maintain deep sleep, reduce neuronal activity, and stimulate metabolism and glutamate release; (2) sufficient sleep time and quality ensure normal wastes and toxin removal by the brain. Adequate sleep can also increase the secretion of BDHF, GH, and IGF‐1 as well as accelerate tissue repair; (3) DEX exhibits neuroprotective effects through several mechanisms, such as improving the activity of SOD and increasing the ratio of GSH to GSSG; DEX can improve the sleep structure of patients and increase NREM sleep time, which may delay the development of ALS; and (4) anticholinergic drugs reduce the side effects of DEX, reduce salivation, excite respiratory centers, decrease SDB events, and improve microcirculation during sleep. The damaged nerve and muscle cells might recover to achieve the expected therapeutic effect (Fig. 1). Meanwhile, nutritional support, intensive care, effective rehabilitation exercise, and close multidisciplinary cooperation are crucial and supplemental for ALS treatment.
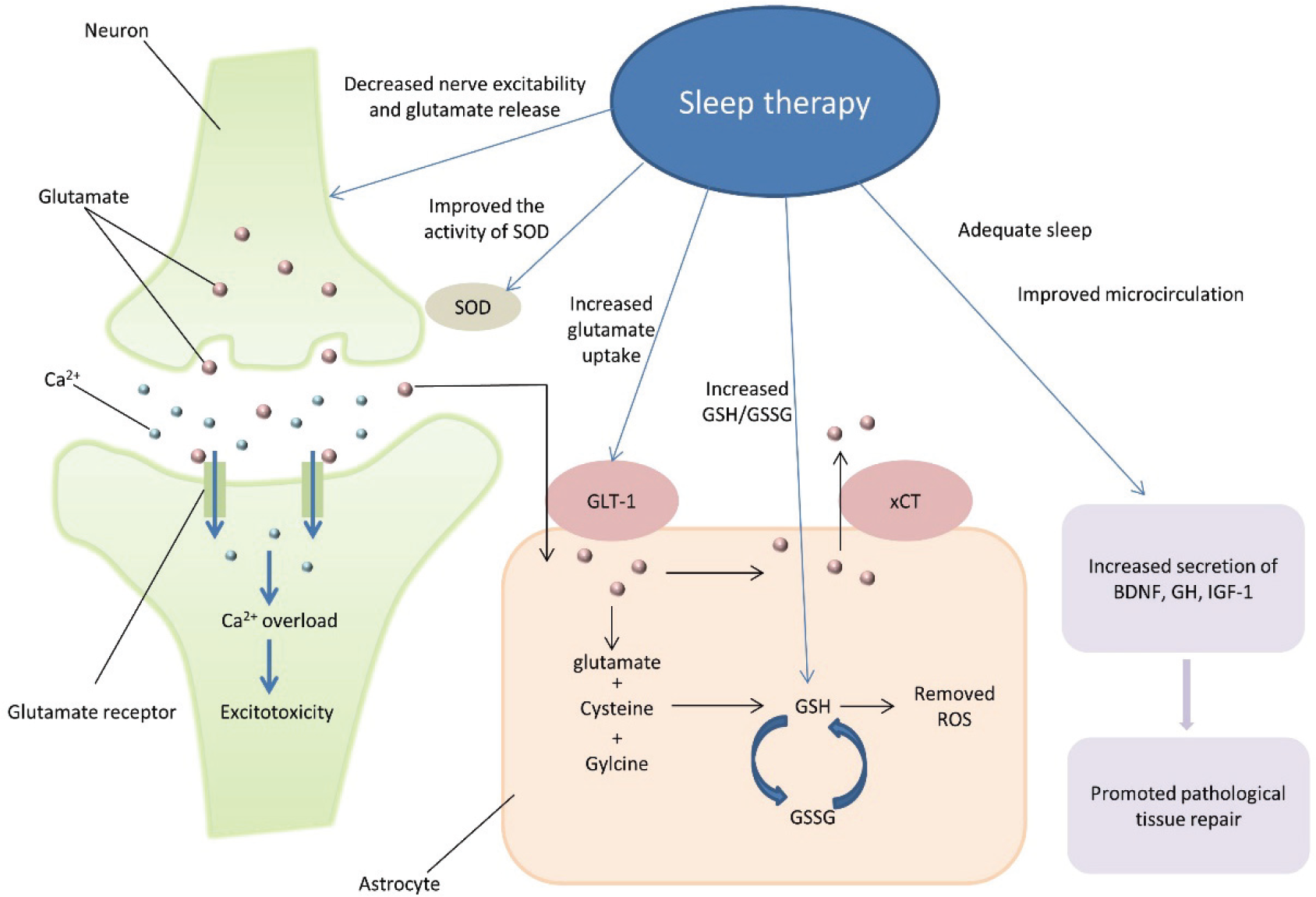
Hypothesis of sleep‐based therapy for ALS treatment. Sleep‐based therapy may reduce the excitability of neurons, reduce the release of glutamate by neurons, and promote the uptake of glutamate by astrocytes to reduce neurotoxicity. It may improve the activity of SOD, increase the ratio of GSH to GSSG, and promote the elimination of ROS to reduce oxidative stress. Adequate sleep can also increase the secretion of BDHF, GH, and IGF‐1 as well as accelerate tissue repair. Abbreviations: SOD, superoxide dismutase; GLT‐1, glutamate transporter‐1; xCT, cysteine–glutamate exchanger; GSH, glutathione; GSSG, oxidized glutathione; ROS, reactive oxygen species; BDNF, brain‐derived neurotrophic factor; GH, growth hormone; IGF‐1, insulin‐like growth factor 1.
5 Summary
At present, the main treatment of ALS is to prolong the patients’ survival time and improve their quality of life. Few treatments can reverse the development of ALS. Adequate sleep is essential for nervous system repair. After reviewing the pathogenesis of ALS and the neuroprotective mechanism of sleep‐based therapy, we found a strong association between the development of ALS and sleep‐based therapy. Sleep‐based therapy is expected to be a novel treatment for ALS and other neurodegenerative diseases. In the future, more research is needed to prove the effectiveness of sleep‐based therapy for ALS and explore the advantages of sleep‐based therapy over existing treatments.
Footnotes
Conflict of interests
All contributing authors report no conflict of interests in this work.
Funding
This work is supported by the National Continuing Education Program of China (Grant No. 2021‐18‐01‐245).