
Abstract
N-methyl-D-aspartate receptors (NMDARs) are a family of ionotropic glutamate receptors mainly known to mediate excitatory synaptic transmission and plasticity. Interestingly, low-dose NMDAR antagonists lead to increased, instead of decreased, functional connectivity; and they could cause schizophrenia- and/or antidepressant-like behavior in both humans and rodents. In addition, human genetic evidences indicate that NMDAR loss of function mutations underlie certain forms of epilepsy, a disease featured with abnormal brain hyperactivity. Together, they all suggest that under certain conditions, NMDAR activation actually lead to inhibition, but not excitation, of the global neuronal network. Apparently, these phenomena are rather counterintuitive to the receptor's basic role in mediating excitatory synaptic transmission. How could it happen? Recently, this has become a crucial question in order to fully understand the complexity of NMDAR function, particularly in disease. Over the past decades, different theories have been proposed to address this question. These include theories of “NMDARs on inhibitory neurons are more sensitive to antagonism”, or “basal NMDAR activity actually inhibits excitatory synapse”, etc. Our review summarizes these efforts, and also provides an introduction of NMDARs, inhibitory neurons, and their relationships with the related diseases. Advances in the development of novel NMDAR pharmacological tools, particularly positive allosteric modulators, are also included to provide insights into potential intervention strategies.
Keywords
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. It acts via activating two types of receptors: metabotropic glutamate receptor (mGluR) and ionotropic glutamate receptor (iGluR). iGluRs include three types: a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors (AMPARs), N-methyl-D-aspartate receptors (NMDARs) and Kainate receptors (KARs) [1]. NMDARs are glutamate-gated cation channels permeable to sodium, calcium, and potassium. Importantly, the calcium influx through NMDARs serves as a trigger for several types of synaptic plasticity and is critical for many other NMDAR-evoked physiological and pathological signaling events [2]. NMDAR dysfunction contributes to various brain disorders, such as schizophrenia, depression and epilepsy, etc. [3]. However, the precise role of NMDARs in these diseases remains elusive.
NMDARs subunit composition and distribution
In the 1980s, based on patch-clamp studies of neuronal activity it was first proposed that NMDARs were composed of different subtypes [1, 3, 4]. Subsequent cloning and expression studies revealed that there are seven different subtypes, including GluN1, four GluN2 (A-D), and two GluN3 (A and B). NMDARs are heterotetramers assembled from two obligatory GluN1 subunits plus two GluN2 or GluN3 subunits. NMDARs containing GluN3 form either di-heteromeric (GluN1/GluN3) or tri-heteromeric (GluN1/GluN2/GluN3) complexes [1]. Among these subunits, GluN1 is encoded by one single gene with 8 different isoforms (GluN1-1a/2a/3a/4a and GluN1-1b/2b/3b/4b) produced by alternative mRNA splicing. The “b” isoforms possess an additional extracellular 21-amino-acid stretch encoded by exon 5. This additional fragment significantly affects gating and pharmacological properties of NMDARs [5]. GluN1 shows about 25%–28% homology to other iGluR subunits [6]. The four GluN2 subunits are encoded by four different genes sharing 18%–20% homology to GluN1 [7]. GluN3A and 3B are also encoded by two different genes. GluN3A shows a higher homology (57%) to GluN3B and a lower homology to GluN1 (27%) and GluN2 (24%–29%) [8]. GluN3B is only 17%–21% homologous to GluN1 and GluN2 [9].
Ligand-binding studies have revealed that NMDARs are distributed throughout the brain with a higher expression level in the forebrain. The distribution patterns of NMDAR subunits in brain have been described with molecular techniques [4, 10, 11]. Specifically, in adult rodents, GluN1 mRNA is expressed ubiquitously throughout the brain. Among eight GluN1 splice variants, GluN1-1(a and b) and GluN1-4 (a and b) show a complementary expression pattern, with the former being concentrated in the hippocampus and cortex whereas the latter primarily in caudal regions such as the thalamus and cerebellum. Notably, the regions expressing GluN1-a and GluN1-b variants are largely overlapping; however, their relative abundances vary across different regions. For instance, the GluN1-a isoform is expressed in all pyramidal neurons in hippocampus, whereas GluN1-b is mainly located in the CA3 region of hippocampus [4, 12].
Rodent data indicate that distributions of GluN2 subunits vary across different regions of the brain. GluN2A exhibits a widespread distribution throughout the adult brain, especially in the cortex, hippocampus, and cerebellum [13]. In contrast, the expression of GluN2B is relatively restricted in the forebrain and mostly distributed in cerebral cortex, hippocampus, olfactory bulb, and caudate putamen. GluN2C is largely restricted to cerebellum and olfactory bulb. GluN2D is weakly expressed and mostly distributed within the diencephalon and mesencephalon.
GluN3A and GluN3B are the two isoforms of the GluN3 subunit, exhibiting restricted spatio-temporal distributions. Previously, it was recognized that GluN3A is more widely expressed than GluN3B [9]. GluN3A exhibits high abundancy in olfactory bulb, cerebellum, CA1 region of the hippocampus, hypothalamus, nuclei of amygdala and certain parts of cortex [14, 15], while GluN3B expression is restricted to hippocampus, spinal cord, brain stem and cerebellum [16, 17]. GluN3B was also found to be expressed in the forebrain, including cerebral cortex, hippocampus, caudoputamen, and nucleus accumbens. [8, 18].
Switching of NMDAR expression pattern during development
The NMDAR subunits undergo changes in their expression patterns during development. Although GluN1 subunit is ubiquitously expressed since the early embryonic stage, its expression level varies. In situ hybridization experiments demonstrate that GluN1 expression starts at embryonic stage 14 (E14), rises to peak around the third postnatal week [19–21], and then decreases to a relatively stable level. All GluN1 isoforms follow the similar expression profile during development [22].
The GluN2 subunits impact functional heterogeneity of NMDARs, and show prominently different spatiotemporal expression profiles [13]. In the embryonic rodent brain, only GluN2B and GluN2D are expressed. Their expression starts as early as E14 and gradually increases with development. GluN2B expression reaches a relatively high level after birth and continues to increase until it peaks around P7-10. Within the first postnatal week, GluN2B becomes widely distributed in the brain, but after P7-10, its expression is restricted to forebrain, including cortex, hippocampus, striatum, and olfactory bulb. GluN2D, in contrast to the widely-distributed and highly-expressed GluN2B, exhibits a striking decrease in the expression after birth and in the adulthood. It only expresses at low levels in diencephalon and brainstem [4]. Both GluN2A and GluN2C start to express in hippocampus and cerebellum after birth. Whereas the expression of the GluN2A increases sharply during the first two postnatal weeks and gradually spreads out to the whole CNS in adulthood, the expression of GluN2C is mainly confined to cerebellum and olfactory bulb.
Among the four GluN2 subunits, GluN2A and GluN2B are the predominant subunits found in the cortex and the hippocampus. During the early developmental stages, GluN2B-containing NMDARs are the primary types at synapses. After birth, during the second postnatal week, the GluN2A subunits start to express and gradually replace GluN2B from the synapses [2, 4].
The GluN3A and GluN3B subunits also have distinct expression patterns. GluN3A starts expressing at around E15 within the spinal cord, tegmentum, hypothalamus, and thalamus. Its expression gradually increases and peaks at around P8, then rapidly declines to the adult level by P20 [14, 23]. GluN3B expression begins during early postnatal stages, peaks at around P14, and then stays plateaus throughout adulthood. GluN3B distribution was initially thought to be confined to brainstem and spinal cord, but it has been lately discovered to be ubiquitously distributed [14, 15, 18].
GABAergic inhibitory interneurons
Neuronal microcircuits are composed of both excitatory and inhibitory neurons. The fine balance of excitation and inhibition is critical for many brain functions. The vast majority of neurons in the cerebral cortex (about 80%) are excitatory glutamatergic neurons, also called principal neurons. These neurons have long axons and can project across different brain regions. Inhibitory neurons account for about 20% of cortical neurons. Compared with principal neurons, inhibitory neurons generally have smaller soma, shorter axons, and project locally; therefore, they are also called interneurons. Interneurons are GABAergic, releasing inhibitory neurotransmitter γ-aminobutyric acid (GABA). GABA acts on GABA-A type ionotropic receptors to mediate fast synaptic inhibition, generating hyperpolarization and, therefore, inhibition in the brain. It is the main source of inhibition in the mammalian brain.
Origin and classification of interneuron
Glutamatergic neurons are originated from the ventricular and sub-ventricular areas of the embryonic brain, while cortical GABAergic neurons are generated in medial ganglionic eminences (MGE) and caudal ganglionic eminences (CGE) in ventral telencephalon [24, 25]. Around E13.5, GABAergic neurons migrate tangentially to proper cortical regions crossing sub-ventricular zone (SVZ) and marginal zone (MZ), and then radially to the cortical plate (CP) [26, 27].
Cortical GABAergic inhibitory neurons exhibit tremendous diversity in morphology, electro-physiological properties, molecular expression profiles, and input and output connectivity [28–30]. Therefore, different criteria has been proposed to classify interneurons [31]. The most widely used classification is based on the expression of specific molecular markers, by which interneurons can be divided into three major types: calcium-binding protein parvalbumin (PV), neuropeptide somatostatin (Sst), or ionotropic serotonin receptor 5HT3a (5HT3aR) positive interneurons [32, 33]. PV interneurons are the most populous GABAergic interneurons in neocortex, accounting for ∼40%. Sst and 5HT3aR interneurons each account for ∼30% [33].
Interneuron function
GABAergic interneurons mainly provide inhibitory input to reduce excitability of principal neurons, which is critical for the maintenance of excitation-inhibition balance [34]. They could also inhibit other interneurons to cause disinhibition [35, 36]. For example, a subtype of vasoactiveintestinal peptide (VIP) interneurons mediates disinhibitory control in mouse auditory cortex (ACx) and medial prefrontal cortex (mPFC) by transiently inhibiting Sst and PV interneurons [37].
Interneurons also contribute to synchronized oscillations [38]. Neural oscillations are rhythmic or repetitive patterns of neural activity in the central nervous system, and are thought to play an important role in processing of neural information, such as perception, motor control and memory [39]. It has been well-established that interneurons are involved in generation and propagation of cortical oscillations [40]. The hypothesis that excitatory-inhibitory feedback loop was first proposed to explain the contribution of interneurons to spike timing during fast oscillations [41, 42], which indicates synchronizing may be partially dependent on the ability of GABAergic interneurons to entrain the firing of principal neurons [43]. Inhibitory loop was later proposed [42]. Interneurons innervate not only the principal neurons, but also themselves and other interneurons. Such interneurons self-innervation can increase the precision and regulate oscillations generation [44]. The propagation of hippocampal oscillation is found to be dependent on excitatory connection onto interneurons, rather than excitatory-excitatory connection [45, 46]. Gamma oscillations, a kind of oscillations in the brain, are thought to underlie cognitive and motor functions [45, 47]. Mounting evidence supports that gamma oscillations critically relies on entrainment by a phasic inhibitory drive [48, 49], and fast-spiking interneurons expressing PV are critical to gamma oscillations [41, 44, 50]. For example, in knock-out (KO) animals of calcium-binding protein parvalbumin (PV), the power of kainate-induced gamma oscillations is approximately twice as much as that in wild-type (WT), suggesting that PV interneuron contributes to gamma oscillations [32].
Interneuronal dysfunctions have been implicated in a number of brain diseases. Schizophrenia is a severe psychiatric disorder, and traditionally thought to be caused by hyperfunction of dopaminergic neurotransmission, which cannot explain the negative symptoms and cognitive deficits of schizophrenia [51]. Pharmacological and genetic evidences suggest that disrupted glutamatergic function may underlie schizophrenia [52–55]. And it was proposed that dysfunction of PV interneurons in cerebral cortex results in glutamatergic deficits as seen in schizophrenia [56–58]. Expression of interneurons marker proteins PV and glutamate decarboxylase 67 kDa (GAD 67) are decreased in the brain of schizophrenia patients [59, 60]. Bipolar disorder (BPD) is a mental disorder and is one of the leading cause of disability [61]. Postmortem studies have reported that the density of hippocampal interneurons is decreased in bipolar disorder patients [61, 62]. Reduced expression of Sst and VIP is also seen in dorsolateral prefrontal cortex [63]. Epilepsy is a group of neurological disorders and thought to be due to the unbalanced excitation-inhibition in the brain. GABAA receptors play critical roles in inhibitory neurotransmission. Clinical and experimental evidences suggest that perturbation of GABAA signaling contributes to epilepsy [64]. In addition, many GABAA receptor mutations are known to cause early-life epilepsy, including loss of function mutations or deletions of GABAA receptor subunit genes [65, 66]. Dysfunction of interneurons is also related to autism spectrum disorders (ASDs). ASDs are neurodevelopmental syndromes characterized by cognitive deficits, impaired language skill and social behavior [34]. Genetic evidence also links ASDs to genes encoding the β3, α5 and γ3 subunits of GABAA receptors [67, 68]. Cortical PV interneurons [69] and GAD67 are both decreased in ASDs [70, 71].
Why NMDAR blockade causes excitation instead of inhibition?
NMDARs are glutamate receptors mediating excitatory synaptic transmission. It is intuitive to predict that NMDAR blockade should inhibit neural circuit activity in the brain. Following this prediction, NMDAR antagonists, phencyclidine and ketamine, have been successfully utilized as anesthetics [72].
Interestingly, however, low doses of NMDAR inhibitors actually cause excitation instead of inhibition in the brain [73]. These observations contradict to the conventional knowledge about NMDAR function. Why does inhibition of an excitatory neurotransmitter receptor cause excitation instead of inhibition? The phenomenon has triggered profound interests ever since it was discovered, as the underlying mechanism might be critical in understanding the role of NMDARs in various brain disorders. Over the past decades, different mechanisms have been proposed to explain this paradox. For example, since both excitatory and inhibitory neurons receive excitatory inputs and have NMDARs at those synapses, one possibility is that NMDAR inhibitors at low doses preferentially act on interneurons in vivo [74], therefore they decrease interneuron activity and result in disinhibition of the neuronal network. An alternative explanation is that basal NMDAR activity suppresses neuronal activity in excitatory neurons; and when NMDAR inhibitors are applied, they alleviate this inhibition, therefore. causing disinhibition [75]. There are also other ways that NMDARs could impact inhibitory circuits. For example, recent studies have revealed that NMDARs on excitatory neurons are critical for the development of inhibitory synapses [76], and NMDARs at GABAergic synapses could impact GABAergic transmission [77, 78]. In addition, NMDARs are critical for the plasticity of GABAergic synapses onto excitatory neurons [79]. Each of these proposed mechanisms is discussed below.
NMDARs promote GABAergic synapse function and development in excitatory neurons to boost inhibition
Glutamate is the major excitatory neurotransmitter in mammalian central nervous system. During the fast synaptic transmission, glutamate activates AMPARs and NMDARs at the postsynaptic membrane. NMDARs are traditionally regarded as postsynaptic receptors; however, both anatomical and physiological evidences suggest existence of presynaptic NMDARs [80, 81]. While postsynaptic NMDARs mediate glutamatergic excitatory neurotransmission, presynaptic NMDARs modulate transmitter release in many brain regions, including cerebellum, entorhinal cortex, neocortex, and spinal cord [3], although the precise function of presynaptic NMDARs in excitatory synapses remains unclear. Interestingly, presynaptic NMDARs also exist at GABAergic presynaptic terminals in cerebellum and neocortex [77]. These presynaptic NMDARs modulate GABAergic synapses by increasing the size of GABAergic synaptic terminals and spontaneous GABA release [82] (Fig. 1, Mechanism B). Presynaptic NMDARs also facilitate GABAergic synaptic transmission in immature neocortex in a developmentally regulated manner, which involves GluN2B-containing NMDARs and impacts neocortical circuit development [78]. Activation of presynaptic NMDARs increases GABA release leading to an enhanced frequency of miniature inhibitory postsynaptic currents (mIPSCs) at cerebellar synapses between basket cells and purkinje cells [83–85].
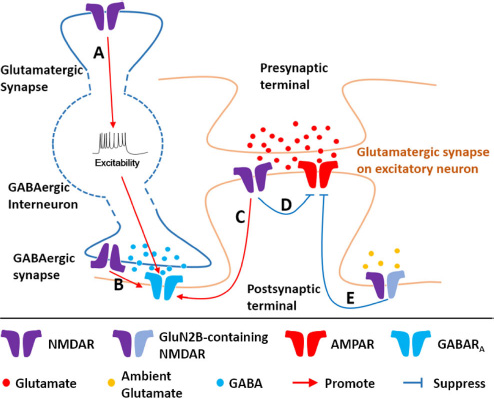
Proposed mechanisms of how NMDARs promote inhibition. Mechanism A: NMDARs at glutamatergic synapse on interneuron promote interneuron excitability. Mechanism B: NMDARs at presynaptic terminal of GABAergic synapse promote development/function of GABAergic synapse. Mechanism C: NMDARs on excitatory neurons promote development of GABAergic synapse or inhibitory long-term potentiation. Mechanism D: Synaptic NMDARs suppress excitatory synaptic transmission. Mechanism E: Ambient glutamate-activated extrasynaptic GluN2B-containing NMDARs suppress excitatory synaptic transmission. GABARA—GABAA receptor.
NMDARs play an important role in mediating plasticity at excitatory synapses. Interestingly, NMDARs are also critical to the plasticity of GABAergic synapses (Fig. 1, Mechanism C). GABAergic synapses can undergo inhibitory long-term potentiation (iLTP) or inhibitory long-term depression (iLTD), depending on the expression and function of GABAA receptors or the GABA release from presynaptic terminal[86]. iLTP or iLTD that relies on postsynaptic NMDARs has been reported in many brain regions, such as hippocampus [87], visual cortex and ventral tegment [88]. In visual cortex, presynaptic NMDARs have been shown to contribute to iLTD in developing optic tectum [79].
NMDARs also play a critical role in the regulation of GABAergic synapse development (Fig. 1, Mechanism C). Indeed, in the superior colliculus activation of NMDARs has been shown to stimulate GABAergic synapse development [89]. In addition, overexpression of a constitutively active NMDAR subunit increases GABAergic transmission in developing hippocampal neurons, indicating a cell-autonomous role of NMDARs in promoting development of GABAergic synapses [76]. On the other hand, genetic deletion of NMDARs in different types of neurons leads to a strong reduction of GABAergic transmission as well as inhibitory synapse density [76, 90], suggesting a necessary role of NMDARs in GABAergic synaptogenesis. Mechanistically, the regulation of formation of GABAergic connections by NMDARs in hippocampal neurons is dependent on the C0 domain of the GluN1 subunit and requires Ca2+-dependent calmodulin binding to the C0 domain [76], showing that the NMDAR acts a key signaling molecule for controlling inhibitory circuit wiring. Consistent with the functional data, a fraction of NMDARs has been found to co-localize with GABAA receptors at GABAergic synapses in developing brain [91–93], providing anatomic evidence for a role of NMDARs in GABAergic synapse development.
Another explanation to the question “Why NMDAR blockade causes excitation?” is that, at low doses, NMDAR antagonists preferentially act on NMDARs on interneurons instead of those on excitatory neurons (Fig. 1, Mechanism A), therefore reducing interneuron activity and leading to a disinhibition effect on the overall network activity. This proposal is supported by the observation that MK-801 treatment leads to reduced activity of GABAergic interneurons and subsequent increased activity of excitatory principal neurons in rodent prefrontal cortex [73, 94]. It has also been reported that hippocampal GABAergic interneurons are more sensitive to NMDAR antagonists than excitatory neurons [95, 96]. One possible mechanism underlying this preferential effect of NMDAR antagonists on interneurons is that interneurons have generally higher frequency of firing compared with pyramidal neurons, which causes a higher NMDAR baseline activity through the depolarization-dependent removal of Mg2+ block. Therefore, more “alert” NMDARs may be more sensitive to antagonist blockade [97–99].
In addition, NMDARs ablation in interneurons has a comparable effect with NMDAR antagonists. Conditional knockout of GluN1 in inter-neurons not only leads to cortical disinhibition but also the schizophrenia-like behaviors, which display novelty-induced hyperlocomotion, deficits in nesting and short-term social memory [56, 100, 101]. These data also support the hypothesis that NMDAR loss-of-function in inter-neurons is important for NMDAR antagonist-induced cortical excitation and psychosis-like behaviors in rodents [102–105].
Basal NMDAR activity suppresses excitatory synaptic transmission
NMDARs are known to cause depolarization and contribute to excitatory synaptic transmission. However, there is also evidence suggesting that basal NMDAR activity suppresses excitatory transmission [75] (Fig. 1, Mechanism D). In this case, NMDAR blockade leads to attenuation of suppression on excitatory synaptic transmission, therefore promoting excitation and global neuronal activity.
In dissociated neuronal cultures, chronic (>12 hours) blockade of action potentials (APs) by tetrodotoxin (TTX) potentiates excitatory synaptic transmission by increasing membrane AMPAR insertion. This is called homeostatic scaling [106]. Interestingly, NMDAR selective antagonist AP5 increases mEPSC amplitude quickly (after one hour) in the presence of TTX. In addition, AP5 alone can also increase mEPSC amplitude after 3 hours treatment in the absence of TTX [75]. Increased miniature excitatory postsynaptic current (mEPSC) amplitude is found to be consistent with increased surface AMPAR insertion; and this increase depends on local protein synthesis [75]. Taking all these data into consideration, it is logical to propose that basal NMDAR activity suppresses excitatory transmission in principal neurons.
In addition, other studies have also found that blockade of NMDARs rapidly potentiates AMPAR-mediated synaptic transmission [107, 108]. In acute hippocampal slices, 30 minutes incubation of another NMDAR antagonist ketamine (20 μM) is sufficient to enhance AMPAR-mediated evoked field excitatory postsynaptic potential (fEPSPs), which sustains even after ketamine wash off [108]. The potentiation is not linked to increased presynaptic release probability because of the unaltered paired-pulse facilitation. A different NMDAR antagonist MK-801 has similar effect as ketamine, supporting that blockade of NMDAR is the underlying mechanism of the ketamine-induced synaptic potentiation [108]. NMDAR blockade-mediated synaptic potentiation requires the deactivation of eukaryotic elongation factor 2 (eEF2) kinase (also called CaMKIII), which triggers protein synthesis of brain-derived neurotrophic factor (BDNF) and increase of surface AMPAR insertion [107, 108]. These suggest that basal NMDAR activity indeed suppresses AMPAR-mediated synaptic transmission, and alleviation of which is sufficient to boost synaptic transmission in a fast and sustainable manner.
Genetic studies have also indicated that NMDARs suppress functional maturation of AMPAR-mediated synaptic transmission. In pyramidal neurons in hippocampus and cerebral cortex, genetic deletion of NMDARs causes a strong increase of AMPA EPSCs [109–113]. Similarly, knockout of the NMDAR GluN1 subunit in midbrain dopaminergic neurons significantly increases AMPAR-mediated transmission [114, 115]. A recent study on hippocampal neurogliaform cells has also demonstrated that NMDARs inhibit excitatory synaptic transmission [116]. Together, these studies show that NMDARs suppress the maturation of AMPAR-mediated synaptic transmission and maintain the silent status of developing glutamatergic synapses [110].
NMDAR dysfunction and diseases
Abnormal NMDAR activity underlies many brain disorders including psychosis, mood disorder, epilepsy and neurodegenerative diseases, etc. [4, 117, 118]. This part of the review will discuss the role of NMDAR in schizophrenia, depression and epilepsy. These three diseases are chosen because both NMDAR dysfunction and abnormalities in inhibition have been strongly implicated in their etiology.
NMDAR, schizophrenia, and anti-NMDAR encephalitis
Schizophrenia is a devastating mental disorder characterized by abnormal social behavior and failure to understand reality. It affects about 1% of the world's population [119]. Its clinical symptoms are summarized into three major categories: positive symptoms, negative symptoms and cognitive deficits. Positive symptoms include delusions, hallucinations, disordered thoughts, and speech. Negative symptoms are deficits of normal emotional response, including lack of motivation, inability to experience pleasure and poverty of speech. Cognitive deficits are also recognized as a core feature of schizophrenia, including dysfunction of work memory, long-term memory, attention, learning, etc. [120, 121].
Studies of schizophrenia pathology have mainly been focused on dopamine hyperfunction theory. This is supported by the finding that the affinity of dopamine D2 receptor antagonist correlates with their anti-psychotic potency [122, 123]. However, this hyperdopaminergic hypothesis can only be best used to explain positive symptoms of schizophrenia. Although the most common antipsychotic drugs nowadays are D2 dopamine receptor antagonists, they are only effective in treating positive symptoms, but not negative symptoms or cognitive deficits [124]. Therefore, it has been a hot topic to search for other hypothesis that could explain all symptoms of schizophrenia.
NMDAR antagonists can induce a psychotic state that resembles all three clusters of schizophrenic symptoms in healthy people and also may exacerbate the symptoms in schizophrenia patients [125, 126]. Reduced NMDAR activity caused by its antagonists results in schizophrenialike behaviors, which suggests that hypofunction of NMDARs might be one of the causes of schizophrenia, leading to the proposal of the NMDAR hypofunction hypothesis for schizophrenia [127, 128]. As NMDARs are expressed in both excitatory glutamatergic and inhibitory GABAergic neurons, it remains largely unclear whether the NMDAR hypofunction in excitatory neurons, inhibitory neurons, or both types of neurons cause schizophrenia.
Hypofunction of inhibitory neurotransmission has been implicated in schizophrenia pathology. Expression of key GABA signaling components is reduced in post-mortem brain of schizophrenia patients including PV and GAD67 [56,129–131]. Deletion of the GluN1 subunit in PV interneurons in mouse mimics some key behavioral features of schizophrenia [56, 100], and these mutant mice are more susceptible to MK-801 induced schizophrenia-like deficits [101]. This supports the idea that NMDAR hypofunction in PV interneurons contributes to schizophrenia.
Another evidence that supports NMDARs hypofunction hypothesis for schizophrenia comes from patients with anti-NMDARs encephalitis. Anti-NMDAR encephalitis is an autoimmune disorder featured with psychosis. This disease is caused by auto-antibodies targeting NMDARs, which could pass through blood-brain barrier (BBB) and lead to removal of NMDARs from synapses [132, 133]. Anti-NMDARs encephalitis has two major triggers, virus and tumors [134]. About 20% of patients infected with herpes simplex encephalitis (HSE) develop antibodies against NMDAR [135, 136]. Approximately 50% of young women with ovarian teratoma develop anti-NMDAR encephalitis. In addition, anti-NMDARs encephalitis patients usually benefit from removal of NMDARs as antigens in peripheral [132, 133]. This line of evidence also suggests that NMDAR loss-of-function contributes to psychosis and strongly supports the NMDA hypofunction hypothesis.
There is also human genetic evidence supporting NMDAR hypofunction hypothesis [137]. A recent genome-wide association study (GWAS) has linked NMDAR gene variances to risk in schizophrenia. Exome sequencing has identified de novo mutations of GluN2A and GluN2B in schizophrenia patients [138–140]. These schizophrenia-related mutations tend to be located at amino and carboxyl-termini [138, 139]. The functional impacts of these variances, although remain mostly unclear, will be critical for dissecting the role of NMDARs in schizophrenia.
NMDAR and depression
Depression is a widespread mental disorder, affecting more than 300 million people worldwide (WHO). It is also the major cause of suicide in adults. The most popular antidepressants are serotonin (5-HT) or norepinephrine (NE) reuptake inhibitors [141–143]. Although these drugs have benefited many, some obvious disadvantages remain. First, the antidepressant effects can only be observed after a few weeks of treatment [144, 145]. This suggests that 5-HT and NE system are unlikely the direct cause of depression. Second, about 30% patients are resistant to these drugs for reasons unknown [146, 147]. Third, selective serotonin reuptake inhibitors [148] antidepressants may increase the risk of suicide [148, 149]. These caveats make current antidepressants unsatisfying.
Recent clinical studies have shown that a low sub-anesthetic dose of NMDAR antagonist ketamine can generate quick and sustainable anti-depressant effects on treatment-resistant depression patients [150–152]. In fact, it was first reported back in 1990 that NMDAR antagonists including competitive AP-7 and non-competitive MK-801 exhibited antidepressant actions in mice [153]. Scientists then proposed that direct NMDAR inhibition might lead to rapid anti-depression actions [153]. This breakthrough suggests a new mechanism of action for fast antidepressants, and indicates that the glutamatergic system malfunction may underlie depression. However, higher dose ketamine is psychogenic and could be abused for entertainment usage causing severe side effects [154]. Therefore, it would be extremely beneficial to understand the mechanism of ketamine antidepressant effect and therefore to instruct development of better antidepressant with fast action and less side effects.
Ketamine is a non-selective NMDAR antagonist, but whether its rapid antidepressant effect depends on NMDARs remains controversial. Zanos and coworkers [155] have indicated that a ketamine metabolite (2R,6R)-HNK (hydroxynorketamine) is essential to produce antidepressant effect in mice by increasing AMPAR-mediated excitatory post-synaptic potentials [156] in CA1 region, independent to its actions on NMDARs. However, Suzuki et al. [157] has reported that slightly higher concentration of (2R,6R)-HNK can significantly impair NMDAR function at rest in hippocampal slices and deactivates eEF2K, eliciting AMPAR-dependent synaptic potentiation to produce the anti-depression effect. Meanwhile, NMDAR-dependent lateral habenula (LHb) burst firing increases significantly in brain slice of depressed rats, and ketamine inhibits bursting in LHb to rapidly relieve depressed behaviors in animal models in an NMDAR-dependent manner [158].
NMDARs are mainly known to mediate excitatory synaptic transmission. However ketamine at sub-anesthetic doses significantly increases extracellular level of glutamate in the PFC in rats [94, 159], and can increase focal prefrontal activity in healthy volunteers [94, 159]. These findings are rather surprising, because one would expect that blockers of excitatory transmission should decrease excitability. These new evidences indicate that low dose ketamine may preferentially suppress NMDARs on inhibitory neurons to reduce principal neurons inhibition from inhibitory neurons and indirectly enhance overall neuronal activity to produce antidepressant effects [94, 99]. However, other studies have proposed that GABA receptor hypofunction may lead to depressive disorders, and enhancing GABAergic neurotransmission can induce antidepressant-like behaviors in rodents [160, 161]. Therefore, the NMDAR dysfunction hypothesis for depression requires further investigations [162].
Different subunits of NMDARs have different physiological and pathological functions. Which NMDAR subtype(s) plays the key role in the ketamine-evoked antidepressant actions is an interesting question. Hypermethylation of the GRIN2A gene has been observed by epigenomewide methylation analysis in human brain specimens [163]. Decreased GluN2A expression level in the prefrontal cortex, but increased in the lateral amygdala of majority depressed patients have been reported [164]. In animal models, the homozygous knock-in mice with a Tyr-1325-Phe mutation in GluN2A to prevent its phosphorylation/activation show antidepressantlike behaviors [165]. GluN2A KO mice also have anxiolytic and antidepressant-like behaviors. However, locomotion of GluN2A KO mice is also enhanced significantly. These observations complicate the interpretation of the role of NMDARs in the regulation of anxiolytic and antidepressantlike behaviors [166]. The administration of selective GluN2A antagonist NVP-AAM077 can elevate the release of glutamate and 5-HT, and rescue depressant-like activity in the forced-swim test [167]. These results support that GluN2A plays an important role in the regulation and treatment of depression. However, poor subtype selectivity of NVP-AAM077 makes it difficult to rule out the potential involvement of GluN2B.
GluN2B can also contribute to ketamine-induced fast antidepressant effect. GluN2B-containing NMDARs are activated under non-stimulated conditions by ambient glutamate and suppress excitatory transmission [168] (Fig. 1, Mechanism E), and its inhibition would promote protein synthesis and trigger antidepressant actions in an mTOR (the mammalian target of rapamycin)-dependent manner. Importantly, ketamine loses its efficacy in inducing anti-depressant-like behaviors in mice with GluN2B knockout in cortical principal neurons [168]. This suggests that GluN2B is necessary for ketamine-induced antidepressant effect. Meanwhile, GluN2B antagonists can also rapidly reverse the depression-like behavioral deficits in chronic unpredictable stress model. The rescue result is long-lasting, similar to the action of ketamine [169]. In addition, fluoxetine, serotonin reuptake inhibitor class of antidepressant, can selectively inhibit GluN2B-containing NMDARs [170]. A recent clinical study has shown that GluN2B selective antagonist CP-101,606 can significantly improve the patients’ mental status, although with a much slower onset after administration compared with ketamine [171]. These data support that GluN2B-containing NMDARs play a role in ketamine-induced antidepressant effect.
NMDAR and epilepsy
Epilepsy is a chronic and recurrent disorder caused by abnormal electrical activity in the brain. Epilepsy affects about 50 million people worldwide (WHO). The imbalance of inhibitory and excitatory neurotransmission [172, 173] plays a pivotal role in the pathogenesis of epilepsy. Abnormal GABAergic function is believed to be the key factor underlying epilepsy. Patients with temporal lobe epilepsy have a region-specific decrease of GABAA receptors [174], reduced hippocampal somatostatin and neuropeptide Y interneurons [175] and altered excitatory and inhibitory neurotransmission [176]. In animal models, GABA-immunoreactive cell density and the number of GABA-positive terminals are reduced during motor focal epilepsy in rats. Grafting of GABAergic progenitors decreases seizure activity and abnormal behaviors in mice [177]. Current GABA-based anti-epileptic drugs work well for many epilepsy patients. However, a significant portion of epilepsy patients are resistant to these treatments [178], suggesting that alternative mechanisms could exist in these treatment-resistant epilepsy patients.
Recently, NMDARs have drawn attention in the pathophysiology of epilepsy. The evidence comes from human genetic studies. Particularly, GRIN2A missense mutations cause epileptic aphasia and idiopathic focal epilepsy [117, 118, 179]. In addition, GluN2 mRNA increases in the hippocampus of temporal lobe epilepsy patients [180]. Blockade of GluN2C overexpression can rescue epileptogenesis in a tuberous sclerosis murine model [181]. These strongly support a critical role of NMDAR in these two forms of epilepsy and raise the possibility of a potential role of NMDAR in epilepsy in general.
The underlying mechanism however remains debatable. A key question is whether it is gain-of-function or loss-of-function of NMDARs that causes the disease. Interestingly, similar disease symptoms can be triggered by both increased and decreased NMDAR function. The missense mutations in GRIN2A, such as A243V [118], P552R [182], N615K [183], and L812M [184], enhance NMDAR function and cause neuronal hyper-excitation. However, other GluN2A variants I184S and R518H are rather loss-of-function mutants by reducing surface expression, current amplitude and opening time, while also increasing deactivation time [185]. Mutations P79R, C231Y, G483R and M705V also reduce the response to the co-agonists [186]. It is unclear why both gain-and loss-of-function mutations of NMDARs could cause epilepsy.
Pharmacological tools for NMDARs
NMDARs are widely distributed in the central nervous system with many critical physiological functions. Their hypo- or hyper-activation are associated with various brain diseases. Properly designed pharmacological tools can help reveal the NMDAR function and the associated mechanisms of related diseases, and will be potentially useful for clinical intervention. A few mostly used or newly developed compounds are discussed below. Structures of the mentioned compounds are summarized in Fig. 2.
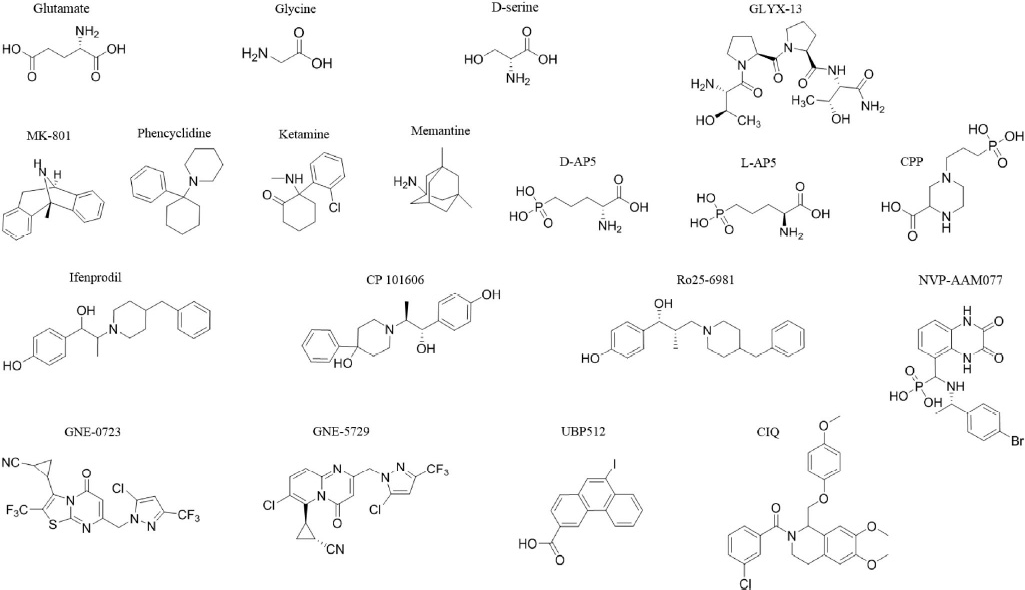
Structures of selected pharmacological tools targeting NMDAR. Row 1: agonists and co-agonists; Row 2: antagonists; Row 3: GluN2A and 2B subtype selective antagonists: Row 4: PAMs.
The activation of NMDA receptors requires not only glutamate binding to GluN2 but also a co-agonist (glycine or D-serine) binding to GluN1 [187, 188]. Agonists acting on the glutamate binding site of NMDARs cause excitotoxicity, therefore not useful to study NMDAR physiological function nor to treat patients [119, 189, 190]. Glycine and D-serine are endogenous co-agonists. They can increase the affinity of glutamate binding on GluN2 subunit [191] and might be useful to treat diseases caused by NMDAR hypofunction, such as schizophrenia [192]. D-serine acts on synaptic NMDARs, while glycine affects extrasynaptic receptors[193]. Synaptic NMDARs play an important role in long-term potentiation, while long-term depression requires both synaptic and extrasynaptic receptors [194].
Compared to agonists at the glutamate site, glycine site co-agonists are safer and do not usually cause excitotoxicity. GLYX-13, which acts at the glycine site, shows fast antidepressant effects and has no psychotomimetic effects [195, 196].
Glycine transporter (GlyT) is responsible to reuptake extracellular glycine and can play a critical role in regulating NMDAR signaling. GlyT has two distinct subtypes, GlyT-1 and GlyT-2. They are sodium-dependent transporters and can maintain local glycine concentrations at sub-saturation levels [197]. Studies on the GlyT-1 knockout mice have shown that GlyT-1 enhances hippocampal NMDAR function and cognition [198]. D-serine and GlyT-1 inhibitors can reverse pre-pulse inhibition [152] and latent inhibition (LI) in schizophrenia animal models [199, 200]. D'Souza et al. [201] have demonstrated that GlyT-1 inhibitor has the antipsychotic potential. However, GlyT-1 inhibitor phase 3 clinical trials on treating schizophrenia failed for lack of efficacy [202].
Antagonists
Excitotoxicity-induced neuronal death is mediated by hyperactivation of iGluRs, particularly NMDARs. It contributes to neuronal damage in many neurological disorders including stroke, traumatic brain injury, epilepsy, and neurodegenerative diseases. NMDAR antagonists could be useful in blocking excitotoxicity and neuronal damage.
MK-801, phencyclidine and ketamine are the non-competitive open channel blockers with limited subunit selectivity [203]. Phencyclidine was introduced as an anesthetic in 1957 but it caused a high rate of acute psychosis reactions and thus has never been approved for clinic application [204]. To reduce the side-effects, its analogue ketamine was developed. Ketamine has been used as the anesthetic for around 50 years and low dose ketamine has rapid and sustained anti-depressant effects in patients with treatment-resistant depression. MK-801, phencyclidine and ketamine have been used to induce animal models of schizophrenia [205, 206]. Low affinity channel blocker memantine has also been used to treat moderate to severe Alzheimer's disease [207].
2-amino-5-phosphonovaleric acid (AP5) and 4-(3-phosphonopropyl) piperazine-2-carboxylic acid (CPP) are common NMDAR competitive antagonists. AP5 is racemic mixture of D- and L-isomers. The D-isomer has higher potency and efficacy [208]. AP5 has been used to study NMDAR-dependent physiology and pathology, including epilepsy and memory impairments in the mammals [209–211], but its penetration of the blood-brain barrier is poor [212]. The CPP, an analogue of AP5, has the higher affinity for [3H]D-AP5 binding sites and is regarded as a potential anticonvulsant [213]. However, its clinical trials have failed [214, 215], as it can impair memory in healthy human [216].
Different NMDAR subtypes have different, sometime opposite physiological functions; therefore subunit-selective antagonists are also developed. The first subunit-selective NMDAR antagonist is GluN2B-selective antagonist ifenprodil. Ifenprodil and its derivatives bind to GluN2B amino terminal domain (ATD) [217, 218]. Ifenprodil has antiparkinsonian effects in the MPTP-Lesioned marmoset model [219]. CP101,606 and Ro25-6981 also are GluN2B selective antagonists [220, 221]. Remarkably, GluN2B-selective antagonists do not induce side effects often triggered by non-selective NMDAR antagonists in humans [222]. NVP-AAM077 preferentially acts on human GluN2A over GluN2B [223] and has been used to study GluN2A [224, 225]. However, selectivity of NVP-AAM077 is poor on rodent GluN2A, which limits its application in research [226]. GluN2A selective antagonists for animal studies will be very useful to demonstrate the role of GluN2A containing NMDARs.
Positive allosteric modulators
Another novel pharmacological tool to enhance NMDAR activity is positive allosteric modulator (PAM). This family of compounds act at distinct sites as compared with agonists and do not directly activate the receptors. Instead, they enhance the receptor response to its agonists. In theory, they have several distinct advantages over agonists. PAMs usually display better subtype selectivity, because they could act on the sites that are less homologous compared with generally more conserved agonist binding pockets among different subtypes [227]. Also, PAM does not directly activate NMDARs, therefore may not cause excitotoxicity as agonists. In addition, PAM's potentiation depends on the endogenous agonists, therefore enhances NMDAR activity in a more physiologically relevant manner. This could be particularly important in maintaining endogenous neuronal activity pattern.
Hackos and colleagues published a family of orally available GluN2A selective PAMs using cell based high throughput screening of random chemical library and following lead optimization [228–231]. These compounds potentiate GluN2A-containing NMDAR response to their co-agonists in both cellular and brain slice assays with up to 20 nanomolar potency, with minimum activity on GluN2B and GluN2C. They bind to the GluN1-GluN2A dimer interface of the extracellular ligand-binding domains (LBD) and can enhance NMDAR-dependent synaptic plasticity [228]. These compounds provide promising tools to study GluN2A function.
Perszyk and colleagues reported a series of subtype nonselective PAMs, which were converted from NAMs (negative allosteric modulator) by subtle structural modifications. The effects of these compounds depend on the subtype of NMDARs and agonists concentration. Interestingly, these PAMs do not act at extracellular ATD of the receptor, suggesting multiple allosteric modification sites exist on NMDARs [232].
Some derivatives of carboxylated naphthalene and phenanthrene can also modulate NMDAR activity [233]. For example, UBP710 preferentially potentiates GluN2A and GluN2B. It also acts on GluN2C and GluN2D, but with a weaker efficacy at lower doses and leads to 2C and 2D inhibition at a higher dose. UBP608 inhibits all GluN2 subtypes. Whereas, UBP512 action is dependent on its dose. It potentiates GluN2A and inhibits GluN2C and 2D at high doses, but weakly inhibits GluN2A and 2B at lower doses. ATD is unnecessary for the action of all these compounds. In addition, S2 domain is important for UBP512 and UBP710, and S1 domain is vital for UBP608-induced inhibition.
Meanwhile, (3-chlorophenyl)(6,7-dimethoxy-1-((4-methoxyphenoxy)methyl)-3, 4-dihydroisoquinolin-2(1H)-yl)methanone (CIQ) can enhance activity of GluN2C/2D by increasing their channel open probability without affecting the open state, and its action requires Thr592 in the M1 region and the linker between the ATD and LBD [234].
NMDAR dysfunction or loss-of-function may be the cause underlying several brain diseases, such as schizophrenia and epilepsy, etc. Pharmacological tools enhancing NMDAR activity will be very useful to compensate the compromised NMDAR activity in these diseases and provide benefits as a novel treatment. However, this kind of drugs had not been available for a long time. As discussed above, NMDAR PAMs represent a new hope to solve this issue, which could serve as an unprecedented opportunity to treat these brain diseases in the future. However, most these compounds are still at their early stage of development. Although a few families of NMDAR PAMs have been developed to enhance NMDAR endogenous activity without evoking excitotoxicity, their behavioral benefits have remained to be addressed.
Summary
Why do NMDAR antagonists cause excitation instead of inhibition in the brain under certain conditions? This represents one of the most critical questions concerning NMDAR function. Its answer would be essential to address etiology of many related brain diseases, including psychosis, depression and certain forms of epilepsy. Our review summarizes recent explanations for this question. However, limited by available tools, the answers remain controversial and largely unclear. New emerging research tools, such as novel subtype selective pharmacological reagents (particularly gain of function tools like PAMs) together with new genetic models would be very useful to clarify this important question in the near future.
Footnotes
Acknowledgements
This work is sponsored by Shanghai Science and Technology Committee (No. 17DZ1205402).
All contributing authors have no conflict of interests.