
Abstract
Introducción
La familia Calliphoridae se considera de importancia en entomología forense debido al comportamiento necrófago de sus especies, las cuales se desarrollan a diferentes tasas de crecimiento sobre cuerpos en descomposición (Anderson 2000). Esto permite utilizar algunas de sus especies como indicadoras del tiempo de muerte y como herramienta para calcular el intervalo post-mortem en casos legales (Marchenko 2001).
En Colombia se han registrado 12 géneros y 29 especies de califóridos, de las cuales por lo menos 13 son de importancia forense (Amat et al. 2008; Amat 2009; Florez y Wolff 2009). De particular interés se considera el género
Aunque se ha avanzado grandemente en la taxonomía del género y en precisar aspectos como su presencia y distribución en el país, la identificación taxonómica de sus especies con base en caracteres morfológicos es difícil. Esta situación es cierta en el caso de los estadios inmaduros debido a la similitud de las características anatómicas. La identificación de éstos con base en herramientas como las moleculares es relevante, dado que son éstos, los estados que predominan en escenas de muerte (Wallman y Donellan 2001; Wells et al. 2007).
Las especies del género
Entre las metodologías más utilizadas actualmente para complementar estudios de taxonomía clásica, se encuentran aquellas de tipificación molecular a partir de secuencias de ADN. En el caso de los insectos y en particular de aquellos de importancia forense, se considera ventajoso el hecho de que las moléculas posibilitan la identificación de especímenes en cualquiera de sus estados, e incluso de partes de ellos (Benecke 1998). Esto es relevante en entomología forense, no solo para corroborar la identidad de las especies, sino también por la información que puede obtenerse aun en aquellos casos en los cuales los insectos se encuentran muertos, o afectados por los procesos de depredación propios de la sucesión y en los que el ADN es la única fuente disponible para su identificación (Sperling et al. 1994).
En este sentido, el ADN mitocondrial ha sido ampliamente sugerido y utilizado como marcador molecular; su condición haploide, herencia matrilineal, altas tasas de evolución y la presencia de regiones conservadas y variables, facilitan la tipificación molecular de individuos y su uso para la diferenciación de los mismos a diferentes escalas taxonómicas, ventajas que han sido aprovechadas en estudios con diversos grupos de insectos, incluyendo aquellos de importancia médica y forense (Sperling et al. 1994; Behura 2006).
Actualmente se han estudiado alrededor de 12 genes mitocondriales para caracterizar insectos de importancia forense (Lessinger y Azeredo-Ezpin 2000), siendo más utilizados los genes que codifican para Citocromo Oxidasa uno y dos (COI y COII) y validados como marcadores consistentes para la diferenciación a nivel de género y entre grupos monofiléticos de la familia Calliphoridae (Nelson et al. 2007; Wells y Williams 2007; Wells et al. 2007). Sin embargo, en este último se han encontrado bajos niveles de divergencia entre especies hermanas particularmente en los géneros
El gen Citocromo b (Cytb) está involucrado en el transporte de electrones en la cadena respiratoria de la mitocondria. La variabilidad exhibida por el gen a diferentes niveles taxonómicos y su uso exitoso como marcador molecular en conjunto con fragmentos de genes mitocondriales vecinos, lo ha convertido en uno de los más usados para análisis filogenéticos (Irwin et al. 1991). Así mismo, éste ha mostrado su utilidad en el estudio de complejos de especie, especies morfológicamente similares e incluso al interior de especies (Meyer 1994). Aunque el Cytb ha sido principalmente usado para estudiar mamíferos, también se conocen estudios de filogenia y sistemática de insectos, y en especial del orden Diptera (Simmons y Weller 2001; Torgerson et al. 2003).
El objetivo del presente trabajo fue estudiar la variabilidad molecular de un segmento de ADN mitocondrial amplificado mediante PCR, que corresponde a un segmento de la región 3' del gen Citocromo b (Cytb), el RNA de transferencia para Serina (ARNt-Ser), el espaciador intergénico (IG2) y un segmento de la región 3' del gen NADH deshidrogenasa 1 (ND1) en
Esta especie de distribución Neotropical, se ha registrado en Colombia en los departamentos de Antioquia, Bolívar, Caquetá, Cundinamarca, Chocó y Santander (Vélez y Wolff 2008), en asocio con estados tempranos de descomposición cadavérica. Estas características la convierten en una especie con gran potencial para ser utilizada en investigaciones legales y en la determinación de intervalos postmortem.
Materiales y Métodos
Obtención de especímenes
Para la obtención de las larvas se realizaron cuatro muestreos con intervalos de dos meses entre Enero y Agosto de 2006, en tres municipios del departamento de Antioquia (Tabla 1). La frecuencia de muestreo se diseñó con el fin de obtener individuos de diferentes cohortes en las poblaciones seleccionadas.
Datos de referencia de los individuos secuenciados
Se utilizó una trampa Van-Someren–Rydon cebada con hígado fresco de cerdo para colectar adultos y huevos de acuerdo a la metodología modificada de Stevens y Wall (1997). Posteriormente se establecieron crías de laboratorio con el fin de obtener adultos para la determinación taxonómica de la especie y larvas para la tipificación molecular. La determinación taxonómica de la especie fue realizada de acuerdo con el trabajo de Florez y Wolff (2009) y verificada por el especialista Eduardo Amat del Instituto de Investigación de Recursos Biológicos - Alexander Von Humboldt (Villa de Leyva, Colombia), siguiendo la propuesta de Whitworth (2010) para individuos adultos.
Como punto de comparación y para evaluar el uso potencial de la secuencias en separar los individuos de otras especies morfológicamente similares y cercanas, se utilizó una secuencia de referencia depositada en genbank correspondiente a
Análisis molecular
Preparación de especímenes
Las secuencias fueron obtenidas a partir de la extracción y amplificación de ADN de larvas en tercer estadío obtenidas a partir de hembras paridas en el laboratorio. Como paso previo al procedimiento de extracción de ADN, cada individuo fue tratado con agua caliente por 10 segundos y posteriormente disectado con el fin de conservar las estructuras consideradas de importancia en la determinación taxonómica como son el esqueleto cefalofaríngeo y el segmento terminal donde se encuentran las placas espiraculares. Estas estructuras, fueron aclaradas en KOH al 10% según la metodología de Stehr (1987) y posteriormente fijadas en placa con bálsamo de Canadá. El resto fue preservado en isopropanol al 100% para la extracción de ADN.
Extracción de ADN
La extracción de ADN se realizó a partir de fragmentos de 4 mm de larvas, siguiendo la metodología de buffer de macerado (Collins y Porter 1990) que ha sido utilizado con modificaciones menores en otros insectos del orden Diptera (Uribe et al. 1998; Uribe 1999). La extracción del ADN y su calidad fueron confirmadas por medio de electroforesis en gel de agarosa y la concentración se estimó utilizando un nanodrop 1000 de Thermo Scientific ®.
Amplificación del ADN
La amplificación se efectuó mediante la reacción en cadena de la polimerasa (PCR) utilizando un termociclador (PTC 100 MJ Researcher). Los oligonucleótidos utilizados para amplificar los fragmentos de los genes fueron CB3FC (CA(T/C)ATTCAACC(A/T)GAATGATA) y NINFR (GGTA(C/T)(A/T)TTGCCTCGA(T/A) TTCG(T/A)TATGA) .
La reacción de PCR se llevó a cabo empleado entre 0,5 y 1,0 mL de ADN al cual se le adicionaron 4mL de MgCl2 (25mM), 0,8 mL (1mM) de cada deoxinucleótido (Adenina, Timina, Citosina y Timina), 0,4 mL de cada primer (50 mM), y 5mL de Buffer 10X (Fermentas), ajustados con agua bidestilada para un volumen final de 50 mL. Después de la fase de desnaturalización del ADN a 94°C durante tres minutos, los parámetros de amplificación para los siguientes 35 ciclos de la reacción de PCR fueron: desnaturalización por 30 segundos a 93°C, alineamiento por un minuto a 48°C y extensión por un minuto a 70°C. Los productos amplificados fueron verificados mediante electroforesis en gel de agarosa al 1% con bromuro de etidio y empleando un marcador de peso molecular de 100 pb. La visualización de los geles se realizó en cámara de U.V y estos fueron analizados mediante el programa BioDocAnalyze de BIOMETRA ®.
Obtención y análisis de secuencias
La secuenciación de ADN se realizó en el Center for Disease Control CDC de Atlanta (Estados Unidos). La edición y el alineamiento de las secuencias se realizó utilizando el programa BIOEDIT Sequence Alignment Editor (Hall 1999). Para la totalidad de las secuencias provenientes de los individuos se estimó, la diversidad haplotípica (h), la diversidad nucleotídica (pi) y el valor neto de sustituciones nucleotídicas (K), por medio del programa DnaSP versión 4 (Rozas et al. 2004).
El análisis de la variabilidad de
Los resultados y análisis relacionados con la variabilidad de las secuencias se realizaron considerando éste como un estudio exploratorio sobre el uso potencial de la región secuenciada para agrupar individuos de la especie de interés (provenientes de diferentes localidades geográficas) y separarlos de otras especies morfológicamente similares con base en las distancias genéticas y no como un estudio de genética de poblaciones. No obstante, la validación del gen como marcador taxonómico, corresponderá a un estudio con mayor número de individuos, localidades y especies similares como punto de comparación y que se deriva de los resultados del presente estudio.
Resultados
Extracción y amplificación
El búfer de macerado permitió obtener ADN de las larvas en cantidad y calidad suficiente para una amplificación mediante PCR de la región de interés, usando entre 0,5 y 1mL de ADN de extracción en una reacción de PCR de 50mL. Además, se logró extraer exitosamente ADN con calidad para PCR de fracciones de 2, 3, y 4 mm de larvas L1, L2, y L3 respectivamente con una concentración promedio de 86ng/uL de ADN por muestra. Como dato adicional y pensando en las muestras frecuentemente encontradas en los cadáveres, también se usaron para la extracción de ADN otras fuentes o partes del insecto disponibles en la cría al momento de los ensayos moleculares. Al respecto se encontraron extracciones exitosas a partir de tres huevos, pupas completas, dos patas y pequeños fragmentos de tórax extraídos de los adultos. No se recuperó ADN de puparios vacíos, (datos no mostrados).
Análisis de secuencias
El segmento amplificado incluyó 324 pares de bases correspondientes a la región 3' del gen Cytb, además de un segmento de 66 pares de bases de ARNtser, 23bp de la región IG2 y un segmento del extremo 3′ del gen ND1 de 66 bp. La identidad de las secuencias se verificó por comparación con la secuencia de referencia de
Al observar la distribución de la variabilidad entre las posiciones del alineamiento, se evidenció que los sitios con mayor cantidad de polimorfismos se encuentran en los extremos del alineamiento. Estas posiciones corresponden a las regiones terminales 3' de los genes Cytb y ND1 respectivamente, por lo cual se realizaron análisis detallados en términos de haplotipos con énfasis especial en la porción de ND1, que fue tomada para caracterizar por tipos los sitios de muestreo. Los sitios conservados, se encontraron distribuidos, en las posiciones centrales del alineamiento.
Los porcentajes de la composición nucleotídica (Tabla 2) mostraron una mayor cantidad de Adenina y Timina (76,1%) con respecto a la cantidad de Citosina y Guanina (23,8%) patrón de composición de bases que corresponde a lo observado en la mayoría de los genomas mitocondriales de insectos, especialmente en las regiones codificantes (Hoy 2003).
Composición nucleotídica expresada en porcentaje.
M= Medellín, I= Itagüí. SR= Santa Rosa.
Respecto a las sustituciones nucleotídicas, se encontró un mayor número de transiciones que de transversiones (Tabla 3).
Número de transiciones y transversiones en un segmento de 480 pb de ADN mitocondrial.
Variabilidad de Cytb
El análisis incluyó 324 nucleótidos de los cuales, 322 sitios fueron conservados y dos polimórficos con mutaciones en las posiciones 23 y 34 de la secuencia que corresponden a las terceras posiciones del codón, este cambio muestra las poblaciones de Medellín e Itagüí con un mismo haplotipo y la población de Santa Rosa como un segundo haplotipo. Los valores de diversidad haplotípica y diversidad nucleotídica para las secuencias de L.eximia (Tabla 4), corresponden con los observados a nivel intraespecífico para datos haploides (Stephan y Langley 1992).
Diversidad nucleotídica para las secuencias amplificadas.
Variabilidad de ND1
El segmento que codifica para ND1 de 66 pb, corresponde a la región 414 - 480 de la matriz. En esta fracción, se encontraron 61 sitios conservados y cinco polimórficos que originaron cambios en las primeras y terceras posiciones del codón. Los sitios variables fueron parsimoniosamente informativos con dos estados en las posiciones dos, tres, 55, 57 (416, 417, 469 у 471 de la matriz general) y un sitio igualmente informativo con tres estados en la posición cuatro (418 de la matriz general) en la primera posición del codón. Este fragmento representó la porción más variable de la región de ADN estudiado y con base en el cual se observó una clara separación de los especímenes por localidad.
Al comparar las distancias genéticas Jukes y Cantor (1969) al interior de la especie con base en las regiones (Cytb, ND1), se encontraron valores entre 0,01 y 0,011, (Tabla 5). Estos resultados son similares a los encontrados a nivel intraespecífico en otras especies del género como
Distancias genéticas intra e interespecíficas obtenidas usando un criterio de máxima verosimilitud
M =Medellín, I= Itagui. SR= Santa Rosa.
De igual forma, los valores de h (Nei 1987) encontrados entre los individuos de las diferentes localidades (0,53 y 0,684) están ligeramente por encima de los valores promedio ya que para marcadores de evolución rápida como el ADN mitocondrial, los valores de diversidad haplotípica intra especies generalmente son menores de 0,5. Así mismo, el número promedio de diferencias nucleotídicas entre los haplotipos (K) (Tajima 1983) indicó una diversidad genética media-baja.
La topología obtenida a partir de distancias genéticas de Jukes y Cantor muestra las relaciones entre haplotipos correspondientes a los sitios de muestreo (Fig. 1). En él se observa un único haplotipo por municipio, así como la correspondencia entre las distancias genéticas y las distancias geográficas. El fragmento completo secuenciado permitió discriminar individuos de los tres municipios separados a una distancia mínima de 8 kilómetros.
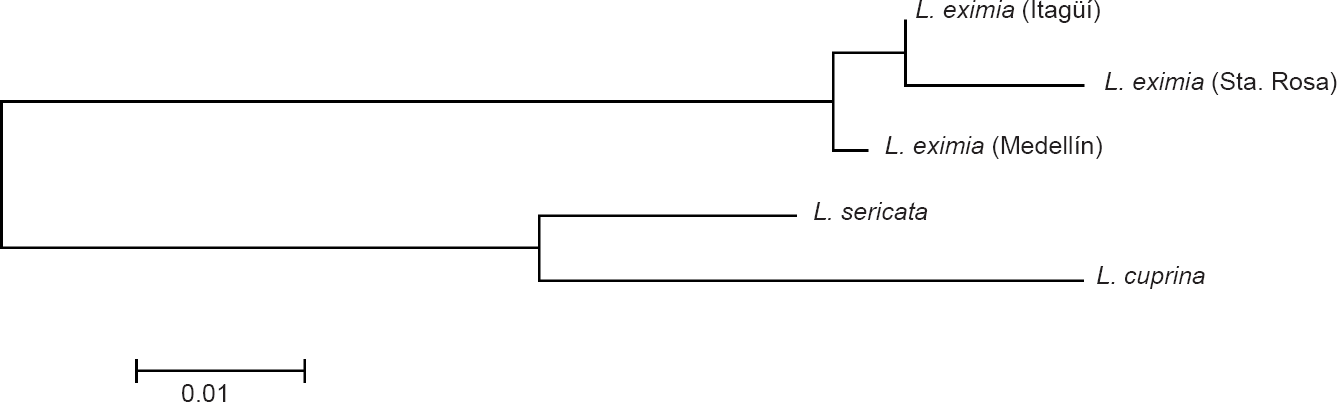
Fenograma de distancias de las muestras de
Discusión
Para las Ciencias Forenses, la aplicabilidad de cualquier método de análisis, depende de varios factores. Entre los más importantes se encuentran la rapidez, eficiencia y costos de la técnica. En cuanto a la obtención y uso de ADN, actualmente se encuentran disponibles y bien documentadas técnicas como RAPDS, RFLPS, y secuenciación de ADN mitocondrial utilizados para análisis de genética de poblaciones, taxonomía molecular y análisis filogenéticos en especies de importancia forense. En la actualidad la obtención directa de secuencias aparece como el método más utilizado y de gran potencial en la sistemática de insectos, mostrando en comparación con otras técnicas excelentes resultados en cuanto a la variabilidad genética de las especies, siendo una información 100% conectable con otros resultados similares y permitiendo a posteriori la implementación de técnicas masivas de caracterización que usen las regiones más variables o conservadas de acuerdo al interés (Infante-Malachias et al. 1999; Litjens et al. 2001; Stevens y Wall 2001).
En este trabajo realizamos una estandarización de la extracción y amplificación de una región del ADN mitocondrial, Cytb y ND1, (480 pb) y verificamos su uso potencial en la asignación de especímenes o sus partes a una especie de interés forense con identificación taxonómica previamente realizada con base en morfología. Esta región además permitió separar los individuos con base en su procedencia. Adicionalmente, exploramos como el polimorfismo en las secuencias de esta región permite separar de forma correcta individuos de otras dos especies cercanas y morfológicamente similares:
A diferencia de la mayoría de los casos reportados para ciencias forenses, en el presente estudio no se usaron kits comerciales de extracción de ADN que suelen ser costosos en comparación con la solución de lisis con la cual se realizó la extracción. La calidad y cantidad de ADN evaluada con el nanodrop y mediante gel de agarosa, así como la amplificación exitosa de PCR es de resaltar, en especial, si se considera que a partir de pequeñas partes o tejidos de los insectos, con menores costos y en tiempos similares se obtienen resultados adecuados y comparables con los que representa el uso de productos comerciales.