
Abstract
Morphological characters have been traditionally used for identification of sand flies of the genus Lutzomyia; however, their utility is limited in some species. In this work, we characterized the mitochondrial DNA region coding for the carboxyl-terminal domain of cytochrome b protein in seven species of Lutzomyia: L. trinidadensis, L. panamensis, L. cayennensis cayennensis, L. dubitans, L. gomezi, L. rangeliana and L. evansi. A total of 134 polymorphic sites were detected in the gene (40.98%) and 29 sites in the protein (26.6%). The very high level of polymorphism observed in cytochrome b, included replacement of amino acids, use of alternative stop codons, and differences in the size of the protein. The utility of the studied region in the identification of Lutzomyia species is discussed.
Introducción
Los flebotomíneos son insectos nematóceros ubicados en la subfamilia Phlebotominae Rondani, 1840, la cual está integrada por los géneros Phlebotomus Rondani y Berté, 1840, Sergentomyia França & Parrot, 1920, Chinius Leng, 1987, Brumptomyia França y Parrot, 1921, Warileya, Hertig, 1948, y Lutzomyia França, 1924 (Bejarano 2006). A la fecha se han descrito casi 500 especies de Lutzomyia que se encuentran distribuidas principalmente en las zonas tropicales y subtropicales de América. Más del 10% de estas especies son transmisores de Leishmania Ross, 1903, protozoo causante de las enfermedades denominadas leishmaniasis (Killick-Kendrick 1999; Lainson y Shaw 2005), las cuales han recobrado su importancia epidemiológica en los últimos años por el aumento del número de casos registrados tanto en áreas rurales como urbanas de Latinoamérica (Bejarano et al. 2002; Shaw 2007).
Un aspecto primordial en el estudio ecoepidemiológico de la leishmaniasis es la identificación de las especies de Lutzomyia presentes en cada foco de transmisión, porque facilita la dirección de las medidas de control vectorial hacia los responsables de la transmisión del parásito. Esta determinación taxonómica se fundamenta en los caracteres morfológicos del insecto adulto debido a las dificultades para localizar los estados preimaginales en la naturaleza (Montoya-Lerma y Ferro 1999). En la actualidad los flebotomíneos americanos se identifican con las descripciones y la clave morfológica de Young y Perkins (1984) y Young y Duncan (1994), basadas en la propuesta de Lewis et al. (1977), que acepta la presencia de tres géneros en el Nuevo Mundo, o la clave de Galati (2003a), que sigue en parte el planteamiento de Forattini (1973), y propone 23 géneros americanos (Galati 2003b; Galati et al. 2003).
Más allá de las divergencias en el tratamiento taxonómico al nivel supraespecífico, estas claves no permiten separar las hembras o los machos de algunas especies con alta similitud morfológica, como en la serie townsendi del grupo verrucarum Theodor, 1965, la serie squamiventris del subgénero Psychodopygus Mangabeira, 1941, y los subgéneros Helcocyrtomyia Barretto, 1962, Trichophoromyia Barretto, 1962, y Trichopygomyia Barretto, 1962, entre otros grupos taxonómicos (Young y Duncan 1994; Galati 2003a). Esto podría ser particularmente problemático para la incriminación de especies simpátricas que presentan la misma morfología pero diferente capacidad vectorial. Las variaciones en los rasgos morfológicos usados tradicionalmente para la determinación taxonómica tornan imperiosa la necesidad de nuevos caracteres con mayor capacidad de discriminar especies.
Las secuencias de nucleótidos son una herramienta molecular que se ha usado en los flebotomíneos para identificar especies indistinguibles en uno de los sexos (Testa et al. 2002), estudiar las relaciones filogenéticas (Torgerson et al. 2003; Beati et al. 2004; Vivero et al. 2007) y analizar la estructura genética de las poblaciones (Ready et al. 1997; Ishikawa et al. 1999), en especial cuando se sospecha la existencia de complejos de especies (Uribe et al. 2001; Arrivillaga et al. 2002; Hodgkinson et al. 2003). Aunque por su costo, las secuencias están aún distantes de implementarse en el diagnóstico rutinario de especie, son una alternativa que permite, además de sortear las limitaciones inherentes a los caracteres morfológicos, establecer las bases para el futuro desarrollo de claves moleculares. El propósito de este estudio fue caracterizar el polimorfismo de la región del genoma mitocondrial que codifica para el extremo carboxilo de la proteína citocromo b en siete especies de flebotomíneos del género Lutzomyia y evaluar su potencial utilidad como carácter taxonómico.
Materiales y Métodos
Flebotomíneos
Los muestreos entomológicos se desarrollaron en la ciudad de Sincelejo (9°18’N, 75°25’W), capital del departamento de Sucre, Colombia, en la que se registra en promedio por año una temperatura de 28ºC y precipitación de 1050 mm, que corresponde, según la clasificación de Holdridge (1967), a bosque seco tropical. Los insectos se recolectaron cuatro veces por semana, entre los meses de febrero y noviembre de 2005, con dos trampas de luz tipo CDC instaladas en el intradomicilio y peridomicilio entre las 18:00 y 06:00 horas.
Cada flebotomíneo se diseccionó separando la cabeza, un ala y los segmentos terminales del abdomen, los cuales fueron aclarados por 24 horas en lactofenol y montados en una lámina portaobjeto con el medio de Hoyer. La determinación de especie se efectúo con las claves taxonómicas de Young y Duncan (1994) y Galati (2003a). Los especímenes testigo están depositados en la “Colección de Artrópodos de Importancia Médica de la Universidad de Sucre - CAIMUS”, en Sincelejo, Colombia.
Extracción del material genético
El ADN se extrajo siguiendo el protocolo de Collins et al. (1987), con algunas modificaciones. El tórax, un ala, las patas y los segmentos iniciales del abdomen se maceraron en 60 μl del tampón de lisis (0,08 NaCl, 0,16 M sacarosa, 0,06 M EDTA, 0,5% SDS, 0,1 M Tris-HCL a pH de 7,5), e incubaron a una temperatura de 65°C por 30 minutos. Las proteínas se precipitaron con 14 μl de acetato de potasio 8M durante 30 minutos sobre hielo, seguido por centrifugación a 12000 x g durante 10 minutos. La precipitación del ADN del sobrenadante se realizó con 200 μl de etanol absoluto e incubación a -20°C durante 24 horas. Las muestras se centrifugaron a 12000 x g durante 20 minutos y el ácido nucleico precipitado se lavó con 200 μl de etanol al 70% y luego con 200 μl de etanol absoluto. Finalmente, el ADN extraído se secó a temperatura ambiente y se resuspendió en 30 μl de agua.
Secuenciación de nucleótidos
La reacción en cadena de la polimerasa (PCR) se desarrolló con los oligonucleótidos sentido (CA(T/C)ATTCAACC(A/T)GAATGATA) y antisentido (GGTA(C/T)(A/T)TTGCCTCGA (T/A)TTCG(T/A)TATGA) (Ready et al. 1997). El gen citocromo b se amplificó en un volumen final de 50 μl que contenían 0,3 mM de cada cebador, buffer de PCR 1x, 3 mM de MgCl2, 0,2 mM de la mezcla de desoxirribonucleósidos trifosfatos (dNTPs), 1,5 U de Taq ADN polimerasa y 8 μl de la solución con el ADN extraído. El perfil térmico incluyó una etapa inicial de desnaturalizacion a 94ºC por 3 minutos, una segunda etapa de 35 ciclos, consistente en desnaturalizacion a 93ºC por 1 minuto, alineamiento a 50ºC por 1 minuto y extension a 72ºC por 1 minuto, y una tercera etapa de elongación a 72ºC durante 10 minutos. El producto amplificado se purificó usando Wizard PCR Preps (Promega) y se secuenció automáticamente por electrofóresis capilar con distintos fluorocromos, para marcar los didesoxinucleótidos terminadores de cadena.
Análisis molecular
La secuencia nucleotídica del gen citocromo b se determinó en ambos sentidos de la doble hebra de ADN mitocondrial. Los cromatogramas fueron editados con el programa Molecular Evolutionary Genetics Analysis (MEGA) versión 3.1 para generar la secuencia consenso de cada espécimen (Kumar et al. 2004). El alineamiento nucleotídico se realizó con el algoritmo Clustal W incorporado en MEGA 3.1, y se usaron los valores por defecto para la penalización por apertura y extensión de gaps (Thompson et al. 1994). A partir del alineamiento se determinó la composición nucleotídica del gen, se tradujeron las secuencias de nucleótidos a aminoácidos y se calcularon las distancias genéticas pareadas bajo el modelo biparamétrico de Kimura (1980) que otorga tasas diferentes de mutación a las transiciones y transversiones. El análisis de la secuencia de aminoácidos de la proteína se realizó con base en el código genético mitocondrial de Drosophila integrado a MEGA 3.1. La distribución de la variabilidad dentro del fragmento mitocondrial secuenciado se examinó determinando la entropía H(x) de cada posición nucleotídica con el programa Bioedit (Hall 1999). Las secuencias de nucleótidos generadas durante la presente investigación están depositadas en Genbank (National Center for Biotechnology Information - NCBI) bajo los códigos de acceso EF012215, EF012216, EF012217, EF012218, EF012219, EF012220, EF012221, EF012222, EF012223, EF012224 y EF012225.
Resultados y Discusión
Se obtuvo un fragmento nucleotídico de 327 pares de bases (pb) del extremo 3’ del gen mitocondrial citocromo b, que codifica para 108 aminoácidos del dominio carboxilo terminal de la proteína homónima localizada en la membrana interna de la mitocondria, la cual es un componente indispensable dentro de la cadena respiratoria (Figs. 1 y 2). En el alineamiento múltiple se incluyeron además nueve posiciones nucleotídicas correspondientes al espaciador intergénico 1 (IG1), que separa, en algunas especies, a los genes citocromo b y ARN de transferencia mitocondrial para serina (UCN) (ARNtSer).
Las secuencias se aislaron a partir de siete especies de Lutzomyia integrantes de igual número de subgéneros o grupos, las cuales fueron identificadas como L. trinidadensis (Newstead, 1922) del grupo oswaldoi, L. panamensis (Shannon, 1926) del subgénero Psychodopygus, L. cayennensis cayennensis (Floch y Abonnenc, 1941) del subgénero Micropygomyia, L. dubitans (Sherlock, 1962) del grupo migonei, L. gomezi (Nitzulescu, 1931) del subgénero Lutzomyia, L. rangeliana (Ortíz, 1952), especie no agrupada y L. evansi (Núñez-Tovar, 1924) del grupo verrucarum. Con la excepción de L. dubitans, todas las especies tienen antecedentes en salud pública. L. evansi es un reconocido vector de Le. infantum Nicolle, 1908, L. panamensis y L. gomezi son transmisoras de Le. panamensis Lainson y Shaw, 1972, L. rangeliana y L. trinidadensis se han encontrado con Leishmania, y L. c. cayennensis ha sido hallada infectada con parásitos tripanosomatídeos (Bonfante-Garrido et al. 1990, 1999; Travi et al. 1990; Santamaría et al. 2006; Cochero et al. 2007).
En total se obtuvieron 11 secuencias de nucleótidos (dos aisladas de igual número de ejemplares de L. panamensis, L. c. cayennensis, L. gomezi y L. rangeliana, y una de L. trinidadensis, L. dubitans y L. evansi), de las cuales se derivaron ocho haplotipos nucleotídicos (Fig. 1) y siete haplotipos aminoacídicos (Fig. 2). La composición nucleotídica estuvo marcada por un alto contenido de adenina y timina equivalente al 73,09% de las bases secuenciadas, mientras que los nucleótidos guanina y citosina alcanzaron sólo el 26,1%. Como era esperarse al nivel proteico prevalecieron los aminoácidos no polares como isoleucina y leucina que representaron en promedio el 16,03% y 15,27%, respectivamente, del contenido aminoacídico, lo cual está relacionado con la hidrofobicidad de la proteína citocromo b.
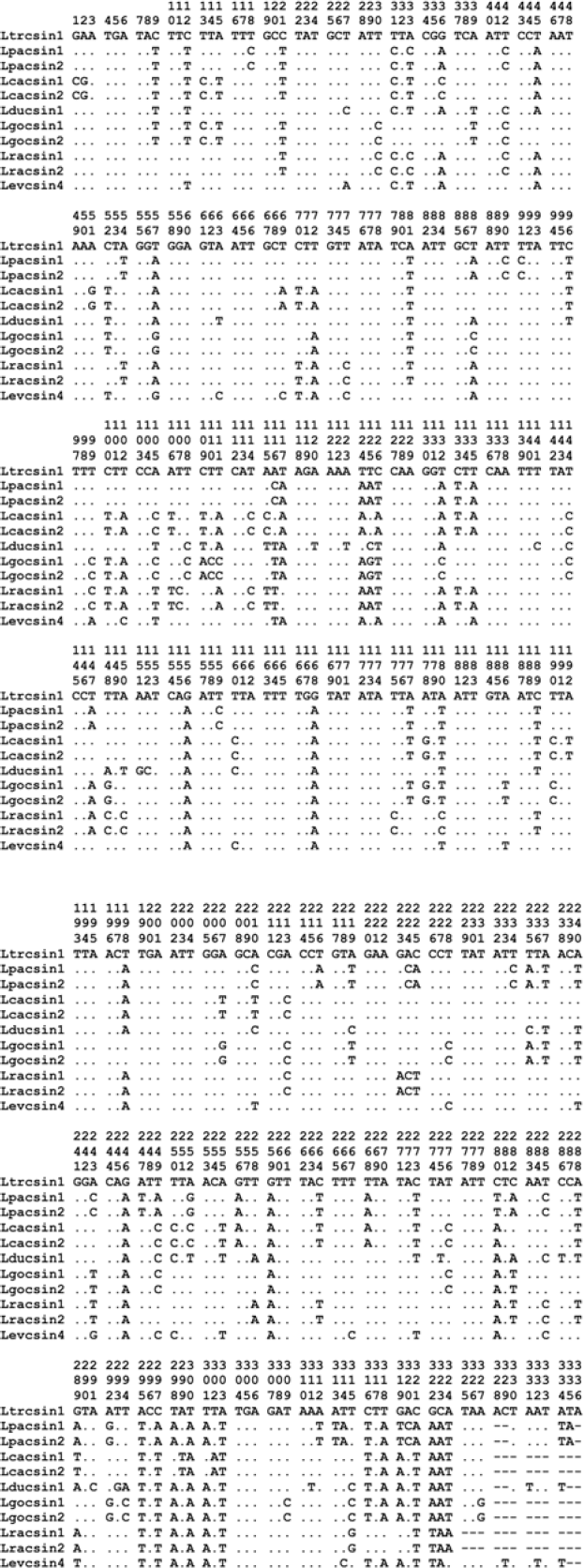
Alineamiento de secuencias nucleotídicas parciales del gen citocromo b (posiciones 1 a 327) y del espaciador intergénico 1 (posiciones 328 a 336) aisladas de L. trinidadensis (Ltrcsin1), L. panamensis (Lpacsin1, Lpacsin2), L. c. cayennensis (Lcacsin1, Lcacsin2), L. dubitans (Lducsin1), L. gomezi (Lgocsin1, Lgocsin2), L. rangeliana (Lracsin1, Lracsin2) y L. evansi (Levcsin4). Los nucleótidos se representan con la primera letra de su respectivo nombre, las homologías con puntos y los eventos indel (inserción-delección) con guiones.

Alineamiento de secuencias aminoacídicas del dominio carboxilo terminal de la proteína citocromo b de L. trinidadensis (Ltrcsin1), L. panamensis (Lpacsin1, Lpacsin2), L. c. cayennensis (Lcacsin1, Lcacsin2), L. dubitans (Lducsin1), L. gomezi (Lgocsin1, Lgocsin2), L. rangeliana (Lracsin1, Lracsin2) y L. evansi (Levcsin4). Los aminoácidos se representan con su código universal de una letra, las homologías con puntos, los eventos indel (inserción-delección) con guiones y los codones de parada con asteriscos.
A lo largo del extremo 3’ del gen analizado se detectaron 134 sitios polimórficos que arrojaron una variación global del 40,98%, con porcentajes de divergencia entre especies del 16,51% al 23,55%. Consecuentemente, los valores de distancia genética de Kimura fluctuaron entre 0.1889 y 0.2868 (Tabla 1). El valor más bajo se registró al comparar L. evansi y L. dubitans, mientras que el más alto se presentó entre L. trinidadensis y L. dubitans. De otro lado, los aislados de L. panamensis mostraron entre sí una transición en la posición 33 del alineamiento nucleotídico (Fig. 1), mientras que en L. c. cayennensis, L. gomezi y L. rangeliana no se observaron sustituciones intraespecíficas. Por lo anterior, las distancias genéticas intraespecíficas oscilaron entre 0 y 0.0031. El análisis de entropía reveló que, salvo por el ligero aumento registrado en algunas posiciones nucleotídicas (Fig. 3), la variabilidad estuvo distribuida casi homogéneamente a lo largo de todo el fragmento secuenciado.
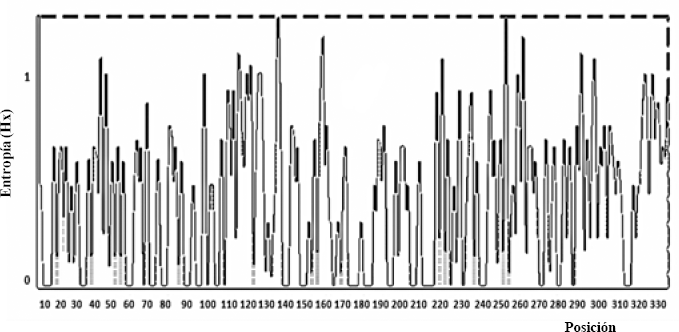
Entropía H(x) en la secuencia de nucleótidos parcial del gen citocromo b de L. trinidadensis, L. panamensis, L. c. cayennensis, L. dubitans, L. gomezi, L. rangeliana y L. evansi.
Es importante anotar que las últimas bases del extremo 3' del gen citocromo b exhibieron polimorfismos importantes en la distinción de cada una de las siete especies de Lutzomyia. L. gomezi mostró la tripleta de nucleótidos timina-adenina-guanina, que corresponde a un codón de parada tipo UAG, en lugar del codón UAA usado por las demás especies. L. evansi y L. panamensis se caracterizaron por tener repetida la secuencia timina-adenina-adenina (Fig. 1), que transcribe para dos codones de parada consecutivos tipo UAA, lo que podría estar asociado a una reconfirmación de la señal de terminación durante la síntesis proteica.
La sustitución nucleotídica de mayor trascendencia se observó en la posición 322 de L. rangeliana y L. evansi (Fig. 1), con el cambio de guanina o adenina por timina, que conlleva a la disminución del tamaño de la proteína citocromo b codificada por estas especies. Es importante resaltar también que en L. gomezi las dos últimas bases del gen citocromo b correspondieron, al mismo tiempo, a las dos primeras del gen ARNtSer, en tanto que en L. c. cayennensis y L. rangeliana estos genes compartieron una sola base. En las otras especies de este estudio los genes citocromo b y ARNtSer, se encuentran separados por el IG1, que está constituido por 10 pb en L. evansi, 9 en L. trinidadensis, 6 en L. panamensis y 5 en L. dubitans (Fig. 1).
La disminución gradual en la longitud del IG1 hasta llegar a la ausencia del mismo y el solapamiento de bases entre los genes citocromo b y ARNtSer, permite inferir que el genoma mitocondrial de estos insectos podría haber experimentado, en algunas especies, un proceso de reducción evolutiva que eliminó aquellos componentes no indispensables para la función que desempeña y disminuyó u optimizó el tamaño de los genes codificadores. Esto se fundamenta, en parte, en la teoría del origen endosimbiótico de la mitocondria (Margulis 1970).
En el extremo carboxilo terminal de la proteína citocromo b se registraron 29 sitios aminoacídicos polimórficos que generaron un porcentaje de divergencia global del 26,6%, útil para la discriminación de especie. Los sitios con mayor variación correspondieron a las posiciones 39, 42 y 97 del alineamiento proteico (Fig. 2). Entre estos se destaca la posición 39 por poseer un aminoácido distinto en cada especie, asparagina en L. trinidadensis, treonina en L. panamensis, glutamina en L. c. cayennensis, leucina en L. dubitans y fenilalanina en L. rangeliana, con la excepción de L. gomezi y L. evansi que presentaron una metionina en la posición referida. No obstante, las dos últimas especies difieren entre sí en otros 10 sitios aminoacídicos distribuidos a lo largo de esta región proteica. El polimorfismo más relevante en la proteína citocromo b fue la disminución del tamaño de la misma en L. rangeliana y L. evansi como producto del reemplazo de un codón con sentido por un codón de parada en la posición 108 del alineamiento aminoacídico (Fig. 2).
Aunque en años recientes el gen citocromo oxidasa I se ha propuesto como el "código de barras del ADN" para la identificación de la diversidad animal (Hebert et al. 2003), los resultados del presente estudio demuestran que la región mitocondrial aquí secuenciada es un excelente candidato para el diagnóstico molecular de especie en flebotomíneos. Los siete taxones del género Lutzomyia analizados exhibieron polimorfismos ostensibles en el extremo 3' del gen citocromo b y la proteína derivada del mismo, que comprendieron sustituciones nucleotídicas, solapamiento de una o más bases con el gen tRNASer, reemplazo de aminoácidos, empleo de codones de parada distintos y diferencias en el tamaño del producto proteico. A lo anterior se suman los notables cambios en la longitud del IG1 que, incluso, desaparece en algunas especies.
Valores de distancia genética según el modelo de dos parámetros de Kimura, entre secuencias nucleotídicas parciales del gen citocromo b de L. trinidadensis (Ltrcsin1), L. panamensis (Lpacsin1, Lpacsin2), L. c. cayennensis (Lcacsin1, Lcacsin2), L. dubitans (Lducsin1), L. gomezi (Lgocsin1, Lgocsin2), L. rangeliana (Lracsin1, Lracsin2) y L. evansi (Levcsin4).
Si bien estos polimorfismos permiten la distinción de cada uno de los taxones estudiados, se requieren más estudios para inferir las posibles relaciones filogenéticas entre tales grupos. Del mismo modo, es necesario continuar con la valoración del marcador en especies morfológicamente similares (Pérez-Doria et al. 2008). Es pertinente mencionar también que el IG1 se encuentra adyacente al gen tRNASer, el cual pese a sus restricciones operacionales, exhibe en flebotomíneos una variabilidad genética interespecífica importante. Por su proximidad, estas regiones pueden secuenciarse en una sola reacción con el segmento codificante para el extremo carboxilo terminal de citocromo b (Ready et al. 1997; Vivero et al. 2007), lo que facilita el análisis integral de esta sección del genoma mitocondrial, con sus diferentes productos y aumenta la utilidad del mismo.
El volumen de secuencias de nucleótidos o aminoácidos derivadas de flebotomíneos del Nuevo Mundo, que reposan en los bancos de datos moleculares, es muy bajo, comparado con el número de especies descritas (Bejarano 2002), por lo tanto se espera que los polimorfismos en la región mitocondrial estudiada puedan servir de base para formular, en el futuro, claves moleculares útiles para la identificación de vectores de Leishmania.
Footnotes
Agradecimientos
Los autores agradecen la colaboración de Alveiro Pérez-Doria y Luz Fernanda Lambraño, para la obtención del material entomológico.