
Abstract
Resistence to Bacillus thuringiensis (Bt) toxins in Lepidoptera and Diptera insects has been showed under laboratory enviroments and in 1990 it was reported by the first time under field conditions. Therefore, it is important to characterize the new toxic proteins and identify their genes in the recently discovered strain of Bacillus thuringiensis ssp. medelllín (Btmed), with mosquito larvae activity. The generation of a genomic library of Btmed allowed the isolation of the toxins genes and their initial characterization. In order to make the genomic library of Btmed in the lambda ZAP II vector, total DNA of Btmed was isolated and a partial restriction was performed with EcoRI. The library was screened using inmunodetection techniques. The purified inmunoreactive clones displayed an insert of approximately 4 Kilobases (Kb) which encoded a protein of a molecular weight of 100 Kilodalton (KDa) that shows toxic activity against mosquito larvae.
Introducción
Bacillus thuringiensis (Bt) es una bacteria Gram positiva que ha sido ampliamente usada en muchos países en el control integrado de plagas agrícolas y de mosquitos vectores de enfermedades. Las cepas más conocidas de Bt para el control de mosquitos pertenecen a las subespecie israeliensis (Bti) (Goldberg y Margalit 1977) y al aislamiento PG-14 de la subespecie morrisoni (Bti) (Padua et al. 1984).
En los últimos seis años se han presentado varios reportes de insectos seleccionados con altos niveles de resistencia a las delta endotoxinas de cepas nativas y recombinantes de Bt, tanto bajo condiciones de campo como de laboratorio. En cada caso, la especie de insecto tiene aparentemente la capacidad de desarrollar resistencia en pocas generaciones. En el laboratorio se han encontrado altos niveles de resistencia a las delta endotoxinas de B. thuringiensis ssp. kurstaki (Btk) en Plodia interpunctella (Hübner) y Cadra cautella (Walker) (McGaughey 1985; McGaughey y Beeman 1988). También, una cepa de Heliothis virescens (Fabricius) ha sido seleccionada como resistente a toxinas de Btk producidas en Pseudomonas fluorescens transformadas genéticamente (Stone et al. 1989). Más significativo aún ha sido el reporte en los últimos cuatro años de poblaciones de campo de Plutella xylostella (L.) de Hawai resistentes a Btk (Tabashnik et al. 1990). Este es el primer ejemplo bien documentado de resistencia ocurrida en el campo, aunque reportes preliminares habían sugerido la posibilidad de resistencia a Bt en poblaciones de P. xylostella en Filipinas (Kirsch y Schmuttere 1988) y en poblaciones de P. interpunctella en granos almacenados en los Estados Unidos (McGaughey 1985; McGaughey y Beeman 1988). Además se ha reportado una cepa de Leptinotarsa decemlineata (Say) seleccionada como resistente a una cepa de Bt activa contra coleópteros (Miller et al. 1990), y Culex quinquefasciatus Say y Aedes aegypti (L.) resistentes a Bti (Georghiou et al. 1983; Goldman et al. 1986).
La aparición de resistencia a las toxinas de Bt en insectos lepidópteros y dípteros ha llevado tanto a los laboratorios de investigación como a las industrias a buscar nuevas moléculas, especialmente de bacterias entomopatógenas, para el desarrollo de nuevas alternativas de control. Orduz et al. (1992) reportaron la subespecie medellin de Bt (Btmed), cepa CIB 163-131 serotipo H-30, un nuevo serotipo de Colombia, el cual es tóxico para larvas de anofelinos y culicinos. Su nivel de toxicidad es diez veces menor que Bti y PG-14. En contraste con Bti y PG-14, Btmed no produce proteínas de tan alto peso molecular (125-135 KDa), pero produce una proteína en el rango de 90-100 kilodalton (KDa) (Orduz et al. 1994). La presencia de una nueva proteína insecticida de aproximadamente 100 KDa en Btmed representa una alternativa para los programas de control de mosquitos en lugares donde la resistencia a las toxinas de Bti se presente.
En este estudio se reporta el aislamiento y la caracterización parcial del gen que codifica la proteína tóxica de Btmed de aproximadamente 100 KDa de peso molecular.
Materiales y Métodos
Generación de la librería genómica de Btmed
Para el aislamiento del ADN genómico de la cepa CIB 163-131 se inoculó, al 5%, un erlenmeyer con 200 ml de medio Luria Bertani (LB) (Sambrook et al. 1989) y se incubó a 30°C, 200 rpm hasta una densidad óptica de 1,5 a 600 nm. Luego, el cultivo se centrifugó por 5 minutos a 5.000 rpm y se procedió al aislamiento del ADN genómico mediante un método modificado de lisis alcalina con SDS, tratamiento con lisozima, RNAsa y proteínasa K, y finalmente extracción con fenol y cloroformo y precipitación con alcohol isopropílico (Delécluse)1
DELÉCLUSE, A. Comunicación personal.
El ADN total del Btmed fue digerido con la enzima EcoRI bajo condiciones que permitieron obtener fragmentos entre 10.000 y 7.000 pares de bases. Se ligaron 600 ng de fragmentos de ADN genómico con 1 µg de los brazos del fago lambda (1), usando el sistema de ligación de ADN (Stratagene, La Jolla, CA); el producto de la ligación (1 recombinante) fue empacado in vitro usando el sistema de clonaje Lambda ZAP II/EcoRI/CIAP (Stratagene, La Jolla, CA) para obtener un «stock» de fagos. Luego se transfirió a una cepa de Escherichia coli hospedante (XL1-BLUE MRF') y se sembró en cajas de petri. Para la siembra de la librería, 1 ml del «stock» de fagos se incubó con 200 ml de células XL1-BLUE MRF' por 15 minutos a 37°C, luego se le agregaron 3 ml de agar semisólido (NZY líquido con 0,7% de agarosa) a 48°C, más 15 ml de IPTG (Isopropiltio-B-D-galactosidasa) 0,5 M y 50 ml de X-GAL (5-Bromo-4- Cloro-3-Indolil-B-D-galactosidasa) a 250 mg/ml, y se virtió en cajas de agar NZY (5 g de NaCl, 2 g de MgSO4. 7H20, 5 g de extracto de levadura, 10 g de caseína hidrolizada y 15 g de agar por litro, pH 7,5) que se incubaron a 37°C. Las placas de lisis se visualizaron luego de 6 a 8 horas de incubación. Una vez obtenidas las placas se procedió a titular la librería usando 200 ml de células XL1-BLUE MRF' y 1 ml de diluciones seriadas del «stock» de fagos de 10-2 a 10-6; para determinar el número de unidades formadoras de placas (ufp).
Para la amplificación de la librería se mezclaron alícuotas de la suspensión de fagos, que contenían aproximadamente 28.000 ufp y 340 ml de células XL1-BLUE MRF' frescas. La mezcla se incubó 15 minutos a 37°C y luego de agregarles 3 ml de agar semisólido, se sembraron en cajas de agar NZY y se incubaron por 8 horas a 37°C. Al comenzar a observar placas de lisis se virtió, en cada caja, 5,1 ml de buffer SM (5,8 g de NaCl, 2 g de MgSO4. 7H20, 50 ml de 1 M de Tris-HCl pH 7,5 y 5 ml de gelatina al 2% por litro) y se incubaron nuevamente a 4°C durante toda la noche para permitir la difusión del fago en el buffer. Luego, la suspensión de fagos se recuperó en un tubo de polipropileno estéril, se centrifugó para remover los desechos celulares y al sobrenadante se le adicionó 0,3% de cloroformo y 7% de dimetil sulfóxido (DMSO) para evitar contaminación y preservar los fagos en la congelación, y se hicieron alícuotas en tubos a -70°C hasta su uso. Al tener la librería amplificada se tituló nuevamente haciendo diluciones del «stock» amplificado como se detalló anteriormente.
Tamizaje de la librería
Para la obtención de antisueros contra las proteínas tóxicas de Btmed, ratones Balb/c de 6-8 semanas de edad se inmunizaron mediante inyecciones intraperitoneales con la proteína de 100 KDa de Btmed, separada en geles de poliacrilamida bajo condiciones denaturantes a partir de un cultivo completo final de Btmed. La primera inyección se hizo con una emulsión del antígeno en adyuvante completo de Freund, la segunda con adyuvante incompleto y los refuerzos posteriores se hicieron con solución salina y antígeno. Sueros de sangrías períodicas se analizaron mediante inmunoblot para determinar su positividad y especificidad.
El procedimiento de inmunoselección consistio en escoger una dilución del «stock» de la librería amplificada que produjera entre 200 y 300 ufp por caja de 90 mm de diámetro. Células XL1-BLUE MRF' se infectaron con 1 ml de una dilución 1:500, se incubaron 15 min a 37°C, se sembraron y se dejaron incubando 4 horas a 42°C. Luego, las cajas se retiraron del incubador y rápidamente se cubrieron en la superficie con membranas de nitrocelulosa secas, previamente impregnadas en IPTG 10 mM, y se incubaron nuevamente a 37°C durante toda la noche para posteriormente retirar la membrana.
Las membranas se procesaron por la técnica de Western blot, usando como anticuerpo el antisuero de ratón previamente preparado contra la proteína de 100 KDa de Btmed, seguido de proteína A conjugada a fosfatasa alcalina y usando como sistema de revelado naftol fosfato y fast red (Sigma Chemical Co). De esta manera se analizaron aproximadamente 1.500 placas.
Para la purificación de clones positivos, una vez obtenidos los clones inmunorreactivos en la membrana, se compararon con su respectiva caja para ubicarlos en el agar. Con la punta gruesa de una pipeta de Pasteur estéril se perforó el agar para aislar nueve clones inmunorreactivos que se resuspendieron independientemente en 500 ml de buffer SM y cloroformo y se almacenaron a 4°C hasta su uso. Estos clones se codificaron como 3A, 3B, 3C, 3D, 4A, 4B, 4C, 4D y 4E. Con estos clones inmunorreactivos se realizaron infecciones sucesivas hasta obtenerlos puros. Al azar se eligió el clon 3A y se utilizaron sus replicaciones para su caracterización y análisis.
Escisión in vivo del fagémido pBluescript SK(-)
Para el proceso de escisión se seleccionaron cinco clones positivos puros que corresponden a replicaciones del clon 3A. (3A1-1, 3A2-1, 3A3-1, 3A4-1 у 3A6-1) y 2 clones negativos (2NA, 2NB). Se infectaron 200 ml de células XL1- BLUE MRF' con 20 ml del «stock» de fagos correspondiente a cada clon y 1 ml del fago ayudador Ex-Assist que ayuda a la escisión y circularización del inserto existente en los clones positivos. Se incubaron 15 minutos a 37°C y luego se les agregó a cada uno 3 ml de medio YT 2X (10 g de NaCl, 10 g de extracto de levadura y 16 g de bactotriptona por litro) y se incubaron nuevamente por 2,5 horas a 37°C en agitación; luego, los tubos se calentaron 20 minutos a 70°C para matar las células, se centrifugaron a 3.000 rpm por 15 minutos y se recolectó el sobrenadante que contiene el plásmido empacado como una partícula de fago filamentoso. Con 1 ml de este stock se infectaron 200 ml de células de E. coli cepa SOLR, se incubaron 15 minutos a 37°C, se sembraron sobre medio LB con 50 µg/ml de ampicilina y se dejaron crecer toda la noche a 37°C. Al obtener el crecimiento de colonias correspondiente a cada clon, se seleccionaron dos colonias de cada clon, las cuales se repicaron nuevamente a LB con ampicilina para obtener cultivos puros. Las colonias que crecieron en el cultivo contienen el fagémido pBluescript SK(-) con el inserto clonado de Btmed. El proceso se realizó siguiendo el protocolo del sistema de clonaje de Lambda ZAP II/Eco RI/CIAP (Stratagene, La Jolla, CA) para aislar el plásmido que contiene la información genética de la proteína aislada.
Análisis de la actividad tóxica de la proteína sintetizada por las replicaciones del clon 3A
Al obtener el crecimiento de las colonias correspondientes a las replicaciones del clon 3A en LB con ampicilina, se procedió a repicar cada una por duplicado en 5 ml de medio LB líquido, se incubaron por 4 horas a 37°C, 150 rpm; luego, a un grupo se le agregó IPTG en una concentración de 5 mM y el otro grupo permaneció sin IPTG. Se incubaron nuevamente hasta cumplir 24 horas en iguales condiciones. A las 24 horas se tomaron 500 ml de cada cultivo; con 100 ml se realizó un bioensayo con 10 larvas de C. quinquefasciatus de tercer instar, en 100 ml de agua destilada y se realizaron lecturas de mortalidad a las 24 y 48 horas. Los 400 ml restantes se usaron para realizar inmunodetección de la proteína producida.
Análisis por Western blot de la proteína recombinante del clon 3A
Material equivalente al utilizado para efectuar el bioensayo se separó por electroforesis en gel de poliacrilamida al 10% con SDS (Dodecyl sulfato de sodio). Las proteínas se transfirieron a una membrana de nitrocelulosa utilizando corriente eléctrica en un buffer de tris, glicina y metanol. La membrana se coloreó con rojo de Ponceau para confirmar que el procedimiento de transferencia funcionó adecuadamente y determinar la localización de los marcadores de peso molecular. La membrana se procesó entonces con el anticuerpo obtenido en ratón contra la proteína de 100 KDa de Btmed, seguido del mismo conjugado y reactivo de color que se usó en el sistema de inmunoselección de la librería.
Mapa de restricción del fragmento del gen de la proteína de 100 KDa
Para determinar el tamaño del inserto de ADN presente en el clon 3A, se realizó análisis del ADN que se aisló a partir de dicho clon con enzimas de restricción. Se realizaron minipreparaciones de ADN por el método de la lisis alcalina (Sambrook et al. 1989), a los cultivos replica del clon positivo y a algunos negativos para que sirvieran de control. Una vez obtenido los plásmidos, se cortaron con las enzimas EcoRI y HindIII, siguiendo las indicaciones del fabricante. Además se escogió un solo clon positivo para hacer una maxipreparación por el método antes mencionado y una vez obtenido el ADN aislado se realizaron cortes con 16 enzimas de restricción y varias mezclas para hacer un mapa con la localización de los sitios de corte de las enzimas de restricción.
Resultados
Al realizar la titulación inicial de la librería se obtuvo un título de librería no amplificada de 1,9 x 106 upf/ml y posteriormente se obtuvo un título de librería amplificada de 5,2 x 108 ufp/ml, con un porcentaje de clones recombinantes mayor del 90%.
Caracterización del clon 3A
Uno de los aspectos más importantes en la caracterización del clon identificado es la demostración de actividad larvicida. La cepa de E. coli que contiene el plásmido con el fragmento de ADN de Btmed tiene la posibilidad de sintetizar la proteína recombinante una vez que se induce su producción con la adición de IPTG y se puede evaluar la actividad de la proteína por medio de un bioensayo.
Los resultados del bioensayo se observan en las Figuras 1 y 2. Estos resultados indican que el clon 3A corresponde a una proteína que es tóxica para larvas de C. quinquefasciatus, con porcentajes de mortalidad hasta del 80% cuando se indujo la expresión de la proteína con IPTG y mortalidad no mayor del 30% cuando no hubo inducción con IPTG. Es interesante anotar que aislamientos independientes de colonias provenientes del clon 3A, aunque expresan niveles similares de la proteína recombinante, presentan niveles de toxicidad variable.
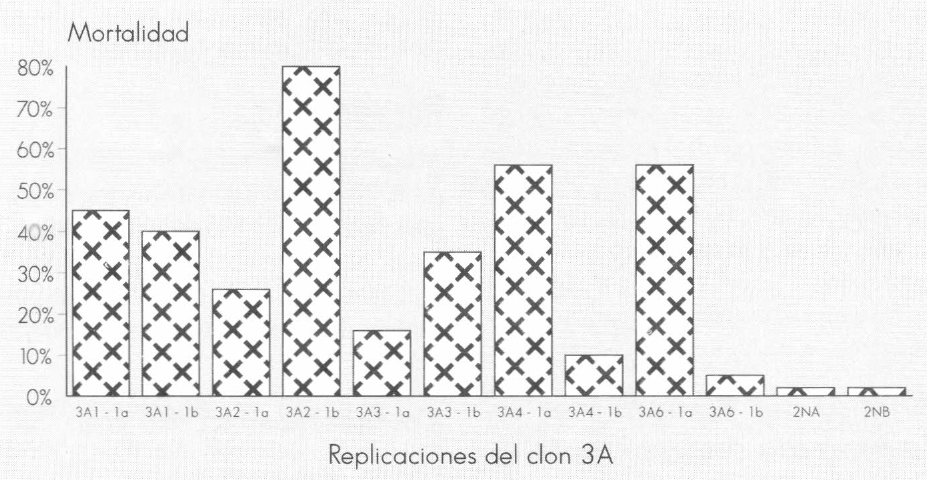
Actividad tóxica de las replicaciones del clon 3A inducidas con IPTG contra larvas de tercer instar de Culex quinquefasciatus. Los clones 2NA y 2NB fueron no inmunorreactivos contra la proteína de 100 KDa y se usaron como controles negativos. Lectura de mortalidad a las 48 horas.
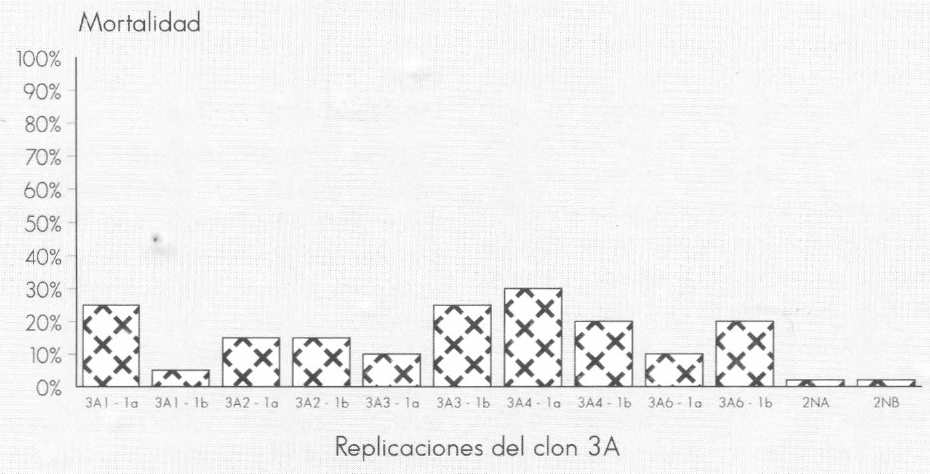
Actividad tóxica de las replicaciones del clon 3A sin inducción con IPTG contra larvas de tercer instar de Culex quinquefasciatus. Lectura de mortalidad a las 48 horas.
Para confirmar que el tamaño de la proteína obtenida correspondiera a un peso molecular de aproximadamente 100 KDa, se procedió a efectuar el análisis por Western blot de la proteína recombinante del clon 3A. Los resultados indican que el clon 3A, una vez que se induce con IPTG, sintetiza una proteína que es reconocida por el antisuero y que comigra con el polipeptido de aproximadamente 100 KDa que se observa en el cultivo completo final de Btmed. Esto, sumado al resultado del bioensayo en el que se demuestra que la proteína recombinante del clon 3A es tóxica para larvas de mosquito, es evidencia clara de que el clon 3A codifica una de las proteínas tóxicas de Btmed con un peso molecular de aproximadamente 100 KDa. Estos resultados sugieren además que la proteína de 100 KDa por sí sola es capaz de ejercer toxicidad contra larvas de C. quinquefasciatus.
El mapa de restricción del fragmento de los genes de la proteína permitió, además, confirmar que la cantidad de ADN presente en el clon 3A tenía información suficiente para codificar una proteína de 100 KDa como la que se identificó en las inmunotransferencias. Con base en los datos que se presentan en el gel de agarosa, se elaboró el mapa de restricción (Fig. 3). Las restricciones con la enzima EcoRI del ADN aislado de las replicaciones del clon 3A revelaron que todos tienen el mismo patrón de corte, liberando un inserto de aproximadamente 4,4 Kb.
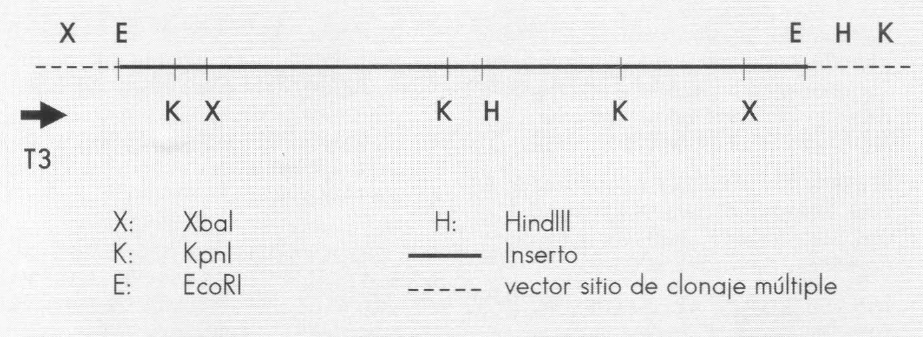
Mapa de restricción parcial del fragmento del clon 3A. La línea gruesa corresponde al inserto de ADN del clon 3A y la línea punteada corresponde al sitio de multiple clonaje del vector pBluescript SK(-). Los sitios de corte de las enzimas de restricción se indican con letras.
Discusión
En este artículo se describe el clonaje y la expresión del gen que codifica la proteína de aproximadamente 100 KDa de Btmed en la cepa de E. coli SOLR. La expresión de este gen cry produjo una significativa mortalidad cuando células completas de E. coli fueron dadas a larvas de tercer instar de C. quinquefasciatus. Resultados similares han sido reportados por varios autores, quienes han usado varias especies de Bacillus y varias cepas de E. coli como huéspedes receptores para la expresión de genes tóxicos tanto de Bt como de B. sphaericus. Estos huéspedes han sido usados para evaluar la toxicidad de proteínas individuales o estudiar su relación estructura-función sin obtener altos niveles de expresión (Baumann y Baumann 1989; Baumann et al. 1987; Baumann et al. 1988; Davidson et al. 1990; De la Torre et al. 1989; Oei et al. 1990; Thanabalu et al. 1991).
Los genes cryIVA, cryIVB, cryIVC, cryIVD y cytA del Bti han sido individualmente expresados en E. coli y las diferencias en la producción de proteína y toxicidad fueron probablemente debidas a diferencias en las células hospedantes, vectores, bioensayos, estabilidad de los polipeptidos y/o a variaciones en la eficiencia del promotor y sitio de unión al ribosoma (Angsuthanasombat et al. 1987, 1992; Chungjatupornchai et al. 1988 Delécluse et al. 1988; Thorne et al. 1986; Waalwijck et al. 1985; Ward et al. 1984; Ward y Ellar 1988).
El gen cryIVB es uno de los pocos genes del cristal de Bti que ha sido bien expresado en E. coli. La proteína de 128 KDa ha sido sintetizada en E. coli TG1 y una vez purificada fue debilmente tóxica a larvas de A. aegypti (Ward y Ellar 1988), y esta misma proteína sobreexpresada en E. coli JM107 fue tóxica a larvas de tercer instar de A. aegypti con una LC50 de 43 ng/ml (Chungjatupornchai et al. 1988).
Ward et al. (1984) clonaron y expresaron en E. coli JM101 un gen de la delta-endotoxina de Bti en el vector pUC12 y se identificaron dos recombinantes produciendo una proteína de 26 KDa. La síntesis de este polipeptido in vivo por E. coli JM101 fue letal a larvas de A. aegypti y citotóxica a células de mosquito in vitro, y recientemente Koni y Ellar (1993) clonaron en E. coli el nuevo gen de la proteína cytB de 29 KDa de Bt ssp. kyushuensis.
La variabilidad en la mortalidad de larvas de C. quinquefasciatus observada por las diferentes colonias del clon 3A puede ser debida a las condiciones de plegamiento de una proteína extraña en la E. coli, en la que no toda la proteína producida es necesariamente doblada de modo que se obtenga un producto funcional.
El tamaño de la proteína tóxica de 100 KDa, reportado en este artículo, coincide con el tamaño de la proteína reportada por Orduz et al. (1994), donde la electroforesis y el Western blot revelaron la presencia en Btmed de proteínas de aproximadamente 100, 80, 75, 67, 65 y dos dobletas de 40-41 y 30-28 KDa y donde la proteína de 100 KDa no presentó reacción cruzada con una mezcla de antisueros (anti-CryIVD, anti-Cry IVA+B y anti-Cyt A) de Bti, confirmando que se trata de una nueva proteína.
Conclusiones
Debido a la presencia de una nueva proteína tóxica en Btmed diferente de las proteínas de Bti, esta subespecie es en el momento el único candidato disponible para reemplazar a Bti en caso de desarrollo de resistencia. Por lo tanto, es muy importante determinar el valor insecticida de las proteínas de Btmed y la caracterización de los genes que las codifican, ya que permitirá el estudio de su estructura y homología en relación con otros genes de proteínas insecticidas ya reportados de las cepas de Bti activas para mosquitos. Esto puede, además, constituir un sistema de estudio muy valioso para evaluar la sinergia de las diferentes proteínas de Btmed en el efecto larvicida y también entre Btmed y otras proteínas tóxicas. Además, el conocimiento genético podría llevar al mejoramiento de las proteínas como plaguicidas durables que sean competitivos con los dañinos plaguicidas químicos.
Agradecimientos
Este trabajo ha sido financiado parcialmente por la Organización Mundial de la Salud y Colciencias.