
Abstract
We report a case of sinonasal phosphaturic mesenchymal tumor (PMT) and conduct a systematic review of the literature to highlight a unique paraneoplastic syndrome associated with PMT. We used English language publications from Medline and Cochrane databases (1970–2013) as data sources. A systematic review of the literature was conducted. All reported cases of head and neck PMTs were included. The presence or absence of the associated paraneoplastic syndrome was noted. We found 33 cases of PMT in the head and neck reported in the literature, 17 of which occurred in the sinonasal area. Approximately 5% of all PMTs are located in the head and neck. Just greater than half are concentrated in the sinonasal area, and the remaining involve various bony and soft tissue structures of the head and neck. PMT is sometimes associated with a paraneoplastic syndrome of tumor-induced (oncogenic) osteomalacia (TIO) causing bone pain, muscle weakness, and pathologic fractures. We present the 18th reported case of sinonasal PMT. A smooth mucosa-covered midline intraseptal mass filling the posterior nasal cavity with destruction and erosion of the skull base was found in an adult male. The patient underwent successful endoscopic resection with wide negative margins and is without recurrence at 24-month follow-up. PMT is a benign, locally aggressive tumor with rare malignant transformation. Knowledge of the bony invasion and destruction caused by this tumor is essential in planning surgical resection with wide negative margins. Familiarity with the associated TIO is essential to investigate for and manage any associated bony morbidity.
Although PMT is rare, awareness of the unique biologic behavior of this entity is essential for management. Surgical resection with wide negative margins should be planned, because the tumor tends to be locally aggressive and invasive. Early curative resection limits destruction of adjacent structures, such as the skull base, orbit and palate. Early evaluation for the associated tumor-induced paraneoplastic syndrome can help identify and limit osteomalacia and its sequelae. The vast majority of reported PMTs are benign, although malignant transformation with distant metastasis has been reported. 2 The sinonasal area is afflicted with a broad range of disparate pathology, and knowledge of the biologic behavior of each rare tumor is essential to the otolaryngologist. We therefore present a brief and pertinent review of PMT.
Materials and Methods
A comprehensive review of the English language literature from 1970 to 2013 was conducted using Medline and Cochrane databases. The following keywords were used: PMT, phosphaturia, head and neck neoplasms, sinonasal, nasopharyngeal neoplasms, nose neoplasms, paranasal sinus neoplasms, oncogenic osteomalacia, TIO, and paraneoplastic syndrome. Bibliographies of obtained reports were also scanned for additional studies. Inclusion criteria included PMTs located in the head and neck, with or without evidence of TIO. Studies that involved PMTs outside the head and neck (e.g., appendicular skeleton and intracranial) were excluded. Head and neck PMTs were then stratified by location as sinonasal versus nonsinonasal.
Results
The comprehensive literature review yielded a total of 404 articles containing PMT cases published between 1970 and 2013, of which 33 were individual case reports of head and neck PMT. Of these, 17 were located in sinonasal area and 16 were in other areas of the head and neck, including mandible, floor of mouth, pharynx, larynx, thyroid, and temporal bone (Table 1).
Case reports of PMT in the head and neck

F = female; M = male; PMT = phosphaturic mesenchymal tumor; TIO = tumor-induced (oncogenic) osteomalacia.
Case Report
A 41-year-old man was referred for management of a nasal mass found during evaluation of progressive nasal obstruction, discolored rhinorrhea, anosmia, dysgeusia, and facial pressure. Nasal endoscopy revealed a large, smooth, mucosa-covered mass splaying the nasal septum posteriorly and filling the nasopharynx (Fig. 1A). A computed tomography (CT) and magnetic resonance imaging (MRI) were obtained and are depicted in Figs. 2 and 3 showing this expansile extracranial mass. Pathological analysis of biopsy tissue showed a sheet-like distribution of monotonous, bland tumor cells in a background of vascular myxoid stroma, consistent with PMT (Fig. 4). Based on this histologic diagnosis, workup for the metabolic derangements of TIO was performed preoperatively. The patient's serum calcium, 1,25-dihydroxyvitamin D, alkaline phosphatase, parathyroid hormone (PTH), and urine phosphate all were normal, suggesting minimal to no tumor secretory function. The patient subsequently underwent successful transnasal endoscopic resection using computerized image guidance. The tumor occupied the nasal septum and was attached broadly to the basisphenoid. It also extended to the ethmoid roof. After tumor resection, the skull base was drilled at the sites of attachment along the planum sphenoidale and ethmoid roof to the posterior frontal table (Fig. 2). No dural penetration or cerebrospinal fluid leak was noted. Final histopathologic diagnosis confirmed negative margins and reconfirmed the diagnosis of PMT (Fig. 4). After 24 months of follow-up, the patient has done well, without evidence of recurrence or development of bony pathology (Fig. 1B).

(A) Rigid 0° endoscopy of left nasal cavity shows a smooth, mucosa-covered mass splaying the posterior septum and filling the entire nasopharynx. (B) Endoscopic 30° view 24 months status after endoscopic resection shows a common nasal cavity after the tumor resection and septectomy, which is well mucosalized and without evidence of recurrence. Solid white arrow shows the base of the drilled-out basi-sphenoid in the midline. Broken white arrow shows the midline ethmoid skull base at attachment of nasal septum, and black arrows depict the most superior vertical attachments of the middle turbinate.
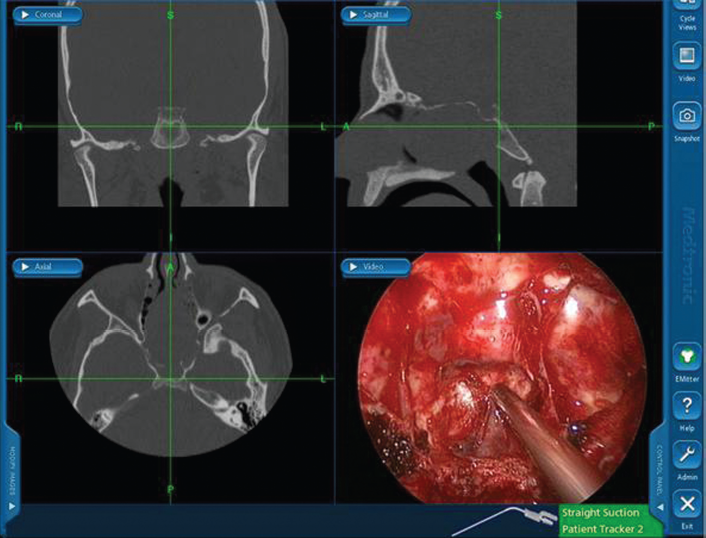
Intraoperative view using the computerized navigation system showing triplanar CT images correlating with tip of suction instrument in intraoperative endoscopic view. The triplanar CT images illustrate the extent of tumor on preoperative imaging, whereas endoscopic image in bottom right shows the sinonasal cavity in real-time with complete tumor resection. Note the splaying of the nasal septum and basi-sphenoid area with tumor abutting ethmoid skull base and extending across the sphenoid rostrum to the clivus on CT images.

Tumor as visualized on MRI. (A) Axial fat-suppressed T2-weighted image demonstrates a heterogeneous mass expanding the posterior right nasal cavity and eroding into the sphenoid sinuses. The mass is predominantly isointense to cerebral gray matter with central areas of T2 hyperintensity (asterisk). (B) Axial fat-suppressed T1-weighted postcontrast image is likewise heterogeneous with central areas of nonenhancement (asterisk). The postcontrast MRI more clearly defines the posterior margins of the tumor (arrows) relative to trapped secretions in the sphenoid sinuses. No cavernous sinus invasion is present.
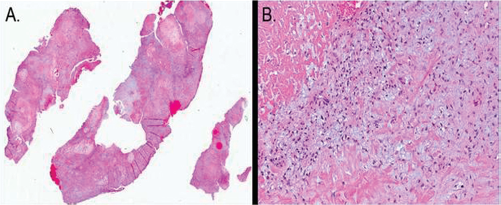
(A) Low power view of sinonasal PMT showing variable cellularity and vascularity. (B) High-power view shows small, stellate cells in a myxoid matrix with a prominent vascular stroma and areas of hyalinization.
Discussion
PMTs of the head and neck are extremely rare. The diagnosis is commonly delayed until late in the course of disease, once signs of osteomalacia are present or local invasion of the tumor has caused symptoms. 3 Patients with the paraneoplastic syndrome of TIO can present with nonspecific, progressive bone pain, loss of height, muscle weakness, and/or fractures. 2 The tumor affects patients within a wide age range of 3 to 73 years, and there appears to be no gender predilection. 2
Ninety-five percent of PMTs are found in the extremities or appendicular skeleton, whereas only 5% originate in the craniofacial region.2,4–6 Historically, it has been reported that, of the PMTs located in the craniofacial region, the vast majority (more than 80%) are found in the nose and paranasal sinuses.2,7 However, this literature review reports all the cases to date of head and neck PMTs, and we report that just greater than half are sinonasal in location, with the remaining located in other bony and soft tissue sites within the head and neck, including the mandible, floor of mouth, pharynx, larynx, thyroid, and temporal bone. There are 17 cases of sinonasal PMT reported in the literature, and these are summarized in Table 1.1,4,7–35 Histopathologically, PMTs are composed of spindle-shaped or stellate cells with low nuclear grade embedded in a mesenchymal myxoid or myxochondroid matrix with “grungy” or flocculent calcification. 2 A notable feature of PMT is the rich intrinsic microvascular supply, which mimics hemangiopericytoma. The osteocytes in PMTs are responsible for causing osteomalacia by producing fibroblast growth factor 23 (FGF23), which acts to inhibit sodium-phosphate cotransport in the proximal tubular cells of the kidney leading to phosphaturia and eventual bone demineralization.36–40 Interestingly, autosomal dominant hypophosphatemic rickets, which is caused by an activating mutation in the FGF23 gene, is clinically indistinguishable from TIO. 41
PMT should be suspected whenever nonfamilial hypophosphatemic osteomalacia is diagnosed. Workup should include a thorough history and physical, followed by radiologic search for tumor in the extremities and head. 10 For the otolaryngologist, patients presenting with a nasal mass and biopsy consistent with a PMT should be evaluated for osteomalacia. Preoperative laboratory values, including serum calcium, 1,25-dihydroxyvitamin D, alkaline phosphatase, PTH, and urine phosphate, should be obtained. If TIO is present, one would expect serum calcium and serum PTH to be normal but 1,25–dihydroxyvitamin D and serum alkaline phosphatase to be diminished. Bone scans and/or FGF23 gene amplification may also be considered in determining secretory status of the tumor. Treatment is surgical excision with wide margins. Tumor resection results in a dramatic reversal of the hypophosphatemia and hyperphosphaturia seen in TIO, with often striking improvement in skeletal mineralization. 2 Persistent metabolic derangements can predict incomplete excision or lesion recurrence. 1 The primary treatment of rare malignant variants is also surgical resection, because a successful chemoradiotherapy program has not been established. 33
The majority of reported PMTs are associated with TIO. However, not all PMTs cause this paraneoplastic syndrome (Table 1).2,14,19,33,34,42,43 In a large review of 109 mesenchymal tumors by Folpe et al., only three PMTs did not demonstrate TIO. 2 Given the sparse case reports of PMTs without TIO in the literature, it is plausible that these cases are underreported. As a result, the precise percentage of PMTs with TIO versus without TIO remains unknown. Current theories suggest that there is a spectrum of FGF23 expression that dictates whether or not the tumor remains clinically silent. Another theory proposes that some patients are able to compensate for the increased production of FGF23 better than others. Alternatively, perhaps the secretory function of the tumor evolves with time and can be prevented by early resection. Further research may help clarify this.
Conclusions
PMTs of the head and neck are usually benign and can be associated with the paraneoplastic syndrome TIO. Approximately half of head and neck PMTs are located in the sinonasal area. Knowledge of this rare entity is important to proactively identify and treat any associated bony morbidities in affected patients. Surgical excision is curative and may prevent or reverse the progression to osteomalacia. A complete resection with wide negative margins is required to limit local destruction, prevent recurrence, and obliterate the source of protein secretion implicated in TIO.
Footnotes
The authors have no conflicts of interest to declare pertaining to this article