
Abstract
Steroid hormone receptor (SHR) expression and changes in SHR expression compared to basal levels, whether upregulated, down-regulated, or mutated, form a distinguishing feature of some breast, ovarian, and prostate cancers. These receptors act to induce tumor proliferation. In the imaging context, total expression together with modulation of expression can yield predictive and prognostic information. Currently, biopsy for histologic assessment of SHR expression is routine for breast and prostate cancer; however, the technique is not well suited to the heterogeneous tumor environment and can lead to incorrect receptor expression assignment, which precludes effective treatment. The development of positron emission tomography (PET) radioligands to image receptor expression may overcome the difficulties associated with tumor heterogeneity and facilitate the assessment of metastatic disease.
STEROID HORMONE RECEPTORS (SHRs), which are frequently overexpressed in tumors, are at the forefront of targeted cancer therapy, with widespread targeting in endocrine-related treatments. The upregulation of these receptors in diseased tissue provides a distinguishing feature of some breast, ovarian, and prostate cancers.1,2 SHRs can provide important information on the prospect of response to endocrine therapy; therefore, assessment of expression provides important clinical data for therapeutic management of patients. Clinically relevant SHRs include estrogen receptor α (ERα) and progesterone receptor (PR), which are often upregulated in breast and ovarian cancers, together with the androgen receptor (AR), an important target in prostate cancer.
Immunohistochemistry (IHC) is currently the gold standard technique for evaluating SHR expression in oncology; an invasive core needle biopsy of the lesion is obtained, which is prepared so that positive cells can be visualized and counted. The SHR status of the tumor is then determined from a given threshold of positivity. Intratumor heterogeneity and the practical impossibility of assessing all metastatic lesions can be a fundamental flaw of the results, leading to inaccurate assignment of SHR status; IHC assay in breast cancer only correctly predicts the response to endocrine therapy in 50 to 60% of cases.3,4 A systematic review by the American Society of Clinical Oncology (ASCO) showed that 20% of all SHR statuses determined by IHC may be inaccurate. 5 Serial biopsy of lesions has been introduced as a means to overcome the problems associated with simple biopsy assessment, particularly for monitoring treatment response. 6 However, serial biopsy sampling may still suffer from the drawback that the results can be confounded by tumor heterogeneity.
The use of positron emission tomography (PET), a sensitive, minimally invasive imaging modality used routinely in the clinic, may overcome the inaccuracies of IHC arising from both intra- and intertumor heterogeneity; whole-body scans can allow the entirety of metastatic and primary lesions to be imaged and are therefore well suited to longitudinal studies. 7 PET has the potential to be used for functional imaging where not only receptor expression but also receptor function can be determined in heterogeneous tissue. 8 This review summarizes the current status and future directions of SHR imaging using PET.
ER Radioligands
Upregulation of SHR can be of prognostic or predictive relevance in selecting patients who may benefit from endocrine therapy; dichotomizing SHR+ and SHR– lesions allows stratification of patients into potential responders and nonresponders of therapy. For example, tamoxifen is a leading antiestrogen prescribed to women with ER+ tumors as a neoadjuvant systemic treatment before surgery or as an adjuvant treatment to reduce the probability of relapse; patients with ER+ tumor status generally have a better prognosis than those with ER– because the tumor growth is driven by ERα signaling, which can be inhibited by tamoxifen. 9 Therefore, assessing ERα function in both primary and metastatic lesions is critical in determining the suitability of endocrine therapy for a given patient.
The ERα has long been of interest as a target for imaging because of its involvement in the proliferation of some breast and ovarian cancers. McElvany and colleagues were instrumental in the synthesis of ER-targeted radioligands, by synthesizing gamma-emitting 16α[125I]iodoestradiol (I-E2) with a specific activity of 33 to 55 GBq/μmol and characterizing its binding and tissue uptake in vivo; the radioligand was found to concentrate in the rat uterus specifically to ER with high affinity. 10 A more detailed biodistribution study of [125I]I-E2in mammary adenocarcinoma tumor–bearing and normal rats showed that although uterus/blood ratios were initially high, [125I]I-E2 was quickly metabolized by the liver within 1 hour. 11 Focus shifted toward the synthesis of more metabolically stable compounds, which allow sufficient time for equilibration and imaging. Brominated estradiol, 16α [77Br] bromoestradiol-17β, showed favorable target tissue uptake in immature and mature female rats, 12 with adequate uterus/blood uptake ratios and limited nontarget tissue/blood ratios. 12 McElvany and colleagues compared 16α [77Br] bromoestradiol-17β and [125I]I-E2 in vivo in immature female rats. 10 Although [125I]I-E2 showed slightly higher uptake in the uterus after 1 hour postinjection, 16α [77Br]bromoestradiol showed significantly greater target uptake at later time points. Thyroid uptake was significantly higher for I-E2 compared to 16α[77Br] bromoestradiol-17β. These radioligands validated the concept that detecting ERα expression was a feasible proposition for in vivo imaging, but the poor resolution of the single-photon emission computed tomography (SPECT) isotopes precluded the accurate quantification necessary for human studies, and attention turned to PET radioligands as potential alternatives. 13
Synthesized by Kiesewetter and colleagues, 16α-[18F]fluoro-17/β-estradiol ([18F]FES), a fluorine-18 (18F) substituted analogue of estradiol (Figure 1), was radiolabeled via nucleophilic displacement of a triflate precursor followed by a hydrolysis and reduction step. 13 [18F]FES was achieved in a 30% radiochemical yield (RCY) with a specific activity of around 7.4 GBq/μmol (Figure 2). Lim and colleagues synthesized [18F]FES from a cyclic sulfone pre-cursor in 30 to 45% RCY with a specific activity of 37 GBq/μmol (see Figure 2). 14 Kil and colleagues developed a novel nosylate precursor to access [18F]FES with a labeling and hydrolysis step in 40 minutes with purification 15 ; [18F]FES was synthesized with a specific activity of 84.2 GBq/μmol and an RCY (decay corrected) of 19 to 24% (see Figure 2). 15
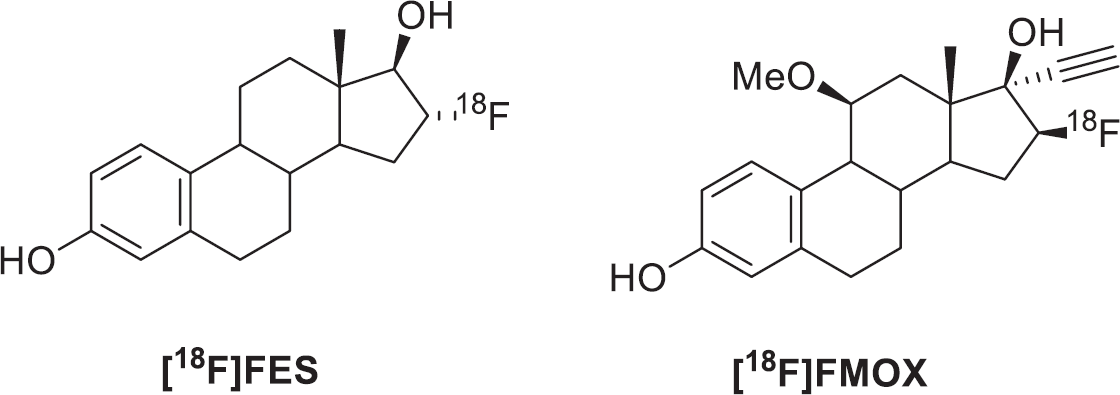
16-α-[18F]Fluoro-17-β-fluoroestradiol ([18F]FES) and 16β-[18F]fluoromoxestrol ([18F]FMOX).

[18F]FES exhibited a relative binding affinity (RBA) of 80% compared to estradiol and proved to be a successful candidate in the development of positron-emitting estrogens. 13 Biodistribution of [18F]FES in immature female rats showed selective uptake in ER+ tissue, which prompted further study into the suitability of PET imaging with this ligand. 13 Mintun and colleagues progressed [18F]FES to a clinical trial to determine if specific uptake into ER+ lesions could be imaged; excellent correlation between ER expression and radioligand uptake was demonstrated. 16 The effectiveness of [18F]FES had been demonstrated and provided a foundation for more detailed clinical evaluation of the radioligand. McGuire and colleagues used [18F]FES in a clinical trial to assess uptake in metastatic breast carcinoma. 17 Increased [18F]FES uptake was seen in 53 of 57 metastatic lesions with only two false positives; this work highlighted the advantage of assessing metastatic lesions by PET imaging that otherwise would have been difficult to assess by other techniques.
Linden and colleagues showed that [18F]FES uptake could predict response to endocrine therapy in breast cancer. 18 The clinical study involved 47 patients with recurrent or metastatic breast cancer; baseline [18F]FES and [18F]fluorodeoxyglucose ([18F]FDG) scans preceded endocrine therapy, and posttreatment [18F]FES-PET followed after sufficient time for tamoxifen washout. Patients with [18F]FES uptake with an initial standardized uptake value (SUV) less than 1.5 did not respond to endocrine therapy; however, 34% of patients with an SUV higher than 1.5 responded to treatment. Patients who did not overexpress HER2/neu (11 of 24) with an SUV higher than 1.5 responded to endocrine therapy. No patients with absent [18F]FES uptake responded to treatment; quantitative assessment of [18F]FES uptake significantly correlated with response. A similar study by Dehdashti and colleagues investigated whether increased tumor uptake of [18F]FDG measured at early time points after administering tamoxifen therapy predicted hormone-responsive breast cancer. 19 [18F]FDG and [18F]FES were used for imaging before and after 7 to 10 days of tamoxifen therapy; a clinical flare response was demonstrated in all responders. In 2009, Dehdashti and colleagues demonstrated the use of [18F]FDG and [18F]FES to determine response to treatment after administering endocrine therapy in 51 postmenopausal women with advanced ERα+ breast cancer. 20 Baseline scans of [18F]FES and [18F]FDG were obtained before administering estradiol (30 mg). Response to endocrine therapy was seen in 17 patients, and 34 did not respond; all responders to endocrine therapy displayed metabolic flare by increased [18F]FDG uptake (> 12% increase in SUV). The uptake of [18F]FES was higher (SUV = 3.5 ± 2.5) compared to that in nonresponders (–4.3 ± 11.0, p < .0001). Ellis and colleagues later showed that administering 6 mg of estradiol was sufficient to provide clinical benefit and that [18F]FDG uptake could be used as a predictive biomarker in patients. 21
[18F]FES PET has also been successful in providing ER status information of tumor lesions where a patient presents a clinical dilemma; that is, patients who have inconclusive or ambiguous pathologic assessment of the lesion or biopsies are not feasible. 22 A clinical trial involved [18F]FES PET measurements of 33 patients; [18F]FES+ uptake was observed in 22 patients. 22 The assessment of metastatic lesions in the bone by [18F]FES proved to be sensitive, identifying 341 lesions compared to 246 by conventional imaging; quantification of liver metastases was poor due to the high physiologic background. This study showed that [18F]FES can help solve a clinical dilemma and support therapy decisions where the standard workup procedure is inconclusive. Although [18F]FES has been proven successful predicting response to endocrine therapy, it is unable to provide prognostic insight into the success of endocrine therapy during treatment as a result of the drug molecule occupancy of the receptor.
The synthesis of estradiol derivatives with improved target affinity and low affinity for off-target sites such as sex hormone-binding globulin (SHBG) ensued to improve on [18F]FES, which exhibits a binding affinity that is only 80% that of estradiol. 23 A [18F]FES derivative, 16β-[18F]fluoromoxestrol ([18F]FMOX), was identified as a selective and metabolically stable estrogen that may be suitable for translation into the clinic for imaging ER expression in breast cancer (see Figure 1). [18F]FMOX appeared to exhibit a decreased rate of metabolism compared to [18F]FES, with nearly a fourfold increase in uterine uptake in an immature rat model.23,24 Despite what appeared to be desirable ligand characteristics, [18F] FMOX was unable to identify ER+ tumors in a clinical study. 25 Jonson and colleagues evaluated the metabolism of [18F]FMOX in comparison with [18F]FES in isolated hepatocytes from rat, baboon, and human species and discovered that SHBG, absent in rat but present in baboon and human, may be responsible. 25 [18F]FMOX was designed to have a low affinity to SHBG with a high affinity for ERα and showed excellent metabolic stability in the rat; however, [18F] FES was demonstrated to experience a protective effect from metabolism as a result of SHBG and was metabolized to a lower extent than [18F]FMOX in primates and humans. 25 The ability of SHBG to improve specific uptake by delivering bound ligand into cells expressing membrane receptors for SHBG may assist by facilitating [18F]FES uptake into target cells. 25
Although 16α-[18F]FES has been the most studied and most successful SHR radioligand, rapid metabolism hinders optimal quantification of ER density. 26 Estradiol derivatives substituted at the C-7α position have been shown to be tolerated by the ER and may provide a radioligand that results in fewer circulating radiometabolites.
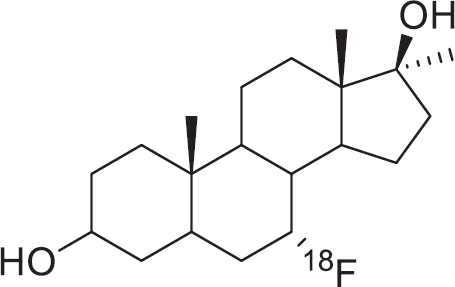
Okamato and colleagues synthesized 7α-(3-[18F]fluoropropyl) estradiol (C3-7α-[18F]FES) and evaluated its potential for detecting ER expression in vitro and in vivo (Figure 3). 27 The radiosynthesis of C3-7α-[18F]FES was achieved from a tosylated precursor using [18F]KF/Kryptofix 222 with an RCY of 30.7% ± 15.1% (decay corrected), and a specific activity of 32.0 ± 18.1 GBq/umol was achieved. 27 The binding affinity of C3-7α-[18F]FES was determined in vitro and was comparable to that of 16α-[18F]FES. The in vivo biodistribution study showed that C3-7α-[18F]FES was selectively taken into the uterus with reversible kinetics. 27 The level of C3-7α-[18F]FES uptake was significantly lower than that of 16α-[18F]FES, but receptor specificity was determined by dose-dependent inhibition of C3-7α-[18F]FES uptake with estradiol coinjections. Defluorination of C3-7α-[18F]FES was speculated by gradual increase in bone radioactivity. 27 Although C3-7α-[18F]FES exhibited favorable receptor specificity, in vivo defluorination of this radioligand led to increased uptake of radioisotope in bone, which is likely to have confounding effects on receptor quantification. Further optimization of this radioligand is required so that receptor specificity is maintained and in vivo defluorination is reduced.

7α-(3-[18F]Fluoropropyl) estradiol (C3-7α-[18F]FES).
PR Radioligands
The PR is a ligand-dependent transcription factor controlled by progesterone binding. 28 In healthy tissue, PR is involved in differentiation of the endometrium, pregnancy, and mammary development 28 ; in breast cancer, PR expression can be used to predict response to endocrine therapy. Functioning ER can upregulate PR; therefore, PR can be used as a surrogate biomarker to indicate the presence of a functioning estrogen response pathway. Around 50% of patients with ER+ status fail to respond to tamoxifen as a result of intrinsic or acquired resistance. 29 Mechanisms of resistance could be attributed to alterations in ER function, drug pharmacology, or genetic/environmental alterations in tumor cells. 29 Patients with ER+/PR+ lesions are likely to respond to endocrine therapy as PR positivity indicates a functioning estrogen response pathway; by contrast, patients with ER–/PR– lesions are unlikely to respond. Patients who respond to endocrine therapy ultimately develop a resistant phenotype. 29 Determining PR expression as well as ER expression allows patients with ER+/PR– and ER–/PR+ lesions to be differentiated; using the PR as a surrogate biomarker for ER expression and function may allow acquired resistance to be monitored, which could aid treatment planning.
The development of PET radioligands to image PR as a surrogate biomarker of ER expression is an attempt to overcome the confounding effects on imaging associated with drug saturation of ER as a result of endocrine therapy. The synthesis of radiolabeled progestins for use in PET imaging of breast cancer has slowly developed in the background of ERα imaging efforts. The first PR PET ligand was 21-fluoroprogesterone; however, it was unable to image PR. 30 Many early attempts at synthesizing PR targeted ligands were unsuccessful due to low affinity and low specific activity of radiolabeled compounds, giving rise to poor tissue selectivity.30–32
A fluoroethyl analogue of ORG2058, 21-fluoro-16α-ethyl-19-norprogeserone (FENP), demonstrated high affinity for PR (RBA of 6,000% relative to progesterone). 33 Pomper and colleagues described the radiosynthesis to obtain [18F] FENP (Figure 4) and showed the ligand to be a suitable candidate for imaging PR+ breast carcinoma by evaluating uterine uptake in estrogen-primed immature rats. 33 [18F]FENP was prepared from a triflate precursor with [18F]tetrabutylammonium fluoride in a 30% RCY (decay corrected) with a specific activity of 25 to 50 GBq/μmol. 33 The promising in vivo results of [18F]FENP resulted in a clinical trial for PET imaging of PR in patients with primary breast carcinoma. However, the ligand was unsuccessful in identifying PR positivity in humans, with only 50% of PR+ tumors identified by PET, from which the investigators concluded that radioligand uptake did not correlate with tumor PR expression. 17 A later study revealed that 20-hydroxysteroid dehydrogenase (20-HSD), present in human blood but absent from rodent blood, was responsible for converting [18F]FFNP into an inactive analogue, which had confounding consequences on imaging PR expression in humans. 34 To overcome the problem of metabolic stability but retain high PR affinity, the 16α, 17α-dioxolane system was identified as a suitable candidate for reducing the sensitivity of metabolism by 20-HSD. The dioxolane system could bear an [18F]fluorophenyl substituent as a convenient location for radiolabeling. Although the ligand was metabolically stable and selective for target tissue, the increased lipophilicity led to nontarget uptake in the fat. 35 Further development of the 16α,17α-dioxolane system was necessary to maintain the high affinity and metabolic stability but reduce the lipophilicity to attenuate uptake in nonspecific tissues.

21-[18F]Fluoro-16α-ethyl-19-norprogesterone ([18F]FENP).
An adaptation of the 16α,17α-dioxolane progestins synthesized by Kochanny and colleagues and Kym and colleagues led to the synthesis of [18F]FFNP (Figure 5).35,36 Buckman and colleagues synthesized 16α,17α-furfural acetal and acetylfuran ketal progestins, which exhibited high affinity and selectivity for PR in target tissue. 37 [18F]FFNP maintained the 16α,17α-dioxolane moiety as reported by Kochanny and colleagues 35 to protect the 20-keto structure against metabolism by 20-HSD; furthermore, the introduction of a less lipophilic moiety (16α,17α-furfural acetal and acetylfuran ketal) reduced nonspecific binding. Radiolabeled compounds were prepared from triflate precursors using [18F]tetrabutylammonium fluoride in 2 to 13% RCY (decay corrected) with specific activities greater than 44 GBq/μmol; favorable biodistribution and selectivity were attained for in vivo target tissue in rats. A clinical trial of [18F]FFNP in women with newly diagnosed breast cancer was designed to evaluate the effectiveness of using [18F]FFNP for PET imaging of PR expression in breast cancer as well as establishing the safety and dosimetry of the compound. Although qualitative assessment of [18F]FFNP uptake showed significant differences in PR+ and PR– patients, the experiment revealed that there was no significant difference between SUVmax (tumor maximum standardized uptake value) in PR+ and PR– tumors. The tumor/normal tissue ratio (T/N) showed significant differences between PR+ and PR– patients with correlation between semiqualitative Allred scoring and T/N ratio. 38 The dosimetry of [18F]FFNP showed the gallbladder as the dose-limiting organ receiving an average radiation dose of 0.113 mGy/MBq; the whole-body dose was 0.015 mGy/MBq, with an effective dose of 0.020 mSv/MBq. 38

21-[18F]Fluoro-16α,17α-[(R)-(1′-a-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione ([18F]FFNP).
Examining the changes in concentration of PR on hormone challenge was initially examined by Howell and colleagues using patient biopsy. 39 A similar experiment using [18F]FFNP to predict response to endocrine therapy was examined by Fowler and colleagues in a preclinical model of breast cancer. 40 Mice were implanted with mammary cell lines (SSM1, SSM2, and SSM3) into the right thoracic mammary fat pad, and an imaging study was devised to determine if changes in ERα/PR expression were of predictive value to tumor response after endocrine therapy. The implanted cell lines had basal SHR levels evaluated using PET; the SSM3 tumor was selected for subsequent evaluation as it showed high [18F]FES, [18F]FFNP, and [18F]FDG uptake. The study showed that response to treatment could be significantly correlated to [18F]FFNP uptake and could be used to determine early effects of treatment even before measurable changes in anatomic tumor growth. 40 Imaging with [18F]FES showed a decrease in uptake after treatment as a result of receptor occupancy with drug molecules. The study also highlighted that a single assessment of PR expression by [18F]FFNP PET had no predictive value in determining responders/nonresponders; however, pretreatment and early posttreatment assessment provided the best predictive value. 40 Using the same animal model, Chan and colleagues evaluated the potential of serial [18F]FFNP imaging to monitor acute functional changes of ERα signaling during estrogen deprivation treatment 41 ; [18F]FES and [18F]FDG were used to monitor tumor glucose metabolism and ERα expression. Mice with estrogen-sensitive tumors showed the same level of [18F]FES and [18F] FDG uptake pre- and post-treatment; however, [18F]FFNP uptake was reduced at an early 3-day posttreatment time point as a result of a reduction in PR expression confirmed by IHC assay. 41 [18F]FFNP uptake remained unchanged in endocrine-resistant tumors. This study proved that acute changes in ERα function can be measured by [18F]FFNP, offering potential as a robust and effective approach to predicting tumor response to endocrine therapy. 41
The range of PR imaging agents was extended by derivatizing Tanaproget, a high-affinity nonsteroidal ligand developed by Wyeth Pharmaceuticals (Collegeville, PA), the potency of which is equivalent to that of steroidal progestins (EC50 = 0.15 nM). 42 Nonsteroidal ligands are more selective than steroidal ligands, limiting cross-reactivity to other SHRs, which may be advantageous in accurately quantifying tracer uptake. Zhou and colleagues synthesized a focused library of Tanaproget derivatives containing fluorine to locate suitable positions for substitution that would not inhibit ligand binding. 43 Substitution was evaluated for both the C4-position (R1) and the N-pyrrole position (R2), with varying length of the fluoroalkyl substituent (Figure 6); affinity was determined by radiometric binding assay using [3H]R5020. Substitution of any moiety larger than a methyl group at the R2 position resulted in a rapid reduction in binding affinity; however, substitutions were well tolerated in the R2 position. 43 Substitution of the R1 position introduced chirality into the molecule, of which the effects were examined computationally. The R-enantiomer and S-enantiomer resulted in changes in dihedral angle between the 5-cyanopyrrole and benzoxazinthione moiety, changing the orientation of the molecule in the binding pocket; it was concluded that the S-enantiomer allowed the molecule to adopt a lower energy dihedral angle and therefore may have a higher affinity. 43 Guided by Structure Activity Relationship (SAR) data, the synthesis of [18F]fluoropropyl-Tanaproget ([18F]FPTP) was described and tissue biodistribution was evaluated in estrogen-primed immature female rats. 44 Radiosynthesis of [18F]FPTP was achieved in three steps, 140 minutes from the end of bombardment from a mesylate precursor with an RCY of 5% (decay corrected); specific activity was reported as > 20 GBq/μmol. The tissue distribution was determined at 1 and 3 hours postinjection in immature female rats that had PR levels induced in the uterus by pretreatment with estrogen; uptake was confirmed at each time point by coinjection with cold compound to saturate the PR and show that uptake was mediated by the PR. 44 The uterus/blood and uterus/muscle biodistribution showed increased uptake of tracer, which could be efficiently reduced by coinjection of cold compound; uptake of radiotracer in the bone remained low, suggesting that defluorination was not occurring. Reduction of tracer uptake in nontarget tissues (kidneys and liver) was not observed on coinjection with cold compound. 44
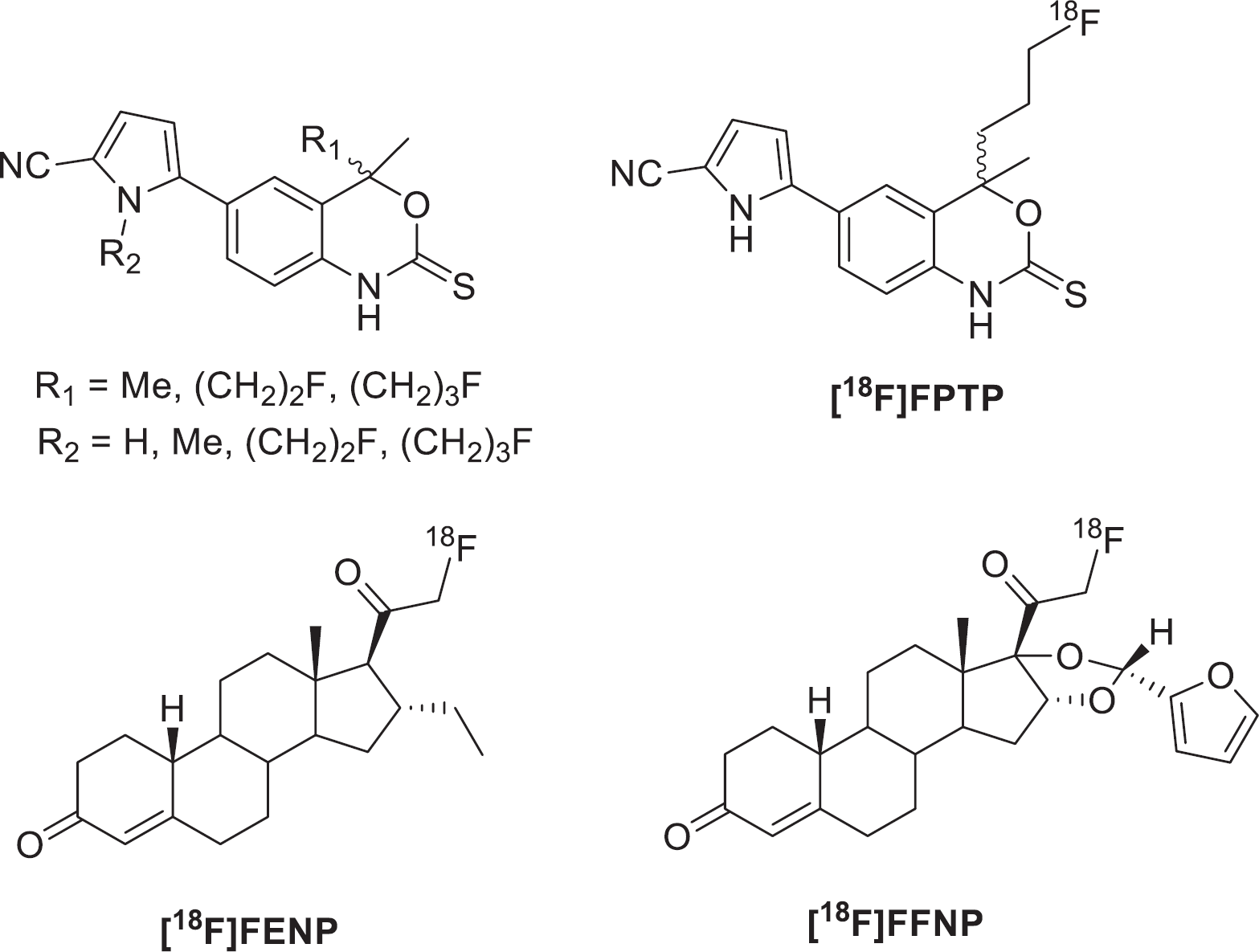
Tanaproget derivatives including [18F]fluoropropyl-Tanaproget ([18F]FPTP) with steroidal progestins 21-[18F]Fluoro-16α-ethyl-19-norprogesterone ([18F]FENP) and 21-[18F]Fluoro-16α,17α-[(R)-(1′-a-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione ([18F]FFNP) R1 = C4 position; R2 = N-pyrrole position.
Lee and colleagues compared the binding affinities, biophysical properties, and tissue biodistribution of [18F]FENP, [18F]FFNP, and [18F]FPTP (Table 1). 44 The highest affinity compound for the PR was [18F]FENP, exhibiting over three times the RBA of FFNP and FPTP. Cross-reactivity of progestins to the glucocorticoid receptor (GR), relative to dexamethasone, and AR, relative to R1881, showed that FPTP had the lowest affinity to both receptors; similar results were obtained for FENP. FFNP was a potent ligand for the GR with an RBA of 365; the AR-RBA of FFNP was 0.53, the highest in the group. The highest uptake of radiotracer in the uterus of immature estrogen-primed female rats was seen for [18F]FENP; however, metabolic instability in human blood prevented further evaluation of this ligand. [18F]FFNP and [18F]FPTP showed comparable tissue distribution in the uterus after 3 hours; however, [18F]FFNP exhibited considerable uptake in the bone at both the 1- and 3-hour time points, suggesting in vivo defluorination. 44
Comparison of Binding Affinities for Three 18F-Labeled Progestins: [18F]FENP, [18F]FFNP, and [18F]FPTP using [3H]R5020 as a reference standard.

AR = androgen receptor; [18F]FENP = 21- [18F]Fluoro-16α-ethyl-19-norprogesterone; [18F]FFNP = 21- [18F]Fluoro-16α,17α-[(R)-(1′-a-urylmethylidene) dioxy]-19-norpregn-4-ene-3,20-dione; [18F]FPTP = [18F]fluoropropyl-Tanaproget; GR = glucocorticoid receptor; PR = progesterone receptor.
RBA is relative binding affinity value relative to R5020 as a standard (KD 0.4 nM).
AR Radioligands
The AR is a ligand-activated transcription factor that mediates normal prostate function; the AR is also implicated in the development and proliferation of prostate cancer. The natural ligands for the AR are testosterone and 5α-dihydrotestosterone (5α-DHT), which bind to receptor and induce transcription; however, this can lead to uncontrolled growth of prostate cells, resulting in a malignant phenotype. 45 The proliferation of some prostate cancers is androgen dependent and will respond to endocrine therapy, which blocks the AR to prevent transcription. Resistance to treatment is ultimately acquired, and endocrine therapy fails. 46 Structural mutation of the AR is one mechanism for ligand-independent activation by reducing the affinity of the ligand for the receptor.45,47
At the initial diagnosis, 80 to 90% of prostate cancers are androgen dependent; treatment with endocrine therapy by blocking AR and reducing androgen concentration is effective for management of prostate cancer. 45 Unlike ER and PR, AR expression does not indicate any predictive or prognostic value in determining the response to endocrine therapy. Ligand-independent activation of AR is known as antiandrogen withdrawal syndrome and affects between 30 and 50% of patients. 48 AR silencing allows ligand-independent activation and reduces the specificity of ligand binding; the receptor is able to bind other members of the SHR family, for example, progestin, estrogen, and antiandrogens, which could result in confounding effects for treatment and imaging as receptor specificity is reduced. 49 There are many mechanisms behind AR silencing; mutations in the ligand-binding domain are most common; however, there are more than 30 substitutions in other parts of the receptor. 50 Mutations are disruptive in the context of therapeutic efficacy because they allow the AR to become promiscuous and bind other circulating steroids. Determining when the AR becomes mutated could provide potential for identifying patients who are developing resistance to treatment; AR mutations are absent from the healthy prostate, but the presence in prostate cancer could give rise to the potential for molecular imaging to identify when patients acquire resistance to treatment and stop responding to treatment so that alternative regimens can be implemented. 47 Identifying a radioligand that will bind to mutated AR selectively may be a challenging feat and has not yet been implemented.
Gamma-emitting radionuclides (77Br, 82Br, 125I, 175Se) have been used to radiolabel androgen derivatives for potential application as AR imaging agents; however, these ligands were problematic, with metabolic elimination of the radionuclide, low affinity, and poor specific activity.51–55 In keeping with previous observations from the development of ER imaging agents, PET isotopes provided the scope for superior quantification compared to SPECT isotopes. Liu and colleagues synthesized six 18F-radiolabeled androgens as potential ligands for imaging prostate cancer and determined their tissue distribution in diethylstilbestrol-treated male rats. 56 These ligands included 16β-fluorine substituted androgens (Table 2), testosterone (16β-FT), DHT (16β-[18F]FDHT), mibolerone (16β-[18F]FMib), and 7α-methyl-19-nortestosterone (16$b-TFMNT); also, the synthesis of 16α-fluoro substituted 7α-methyl-19-nortestosterone (16α-FMNT) and 20-fluoro substituted metribolone (16α-FR1881). Specific binding to the AR was determined by administering a saturating dose of testosterone. The six compounds exhibited relatively high affinity for the AR and selective in vivo uptake in target tissue. 56 This research represents the first time that relatively high affinity and selective 18F-radiolabeled androgens had been synthesized and presented many promising candidates for further development and validation. The six compounds were synthesized by SN2 displacement using triflate precursors or cyclic sulfate derivative with [18F]tetrabutylammonium fluoride; specific activity of these compounds ranged from 11 to 63GBq/μmol apart from the synthesis of 16α-FMNT, which had low specific activity of 0.57 GBq/μmol as a result of an unknown coeluting contamination. 56
Comparison of Binding Affinities of 18F-Substituted Androgens for AR, PR, and SHBG

AR = androgen receptor; 16β-[18F]FDHT =16β-fluorine substituted [18F]-fluoro-5α dihydrotestosterone; 16β-[18F]FMib = 16β-fluorine substituted [18F]-fluoro-5α mibolerone; 16α-[18F]FMNT = 16α-fluorine substituted [18F]-fluoro-5α 7-methyl-19-nortestosterone; 16β-[18F]FMNT = 16β-fluorine substituted [18F]-fluoro-5α 7-methyl-19-nortestosterone; 16β-FT = 16β-fluorine substituted [18F]-fluoro-5α testosterone; PR = progesterone receptor; SHBR = sex hormone-binding globulin.
RBA is relative binding affinity value relative to R1881 as a standard (KD 0.6 nM).
Relative to R5020 (KD 0.4 nM).
Relative to estradiol (KD 1.6 nM).
The highest affinity 16β-fluoro substituted androgen was 16β-[18F]FDHT, with an RBA of 42.7 for the AR (Figure 7); this ligand also exhibited the lowest cross-reactivity for the PR with an RBA of 0.12 with a strong affinity for SHBG. Evaluation in vivo with a baboon animal model expressing SHBG showed that 16β-[18F]FDHT remained unmetabolized and at a concentration sixfold higher than 16β-[18F]FMib and 20-[18F]FMib; it was speculated that stability provided by SHBG may increase uptake of the radioligand. 57 The lowest affinity 16β-fluoro substituted androgen was 16β-FT, which only exhibited an RBA of 2.1 for AR. The prostate/background ratio of 16β-fluorine androgens was relatively high; however, rapid in vivo defluorination of 16β-FMNT, 16β-[18F]FDHT, and 16β-FT resulted in poor prostate uptake (%ID/g) despite 16β-FMNT and 16β-[18F]FDHT having relatively high binding affinities; it was assumed that 16β-FT may be converted to 16β-[18F]FDHT in vivo. It was speculated that the phenomenon of poor prostate uptake (%ID/g) but a high prostate/background ratio may be accounted for by the defluorination process, leading to rapid clearance of radioactivity from the blood and nontarget tissues.

(A) 16β-[18F]FDHT, (B) 16β-[18F]FMib, (C) 20-[18F]FMib, (D) 16β-FMNT, (E) 16β-F. 16β-[18F]FDHT = 16β-fluorine substituted [18F]-fluoro-5α dihydrotestosterone; 16β-[18F]FMib = 16β-fluorine substituted [18F]-fluoro-5α mibolerone; 20-[18F]FMib = 20-fluorine substituted [18F]-fluoro-5α mibolerone; 16β-[18F]FMNT = 16β-fluorine substituted [18F]-fluoro-5α 7-methyl-19-nortestosterone.
Compound 16α-FMNT exhibited an RBA of 21.9 for the AR with low RBA for PR (5.7) and SHBG (4.0). Defluorination in vivo remained very low, and the radioligand showed a very high prostate/muscle ratio after 4 hours. Interestingly, 20-FR1881, which exhibits an RBA for the AR similar to that for 16α-F-MNT and low defluorination, showed the lowest target tissue uptake of all six ligands, suggesting that the relationship between RBA and tissue uptake is tenuous.
A clinical trial was established to evaluate the viability of in vivo targeting of AR using 16β-[18F]FDHT in patients with metastatic prostate cancer. The clinical trial was composed of seven patients with 57 lesions in bone and soft tissue identified by [18F]FDG and correlated with bone scintigraphy, computed tomography (CT), or magnetic resonance imaging (MRI). 58 Of these 57 lesions, 79% took up 16β-[18F]FDHT; importantly, 16β-[18F]FDHT taken up into the tumor bound with greater affinity than normal AR-expressing tissue, allowing the identification of active disease. Pharmacokinetic assessment of 16β-[18F]FDHT in tumors was studied to develop a clinically applicable method for assessing changes in AR levels by PET in patients undergoing therapy. 59 The study concluded that 16β-[18F]FDHT had a blood half-life between 6 and 7 minutes, resulting in prostate cancer lesions reaching a plateau in uptake within 20 minutes; a simple body-mass normalized SUV was deemed reasonable as a measure of AR expression. The success of 16β-[18F]FDHT in identifying AR-expressing lesions in metastatic prostate cancer led to the evaluation of radiation dosimetry in humans in anticipation of clinical use; organ exposure was estimated conservatively, and a recommended 331 MBq of activity was elucidated for diagnostic studies. 60 The administered dose of [18F]FDHT is comparable to routinely used [18F]FDG and therefore could be suitable for routine use in the clinic.
Although 16β-[18F]FDHT appeared to be a promising candidate for assessing AR expression in prostate cancer, it is primarily used to report receptor occupancy. 61 In a phase I to II clinical study to determine the dosing and efficacy of the AR antagonist MDV3100, 16β-[18F]FDHT and [18F]FDG were used in addition to conventional clinical end points. 61 MDV3100 displaced 16β-[18F]FDHT (where uptake occurred in all patients) from the AR at all dose levels; in the same patients, 45% showed a decrease in [18F]FDG SUV greater than 25%, suggesting that 16β-[18F]FDHT uptake does not correlate with [18F]FDG measurement of response. Although 16β-[18F]FDHT is unable to provide a readout of treatment response, it has been valuable in highlighting the discordance of tumor biology between lesions in metastatic bone cancer; [18F]FDG and 16β-[18F]FDHT uptake is concordant in some sites and discordant in others, emphasizing the need for techniques that characterize tumors that accounts for intra-and intertumoral heterogeneity. 62 The AR does not have an associated SHR cognate biomarker as seen with ER and PR; however, prostate-specific membrane antigen (PSMA) can be used to report on treatment response. Evans and colleagues evaluated the use of [64Cu]J591 antibody to image PSMA; expression in the presence of AR blockade would identify patients who retained AR activity and therefore may benefit from full inhibition of AR signaling. 63
Labaree and colleagues synthesized 7α-fluoro and 7α-iodo analogues of 5α-DHT and 19-nor-5α-dihydrotestosterone (5α-NDHT) (Figure 8); binding affinities were evaluated using in vitro cell assays. 64 Compound 17α-methyl fluoro analogue [18F]FMDHT (see Figure 8) exhibited an RBA of 123 when compared against [3H]R1881 in a radiometric binding assay, standing out as the highest affinity compound in the series; the 17α-methyl group protects the 17β-hydroxyl against metabolism. All fluorine-containing ligands exhibited higher binding affinities than the iodo-containing counterparts. Tissue biodistribution of the 17α-methyl iodo [125I] analogue in castrated male rats showed low uptake in the prostate, which was confirmed to be specific by administering a blocking dose of 5α-DHT. The low uptake was speculated to be a result of rapid catabolism of the steroid. An in vitro study into the metabolism of the 17α-methyl iodo analogue revealed that elimination of [125I] under physiologic conditions resulted in poor target tissue uptake; this result would seemingly account for large uptake of radioactivity in the thyroid at the 4-hour time point. The fluoro-substituted analogues were resistant to elimination in chemical and biological studies due to the increased stability of the C-F bond compared to C-I; this study showed that 18F-labeled ligands exhibited the potential to be investigated further as AR imaging agents as a result of high in vivo affinity and stability.
7α-Fluoro-17β-hydroxy-17α-methyl-5α-estran-3-one ([18F] FMDHT).
Garg and colleagues further evaluated the 17α-methyl fluoro analogue for potential application in imaging AR in prostate cancer. 65 [18F]FMDHT exhibited highly specific ligand binding for the AR with negligible binding to other SHRs, such as ER, PR and GR (RBA < 0.1). The radiosynthesis of [18F]FMDHT was achieved from a tosylate precursor using [18F]tetrabutylammonium fluoride in 5 to 9% RCY with a mean specific activity of 11.6 GBq/μmol End of Synthesis (EOS). The biodistribution of [18F] FMDHT was determined in the prostate of intact male rats with supressed testosterone secretion; uptake was high in the prostate (prostate/blood 9.06 ± 3.55 at 1 hour), which could be effectively blocked by administering a saturating dose of 5α-DHT. The ligand was both chemically and metabolically stable, reflected by the high target tissue uptake and low activity in the bone. The radiosynthesis of [18F] FMDHT has been translated onto a remote-controlled automatic system to improve RCY (20–30%), purity (> 99%), and synthesis time (60–70 minutes). 66 Further investigation into the specificity of [18F] FMDHT to AR-rich tissues in chemically castrated rats showed favourable biodistribution that can be blocked with 5α-DHT. 67 The authors’ final statement suggests that the full potential of [18F] FMDHT as a radioligand should be sought by evaluating uptake in mice bearing tumor xenografts and imaging characteristics in higher species. 67
There have been attempts to synthesize nonsteroidal AR ligands for imaging applications; however, these have been largely unsuccessful due to rapid in vivo metabolism.
Parent and colleagues described the synthesis of the nonsteroidal AR ligand 3[76Br]-bromohydroxyflutamide, radiolabeled with bromine-76; the nonsteroidal compound was a derivative of flutamide, an antiandrogen drug. 68 Radiochemical synthesis resulted in a high RCY of 50 ± 8% (decay corrected) with specific activity > 6 GBq/μmol (Figure 9). 68 Tissue distribution studies appeared to indicate that there was no uptake by the prostate, which could be accounted for either by low binding affinity or the metabolic instability of the bromine radiolabel.
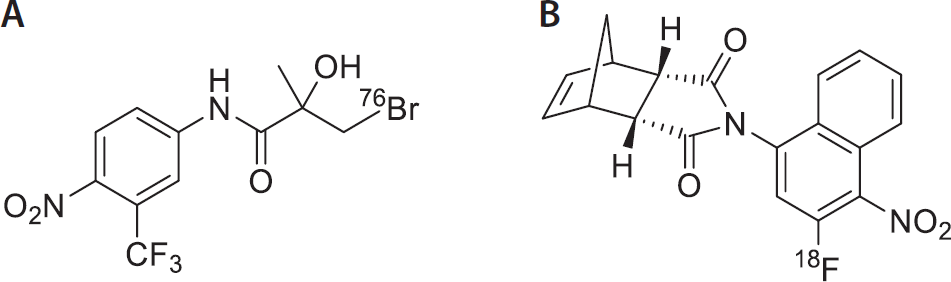
3[76Br]Bromohydroxyflutamide (A) and N-(3-[18F]fluoro-4-nitronapthyl-cis-5-norbornene-endo-2,3-dicarboxylicimide (3-F-NNDI) (B).
Parent and colleagues also examined another nonsteroidal AR agonist, N-(3-[18F]fluoro-4-nitronapthyl-cis-5-norbornene-endo-2,3-dicarboxylic imide (3-F-NNDI), which exhibited a relatively low RBA of 0.2 compared to R1881; however, it appeared to be stable in vitro. 69 Radiolabeled 3-[18F]F-NNDI was synthesized from an ammonium precursor in an RCY of 81%. Bio distribution studies showed limited uptake of activity in the prostate; rapid in vivo metabolism resulted in increased fluoride uptake by the bones before significant levels of tracer could accumulate in target tissue 69 ; the rapid in vivo metabolism in the absence of in vitro metabolism was speculated to be enzymatic in nature.
Discussion
Medical imaging modalities are improving by advances in physics, electronics, and material sciences; advances in PET imaging have further dependence on the development of suitable radiotracers, which interact with the pathologic features of the disease. The unparalleled sensitivity of PET has influenced the development of radiotracers for imaging low–receptor density sites, which have a great clinical impact. 70 SHR expression is an attractive target for imaging because of the association with cancer prognosis together with the caveats of pathologic assessment by IHC. 5 The suitability of an SHR imaging agent to provide quantitative information on receptor expression is very dependent on the specific activity of the radioligand, circulating protein binding, and metabolism; therefore, there is little wonder that the development of SHR imaging agents for ER, PR, and AR has resulted in four decades of work to progress the field.
Exemplary clinical investigation into the utility of [18F]FES PET for imaging ER expression in both primary and metastatic disease as well as identifying response to treatment has demonstrated not only the rigor by which new radiotracers should be evaluated but also the advantage of using PET in a standard clinical workup in patients presenting a clinical dilemma. The use of SHR imaging agents has also played a crucial role in the clinical development of novel endocrine therapies; [18F]FES PET has been used to show differences in the pharmacodynamics of the aromatase inhibitors tamoxifen and fulvestrant in metastatic breast cancer. 71 Investigation into PR radioligands such as [18F]FFNP is in its infancy; however, clinical evaluation has proven the concept. 38 The use of PR expression as a surrogate biomarker for treatment response in breast cancer animal models will no doubt drive the progression of [18F]FFNP into the clinic for imaging estrogen challenge or treatment response.40,41 Although [18F]FFNP has the potential to be a promising radioligand in the clinic, there may be further need for structural refinement or a shift to nonsteroidal PR radioligands to overcome nonspecific interactions by reducing ligand lipophilicity; some work toward nonsteroidal PR ligands appears in the literature; however, it has not been taken any further.43,44
The development of AR PET ligands has been extensive, with [18F]FDHT being evaluated in the clinic. Unlike ER and PR, the expression of the AR remains unchanged from normal to diseased target tissue, which hinders the use of [18F]FDHT to evaluate treatment response in prostate cancer; however, one study highlights the potential use of [18F]FDHT for quantifying changes in AR expression in patients undergoing therapy. [18F]FDHT has still proven to be useful in the clinic for imaging receptor occupancy by testosterone and novel antiandrogens (MDV3100 and ARN-509).61,72 It appears that AR PET may benefit from a new selection of radioligands that can inform when resistance to treatment ensues rather than AR expression alone; acquired resistance is an inevitable characteristic of endocrine therapy in prostate cancer, and PET could be used to inform clinical decision making when resistance is acquired. Abiraterone is a treatment for castration-resistant prostate cancer, which works by irreversibly inhibiting the production of androgens. 73 Assessing AR occupancy with a suitable radioligand could result in the status of AR occupancy to be used as a pharmacodynamic biomarker; pretreatment PET scans to assess basal AR occupancy with follow-up scans after treatment could provide information on treatment efficacy. It would be expected that an increase in radioligand uptake in the tumor compared to basal AR occupancy could correlate with response to treatment.
A lesson learned from the development of SHR imaging agents is that validation is key for successful translation of a radiotracer into the clinic. Careful evaluation of radioligand cross-reactivity and metabolism should always be at the forefront of radiotracer development, particularly with steroidal radioligands that may be protected against metabolism by circulating proteins such as SHBG. PET is a sensitive and versatile technique that can be used far beyond receptor occupancy studies; future development of novel radioligands that report on mutant receptors should be pursued despite the synthetic challenges posed, to meet the challenge of early detection of acquired resistance to treatment.
Table 3 summarizes reported clinical trials to date for SHR imaging agents, chiefly involving imaging of ER status using [18F]FES. Clinical investigation into the efficacy of [18F]FES in primary and metastatic breast cancer for determining treatment response, aggressiveness of disease, and pathologic features and providing insight into appropriate treatment regimens when presented with a clinical dilemma are just some of the studies that have included [18F]FES PET.20,22,74,75 A much smaller number of PR imaging trials are reported, reflecting the more recent interest in imaging of this receptor. As this interest grows, more sophisticated clinical trials involving assessment of treatment response by measurement of PR expression are anticipated.17,38 The growing interest in AR imaging using [18F]FDHT is presumably a result of the new prostate cancer treatments, such as MV3100 (enzalutamide) and abiraterone, that are becoming widely available. These agents require companion diagnostics to assess drug pharmacokinetics (AR occupancy) and treatment response/resistance, which can be achieved through imaging cognate biochemical affected by AR modulation.58,61,72,76
Comparison of Clinical Trials for ER, PR, and AR Radiotracers

AR = androgen receptor; CT = computed tomography; ER = estrogen receptor; [18F]FES = 16-α-[18F]Fluoro-17-β-fluoroestradiol; [18F]FDHT= 16-α-[18F]-fluoro-5α dihydrotestosterone; [18F]FENP = 21-[18F]Fluoro-16α-ethyl-19-norprogesterone; [18F]FMOX = 16-α-[18F]Fluoro-17-β-fluoroestradiol; MRI = magnetic resonance imaging; PET = positron emission tomography; PR = progesterone receptor; SUV = standardized uptake value; T/N = tumor/normal tissue.
Conclusion
The drive toward so-called personalized medicine has promoted imaging techniques such as PET to the forefront of clinical practice; the ability to assess the heterogeneous tumor environment with a simple, minimally invasive process encouraged the development of SHR imaging agents. This development has progressed further for imaging ERα and PR compared to the AR, which may be the result of more detailed characterization and validation of these targets in the pathologic state. [18F]FES for imaging ERα has been validated and proved to be successful in identifying ER+ lesions. Although successful [18F]FES is not infallible, it is encouraging to see continued optimization of estradiol derivatives to address the formation of radiometabolites and to overcome localization in liver—a common metastatic site. The future of SHR imaging is likely to continue in the direction of receptor function, as seen in the case of PR imaging with [18F]FFNP. The strong correlation between [18F]FFNP uptake and response to endocrine therapy has confirmed that the PR is a promising biological target to monitor treatment efficacy. Taking [18F]FFNP into the clinic to determine if the response to treatment after dosing in humans correlates with results from animal models is likely to be the next logical step in the transition from bench to bedside. There appears to be space for further development of high-affinity, nonsteroidal PR imaging agents that exhibit less cross-reactivity to other members of the SHR family. Greater specificity may increase the sensitivity of receptor quantification and provide improved stratification of patients.
Footnotes
Acknowledgments
Financial disclosure of authors and reviewers: None reported.