
Abstract
Tumor-initiating cells, also designated as cancer stem cells, are proposed to constitute a subpopulation of malignant cells central to tumorigenesis, metastasis, and treatment resistance. We analyzed the activity of the proteasome, the primary organelle for targeted protein degradation, as a marker of tumor- and metastasis-initiating cells. Using human and mouse breast cancer cells expressing a validated fluorescent reporter, we found a small subpopulation of cells with low proteasome activity that divided asymmetrically to produce daughter cells with low or high proteasome activity. Breast cancer cells with low proteasome activity had greater local tumor formation and metastasis in immunocompromised and immunocompetent mice. To allow flexible labeling of cells, we also developed a new proteasome substrate based on HaloTag technology. Patient-derived glioblastoma cells with low proteasome activity measured by the HaloTag reporter show key phenotypes associated with tumor-initiating cells, including expression of a stem cell transcription factor, reconstitution of the original starting population, and enhanced neurosphere formation. We also show that patient-derived glioblastoma cells with low proteasome activity have higher frequency of tumor formation in mouse xenografts. These studies support proteasome function as a tool to investigate tumor- and metastasis-initiating cancer cells and a potential biomarker for outcomes in patients with several different cancers.
ALTHOUGH HETEROGENEITY has long been recognized as a defining feature of cancer, more recent studies indicate that tumor heterogeneity extends to the differentiation state of cancer cells. 1 Cancers are proposed to contain subsets of cells, only some of which can initiate and sustain tumor growth. These tumor-initiating cells, also referred to as cancer stem cells, share many features of normal stem cells in tissues, including self-renewal, asymmetric division into stem and nonstem daughter cells, and progression to more terminally differentiated cells with limited replicative capacity. 2 Tumor-initiating cells express transcription factors and other proteins characteristic of normal stem cells. Relative to more differentiated cancer cells, the subset of tumor-initiating cells also resists treatment with standard chemotherapeutic drugs, so tumor-initiating cells are commonly enriched in the population of cells that survive conventional chemotherapy and radiation therapy. 3 Tumor-initiating cells that survive therapy are believed to cause recurrent cancer because only small numbers of these cells are typically needed to produce a tumor. Therefore, considerable attention has been focused on identifying improved methods to identify tumor-initiating cells and establish methods to eliminate these cells with targeted drugs.
Beyond initiating tumors, studies indicate that only a subset of cancer cells within a primary tumor have the ability to produce metastases. In breast cancer, cells with markers of tumor-initiating cells have been identified in bone marrow, suggesting that tumor-initiating cells disseminate systemically to produce metastases. 4 Only a subset of tumor-initiating cells in some malignancies have acquired additional proteins and signaling molecules necessary to produce metastases. For both pancreatic and colon cancer, metastases arise only from tumor-initiating cells that coexpress chemokine receptor CXCR4.5,6 Identifying markers that define the metastasis-initiating population of malignant cells in a tumor is critical to blocking and/or treating metastatic disease, the cause of death for 90% of patients with cancer. 7
A large number of markers, including cell surface molecules, transcription factors, and intracellular enzymes, have been used to identify tumor-initiating cells in different types of cancer. 8 To improve specificity for cancer cells versus other cell types, some methods define tumor-initiating cells based on combinations of two or more markers.9,10 Although potentially improving enrichment for tumor-initiating cells, multiparameter methods may incorrectly classify cells if only specific splice variants of a protein characterize tumor-initiating cells. 11 Combinations of markers can be applied readily to flow cytometry analyses, but these algorithms may be difficult to implement for other approaches, such as immunohistochemistry of fixed tissues. Additionally, markers of tumor-initiating cells are often tissue and cancer specific, which may limit the ability to detect metastatic cells in sites with high expression of the same marker in normal tissue. 12 Therefore, there is an ongoing need for alternative markers of tumor- and/or metastasis-initiating cells to overcome the limitations of current approaches. 8
Recent research has identified the proteasome as a promising marker of tumor-initiating cells in a wide spectrum of cancers, including brain, head and neck, lung, and breast.13–16 The proteasome is a large, multisubunit complex present in all cells, functioning primarily to degrade proteins as part of cell signaling and/or homeostasis. 17 Although a ubiquitous cellular organelle would seemingly have limited utility as a marker of cellular heterogeneity in a tumor, the subset of cancer cells with low proteasome activity show phenotypes of tumor-initiating cells, including resistance to drug and radiation therapy and enhanced tumor formation in mouse models.14,18 In head and neck cancer, patients with low expression of proteasome subunits by immunohistochemistry had significantly reduced survival, linking data from preclinical cell-based and animal models to patient outcomes. 16 Low proteasome abundance and activity in tumor-initiating activity in cancer cells have been defined through methods including degradation of reporter substrates, Western blotting, immunohistochemistry, and direct assays of enzyme activity.15,16,19 The wide range of assay formats for proteasome activity provides flexibility to identify these cells at single-cell and population levels. Despite potential advantages of the proteasome as a marker of tumor-initiating cells, current research has focused predominantly on localized tumor formation by human cancer cell lines, leaving uncertainties about the applicability of this marker to human cancers and metastatic disease.
In this study, we expanded applications of proteasome activity as a marker of cancer cells with tumor-initiating activity. Using both human and mouse breast cancer cell lines, we determined that cells with low proteasome activity have greatly enhanced potential for producing metastases in addition to initiating the growth of localized tumors. We also discovered that low proteasome activity enriches for tumor-initiating cells in xenografts of primary cancer cells from patients, extending applicability of the marker beyond standard cell lines. Finally, we developed a new reporter of proteasome activity with flexibility for labeling with different fluorescent tags, which will increase options for cell-based and animal imaging. Overall, these data support future studies of the proteasome as a marker for tumor- and metastasis-initiating cells in preclinical models and suggest that these critical cell populations may be identified clinically through assays for proteasome activity.
Methods
Cells
We used MDA-MB-231 and SKBR3 human breast cancer cells (ATCC) and AT-3 breast cancer cells derived from a C57BL/6 mouse (gift of Dr. Abrams, Roswell Park Cancer Institute, Buffalo, NY). Breast cancer cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum and 1% glutamine/penicillin/streptomycin (Life Technologies, Carlsbad, CA). Patient-derived glioblastoma multiforme (GBM) primary cells (HF2303 and HF3106) were a gift of Dr. Mikkelsen and Dr. deCarvalho (Henry Ford Hospital, Detroit, MI). Details of the procedure for obtaining these patient-derived neurospheres have been described previously. 20 Briefly, a patient tumor sample was cut into 1 mm3 pieces, triturated, dissociated, and cultured in neurosphere medium (DMEM/F-12 supplemented with N2 (Life Technologies), 0.5 mg/mL bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO), 25 µg/mL gentamicin (Life Technologies), 0.5% antibiotic/antimycotic (Life Technologies), 20 ng/mL basic fibroblast growth factor, and 20 ng/mL epidermal growth factor (Peprotech, Rocky Hill, NJ). 21 GBM cells were maintained in culture up to passage 20 (low passage). All experiments were performed using cells in exponential growth.
Lentiviruses
We used a fluorescent reporter of proteasome activity consisting of ZsGreen fused to the degradation domain of mouse ornithine decarboxylase (ZsGreen-mODC) (pZsProSensor, Clontech, Mountain View, CA). To generate lentiviruses with the proteasome reporter, we digested pZsProSensor with BglII, blunted this site with Klenow, removed ZsGreen-mODC with NotI, and ligated the insert into EcoRI (blunt) and NotI in lentiviral vector pLVX-IRES-Cherry (Clontech) (enzymes from New England Biolabs, Ipswich, MA). To generate the HaloTag ProSensor fusion construct, we excised ZsGreen-mODC with restriction enzymes SacI and NotI and inserted it into the same sites of the pHTN HaloTag CMV-neo vector (Promega, Madison, WI). We amplified the HaloTag-ZsGreen-mODC fusion gene (designated as HaloTag ProSensor) by polymerase chain reaction, including ends compatible with the Gateway system vector pENTR-D-TOPO (Life Technologies). An LR Gateway cloning reaction was used with pENTR 5′-EFalpha promoter, pENTR-HaloTag-ZsGreen-mODC, and pLenti6.4/R4R2/V5-DEST to create the full lentiviral vector. We used MDA-MB-231 cells stably transduced with firefly luciferase and transduced SKBR3 and AT-3 cells with a firefly luciferase lentiviral vector. 22 We generated recombinant lentiviruses as described previously and transduced luciferase-expressing breast cancer cells with ZsGreen-ODC-IRES-Cherry and GBM cells with HaloTag ProSensor.23,24
HaloTag Labeling
HaloTag labeling was performed according to the manufacturer's directions (Promega). Briefly, HaloTag Direct TMR ligand or HaloTag Oregon Green ligand was diluted into 5× stock solution in fresh medium for GBM cells. We replaced one-fifth of the culture medium with HaloTag ligand stock solution. For HaloTag Oregon Green ligand, cells were incubated for 15 minutes, washed with medium twice, and then incubated in fresh medium for another 30 minutes. For HaloTag Direct TMR ligand, cells were incubated with TMR ligand overnight. We analyzed labeled cells by epifluorescence microscopy (Olympus BX-51 microscope with Olympus DF-70 high-resolution digital camera, Olympus, Tokyo, Japan) or flow cytometry (BD FACSCanto, BD Biosciences, San Diego, CA).
Flow Cytometry
We sorted breast cancer cells for equal numbers of cells positive for mCherry and ZsGreen (mCherry+/green fluorescent protein [GFP]+) or mCherry only (mCherry+) on a FACSAria (BD Biosciences). Similarly, we sorted GBM cells into HaloTag ProSensor+ or HaloTag ProSensor− cells based on fluorescence from the HaloTag label. We also analyzed HaloTag fluorescence in cells after treatment with MG132 or in sorted HaloTag ProSensor+ cells after 2 weeks in culture.
Proteasome Inhibition
We added the proteasome inhibitor MG132 (Sigma) to cell cultures at a final concentration of 10 µM for 1 hour before analyzing fluorescence by epifluorescence microscopy or flow cytometry.
Two-Dimensional Cell Growth Assay
We plated mCherry+/GFP+ or mCherry+ MDA-MB-231 cells at 1 × 103 cells per well in black wall 96-well plates. For experiments using 1% serum, we switched wells from medium with 10% serum to medium with 1% serum 4 hours after plating. We then imaged bioluminescence in wells (n = 4 per cell type) to define baseline numbers of cells on day 0 for assays with 1% or 10% serum. 25 To measure cell growth over time, we quantified bioluminescence in a parallel set of wells for each condition on days 1 to 5. We normalized bioluminescence data for each cell population and serum condition to corresponding bioluminescence on day 0 and expressed data as mean values for fold change ± SEM. Presented data are representative of two independent experiments.
Cytotoxicity Assays
We plated mCherry+/GFP+ or mCherry+ MDA-MB-231 cells at 5 × 103 cells per well in black wall 96-well plates. All assays used standard growth medium with 10% serum. One day after seeding cells, we exchanged medium for fresh medium containing increasing concentrations of doxorubicin, paclitaxel, or cisplatin (University of Michigan Hospital Pharmacy) or vehicle control (n = 4 per condition). We measured bioluminescence to assess cell viability after 3 days of incubation in drug. We normalized data to bioluminescence from vehicle control and expressed data as mean values for percentage of vehicle control ± SEM. Presented data are representative of three independent experiments.
Neurosphere Formation Assay
We plated equal numbers of HaloTag ProSensor+ cells and HaloTag ProSensor− cells in ultralow attachment 96-well plates (Corning Costar, Corning, NY) after fluorescence-activated cell sorting (FACS). Total numbers of spheres (spheres with more than eight cells) were counted in nine different fields after 10 days in culture. Data were quantified as mean values ± SEM for spheres from three independent trials.
Immunostaining
Neurospheres were dissociated into single cells, stained with HaloTag TMR ligand, and sorted into HaloTag ProSensor+ cells and HaloTag ProSensor− cells. Those cells were then collected by centrifugation and cytospun onto glass slides. Slides were fixed in 4% paraformaldehyde for 1 hour at room temperature in a humidified chamber, washed twice in phosphate-buffered saline (PBS), and permeabilized for 1 hour at room temperature with 0.3% Triton X-100 in PBS. Slides were then blocked with 5% BSA and then incubated with a 1:400 dilution Sox2 primary antibody (#3579, Cell Signaling Technologies, Danvers, MA) overnight at 4°C. Slides were washed twice in PBS before adding Cy5-labeled secondary antibody at a 1:250 dilution (#711-175-152, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). We used DAPI (4′,6-diamidino-2-phenylindole) to stain nuclei before acquiring epifluorescence images on an Olympus BX-51 upright light microscope and an Olympus DP-70 high-resolution digital camera.
We fixed tumors from MDA-MB-231 cells in 4% buffered formalin overnight and then embedded tissues in paraffin for sectioning. Immunohistochemistry for Ki-67 (Cell Signaling Technologies) was performed by the UM Cancer Center Pathology Core. 26 A person blinded to the identity of each slide manually quantified the percentage of cells positive for Ki-67 staining. Data for Ki-67 staining were graphed as mean values ± SEM, averaging four random sections from each tumor from five different mice per tumor group.
Mouse Experiments
We established orthotopic tumors by implanting 100 MDA-MB-231 or 500 SKBR3 breast cancer cells sorted for mCherry+/GFP+ or mCherry+ into the fourth inguinal mammary fat pads of age-matched, 5- to 8-week-old female NSG (NOD scid gamma) mice (Jackson Laboratory, Bar Harbor, ME). To generate systemic metastases, we injected 100 AT-3 cells sorted for mCherry+/GFP+ or mCherry+ directly into the left ventricle of the heart. We imaged tumor burden and metastases in vivo and ex vivo by bioluminescence imaging with an IVIS Spectrum (PerkinElmer, Waltham, MA) as previously described. 27 To quantify viable tumor-circulating tumor cells, we collected 300 µL blood samples from the right ventricle of mice at the time of euthanization and cultured blood samples in standard growth medium for 1 week before measuring bioluminescence.
To assess the tumorigenicity of HaloTag ProSensor+ or HaloTag ProSensor− cells, we injected 5 × 103 sorted cells mixed with 100 µL of BD Matrigel Basement Membrane Matrix (BD Sciences) into the left or right flank of 4- to 6-week-old athymic female mice (CD-1 nu/nu) (Charles River, Portage, MI) (n = 4 per group). We monitored mice weekly for up to 16 weeks to determine the formation of palpable tumors.
Statistical Analysis
We analyzed cell-based assays by t-test and animal data using the Mann-Whitney test (GraphPad Prism, San Diego, CA). We used Mann-Whitney testing for animal studies because the data were not distributed normally. A p value ≤ .05 defined statistically significant differences.
Results
Breast Cancer Cells with Low Proteasome Activity Show In Vitro Phenotypes of Tumor-Initiating Cells
To investigate functional differences between cancer cells with low and high proteasome activity, we used a validated proteasome sensor composed of a GFP (ZsGreen) fused to the ubiquitin-independent degradation domain of mouse ornithine decarboxylase (Figure S1, online version only).13,28,29 This construct is normally degraded rapidly in the proteasome, so most cells expressing the proteasome sensor have minimal green fluorescence. However, the fusion protein is stabilized in cells with low proteasome activity, allowing identification of these cells based on higher levels of green fluorescence (Figure S2, online version only). 30 We stably transduced MDA-MB-231 human breast cancer cells with the proteasome sensor. Since the lentiviral vector constitutively coexpresses fluorescent protein mCherry through an IRES linkage, we used this protein to define transduced cells independent of proteasome activity. MDA-MB-231 cells also expressed firefly luciferase for bioluminescence imaging.
Based on flow cytometry, we sorted MDA-MB-231 cells into populations with low and high proteasome activity, respectively, based on green fluorescence. The population with low proteasome activity (mCherry+/GFP+) comprised approximately 1% of the total population, with the remaining cells having higher proteasome function (mCherry+) (Figure 1A). We compared the growth of both populations in cell culture under two conditions: 10% serum to model a growth factor–enriched condition and 1% serum to reproduce growth under nutrient-limited conditions as may exist in a tumor. 31 In 10% serum, both cell populations proliferated comparably through 5 days, with mCherry+/GFP+ cells showing significantly greater growth only on the final day of the assay (p < .05) (Figure 1B). By comparison, MDA-MB-231 cells with low proteasome activity exhibited markedly greater growth in 1% serum (Figure 1C). After 5 days in culture with 1% serum, mCherry+/GFP+ cells grew almost fourfold more than mCherry+ cells.
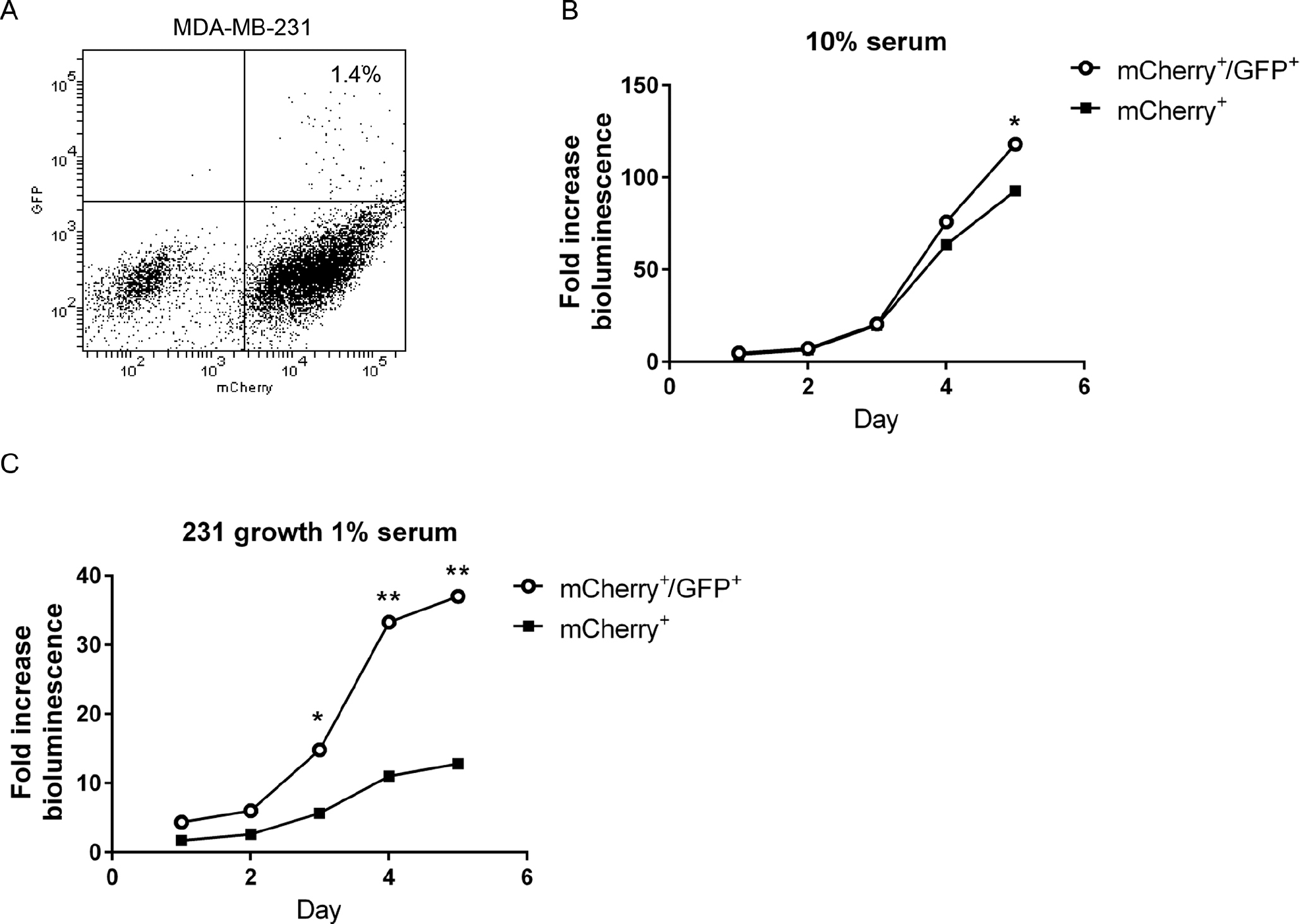
MDA-MB-231 breast cancer cells with low proteasome activity show greater proliferation under low serum conditions. A, Dot plot from flow cytometry of MDA-MB-231 cells sorted for low (mCherry+/GFP+) versus high proteasome activity (mCherry+) based on the ZsGreen proteasome reporter and coexpressed mCherry. The plot shows the percentage of cells with low proteasome activity. B and C, Sorted mCherry+/GFP+ and mCherry+ cells were cultured for 5 days in medium with 10% (B) or 1% (C) serum. Cell proliferation was quantified by daily bioluminescence imaging of parallel cultures of cells. Graphs show mean values ± SEM for fold change in photon flux relative to day 0. Error bars are smaller than symbols in B and C. *p < .05; **p < .01.
We also tested sorted cells for other phenotypes associated with tumor-initiating cells, such as drug resistance and asymmetric cell divisions.3,32–34 Cells with low proteasome activity (mCherry+/GFP+) were modestly resistant to treatment with standard drugs used in cancer chemotherapy: paclitaxel, doxorubicin, and cisplatin (p < .05 by area under the curve analysis of cytotoxicity curves for each drug) (Figure S3, online version only). After 1 week in culture, we observed asymmetric division of mCherry+/GFP+ cells at single-cell and population levels of resolution (Figure S4, online version only). We observed single mCherry+/GFP+ cells dividing into two daughter cells, only one of which retained green fluorescence from low proteasome activity (see Figure S4, A and B). Interestingly, we identified asymmetric divisions in which a GFP+ daughter cell with low proteasome activity segregated with relatively higher or lower fluorescence from mCherry. These states of daughter cells suggest unequal partitioning of messenger ribonucleic acid (mRNA) for the reporter in addition to asymmetric segregation of proteasome protein. Previous studies have reported asymmetric partitioning of mRNA and the proteasome during cell division.35–37 From cultures sorted for mCherry+/GFP+ cells, we also observed populations of cells that converted to a state of high proteasome activity with only fluorescence from mCherry (see Figure S4C). Overall, these data show that mCherry+/GFP+ cells with low proteasome activity exhibit additional in vitro phenotypes previously reported for tumor-initiating cells.
Human Breast Cancer Cells with Low Proteasome Activity Have Enhanced Tumor- and Metastasis-Initiating Capabilities
To investigate to what extent low proteasome activity identifies cells with enhanced potential to form localized tumors and metastases, we implanted 100 mCherry+/GFP+ or mCherry+ MDA-MB-231 cells orthotopically into the mammary fat pads of female NSG mice. We observed greater growth of mCherry+/GFP+ tumors over time using bioluminescence imaging to quantify relative numbers of viable tumor cells (p < .01–.05) (Figure 2, A and B). Mice implanted with mCherry+/GFP+ cells also had greater tumor weight after 65 days (Figure 2C). We noted that tumors formed in 20 of 20 injections with 100 mCherry+/GFP+ cells, whereas only 14 of 20 injections with 100 mCherry+ cells produced a tumor. mCherry+/GFP+ tumors had more proliferating cells as determined by staining for Ki-67, accounting at least in part for greater growth of these cancer cells in orthotopic implants (Figure 2D).

Low proteasome activity identifies human breast cancer cells with enhanced tumor-initiating potential. A and B, Representative bioluminescence images (A) from day 62 and quantified imaging data (B) for mice implanted orthotopically with 100 mCherry+/GFP+ or mCherry+ MDA-MB-231 cells with low and high proteasome activity, respectively. Graph in B shows mean values ± SEM for radiance from firefly luciferase in breast cancer cells on listed days (n = 10 mice with two tumor implants per mouse). The scale bar in A shows the range of pseudocolor display for radiance values. C, The graph shows the weights of individual tumors and mean values ± SEM for orthotopic mammary tumors when mice were euthanized for tumor burden after 65 days (n = 20 tumor implants per group). The dashed line demarcates the weight of a normal mammary fat pad. Mice with excised fat pads weighing less than this amount were considered to have no tumor growth. D, Representative images of Ki-67 immunohistochemistry in mCherry+/GFP+ and mCherry+ tumors. The graph shows mean values for the percentage of cells positive for Ki-67 staining in each group. E, Data show mean values ± SEM for total body metastases quantified by bioluminescence imaging. *p < .05; **p < .01.
Although previous studies with xenografts of human cancer cell lines have shown greater tumor-initiating capability of cells with low proteasome activity, the effects on metastasis have not been established.13,16 When we euthanized mice with MDA-MB-231 tumor implants, we analyzed spontaneous metastases by bioluminescence imaging. Mice implanted with orthotopic tumors of mCherry+/GFP+ cells with low proteasome activity had significantly greater total metastases (Figure 2E). When quantified as metastatic burden in defined anatomic sites, mice with mCherry+/GFP+ tumor implants also had more extensive metastases in all regions, although only differences in the abdomen were significant between groups (p < .05) (Figure S5, online version only).
We verified the effects of low proteasome activity on spontaneous metastasis of breast cancer cells using human SKRB3 cells. Following implantation of 500 cells sorted for mCherry+/GFP+ or mCherry+ only, we observed greater growth of orthotopic tumors by bioluminescence imaging and final tumor weights in mice with mCherry+/GFP+ cells with low proteasome activity (Figure 3, A and B). Most notably, these mice also had greater spontaneous metastases based on bioluminescence imaging (p < .05) (Figure 3, C and D). Collectively, our data from two different human breast cancer cell lines show that malignant cells with low proteasome activity have enhanced tumor- and spontaneous metastasis-initiating capabilities.

SKBR3 breast cancer cells with low proteasome activity show greater growth of orthotopic tumors and more spontaneous metastases. A, The graph displays mean values ± SEM for bioluminescence from orthotopic tumor implants of SKBR3 cells with low (mCherry+/GFP+) versus high (mCherry+) proteasome activity (n = 5 mice with 2 tumors/mouse). *p < .05. B, Weights of excised tumors when mice were euthanized for humane end points. Each dot denotes one tumor, and the horizontal line shows mean values for each group. C, Representative bioluminescence images of spontaneous metastases in each group. The scale bar displays the pseudocolor scale for the range of photon flux values. D, Mean values ± SEM for total metastases in each group measured by bioluminescence imaging.
Breast Cancer Cells with Low Proteasome Activity Produce Greater Experimental Metastases in Immunocompetent Mice
Previous mouse studies of low proteasome activity as a marker of tumor-initiating cells have used human cancer xenografts in immunocompromised mice, largely negating the effects of host immunity. To determine metastasis-initiating potential of cancer cells with low proteasome activity in the setting of an intact immune system, we used AT-3 breast cancer cells, a triple-negative cell line originally derived from a C57BL/6 mouse. 38 We also used the AT-3 breast cancer model to overcome a potential limitation of our orthotopic MDA-MB-231 and SKBR3 models: the fact that mCherry+/GFP+ cells with low proteasome activity formed larger tumors. Although we previously have established that the size of orthotopic tumors does not correlate with numbers of circulating tumor cells, it is possible that tumor size affects overall metastases. 39 We bypassed differences in the growth of orthotopic tumors and vascular intravasation by injecting AT-3 cells directly into the left ventricle of the heart to disseminate cells systemically.
After left ventricular injection of 100 mCherry+/GFP+ or mCherry+ AT-3 cells into C57BL/6 mice, bioluminescence imaging revealed significantly greater growth of mCherry+/GFP+ cells over approximately 3 weeks (p = .01) (Figure 4A). Mice injected with mCherry+/GFP+ cells had more circulating tumor cells in blood samples collected when mice were euthanized at approximately 4 weeks (Figure 4B). The total overall burden of metastatic AT-3 cells at the time of euthanization was dramatically higher in mice with mCherry+/GFP+ cells. Based on bioluminescence imaging, total and site-specific metastases in these mice were more than two logs greater than mice injected with mCherry+ cells (p < .05) (Figure 4, C to E). In a separate experiment in which we followed mice until they had to be euthanized for humane end points rather than euthanizing all animals on the same day, mice injected systemically with mCherry+/GFP+ cells had markedly reduced survival. Only 1 of 10 mice injected with mCherry+/GFP+ cells survived through 100 days, whereas all 10 animals with mCherry+ cells survived through this time (Figure S6, online version only). These data solidify our conclusion that breast cancer cells with low proteasome activity have increased metastatic potential and extend this conclusion to immunocompetent mice.

Greater experimental metastases in immunocompetent mice from breast cancer cells with low proteasome activity. A, C57Bl/6 mice were injected with 100 syngeneic AT-3 breast cancer cells sorted for either low (mCherry+/GFP+) or high (mCherry+) proteasome activity. The graph displays mean values ± SEM for whole animal bioluminescence on listed days after left ventricular injection of cancer cells (n = 7 per group). B, Circulating tumor cells were collected from a subset of mice from each group when animals were euthanized for humane end points. The presented graph shows individual data points, mean values, and SEM for photon flux in blood samples measured ex vivo by bioluminescence imaging. C and D, Representative images (C) and quantified bioluminescence data (D) for total metastases in mCherry+/GFP+ and mCherry+ mice. Mean values + SEM for photon flux are plotted. E, The graph depicts mean values ± SEM for metastases in listed anatomic sites based on bioluminescence imaging. *p < .05;**p < .01.
New Proteasome Imaging Reporter Identifies Primary Glioblastoma Cells with Enhanced Tumor-Initiating Potential
Although the proteasome reporter based on ZsGreen enables facile identification of proteasome activity by microscopy and flow cytometry, use of a GFP limits other potential methods to label and detect cells in vitro and in vivo. To overcome this limitation, we developed a new proteasome reporter by fusing the degradation domain of mouse ornithine decarboxylase to an intracellular HaloTag protein (see Figure S1). The HaloTag system allows covalent labeling of the cell-expressed HaloTag protein with a variety of different probes. 40 We used this system to test the hypothesis that low proteasome activity identifies a population of tumor-initiating cells in primary glioblastoma cells from patients.
We transduced neurospheres of a patient-derived glioblastoma with the HaloTag ProSensor reporter to generate a population of cells for subsequent assays. We incubated parallel cultures of neurospheres with the proteasome inhibitor MG132 or vehicle control, followed by labeling of the reporter with a HaloTag Oregon green ligand. Fluorescence microscopy showed green fluorescence in a small population of neurosphere glioblastoma cells treated with vehicle control, whereas both the number and qualitative intensity of fluorescent cells increased following treatment with MG132 (Figure 5A). Flow cytometry of dissociated neurospheres from a separate experiment using tetramethylrhodamine as the label confirmed these qualitative observations (see Figure 5B). Under baseline conditions, approximately 5% of glioblastoma cells labeled with the HaloTag ligand. After inhibiting the proteasome with MG132, the percentage of labeled cells increased to 15% with a 1.7-fold increase in mean fluorescence intensity for the labeled population. These data demonstrate proteasome-dependent degradation of the HaloTag ProSensor protein and the flexibility of the system for measuring proteasome activity with different fluorescent labels. Moreover, we determined that primary glioblastoma cells also have a subpopulation with low proteasome activity.

HaloTag ProSensor reporter measures proteasome activity in primary glioblastoma neurosphere cells. A, Representative bright field and fluorescence images of neurosphere cells transduced with the HaloTag ProSensor reporter. Neurospheres were incubated with MG132 or vehicle control for 1 hour before labeling with HaloTag Oregon green ligand. B, Dot plots from flow cytometry of primary glioblastoma cells incubated with HaloTag TMR direct ligand (TMR), cells expressing the HaloTag ProSensor reporter and labeled with HaloTag TMR ligand (Control), or reporter cells incubated with MG132 followed by TMR ligand (MG132). The box defines the region used to classify the listed percentage of cells as ProSensor+ under various conditions.
We further investigated the tumor-initiating phenotypes of HaloTag ProSensor+ cells through several complementary methods. Previous studies have shown that transcription factor Sox2 is essential for the self-renewal and tumorigenicity of tumor-initiating cells in glioblastoma.41–43 In two different primary glioblastoma patient samples, immunofluorescence staining revealed that expression of Sox2 is enriched markedly in HaloTag ProSensor+ cells (Figure 6A and Figure S7, online version only). Tumor-initiating cells enriched from a variety of different malignancies subsequently reconstitute the heterogeneity of the original cell population.44,45 We demonstrated that HaloTag ProSensor+ cells exhibit this same phenotype. After culturing sorted HaloTag ProSensor+ cells for 2 weeks, these cells largely converted to a population with high proteasome activity, restoring the relative initial percentages of HaloTag ProSensor+ and ProSensor− cells (Figure 6B). The ability of cancer cells to form spheres under conditions of anchorage-independent growth defines another property of tumor-initiating cells.46–48 Using two different seeding numbers of cells to initiate sphere formation, HaloTag ProSensor+ cells formed significantly more neurospheres than the ProSensor− population (p < .01) (Figure 6C). Finally, we tested the tumorigenicity of HaloTag ProSensor+ and ProSensor− cells by implanting 5 × 103 cells of each type as subcutaneous xenografts in nude mice. Three of four mice implanted with HaloTag ProSensor+ patient-derived glioblastoma cells formed palpable tumors within 16 weeks compared to only one of four mice implanted with ProSensor− cells (Figure 6D). Overall, these data establish that the subpopulation of primary glioblastoma cells with low proteasome activity defined by our HaloTag ProSensor reporter is enriched for cells with enhanced tumor-initiating potential.
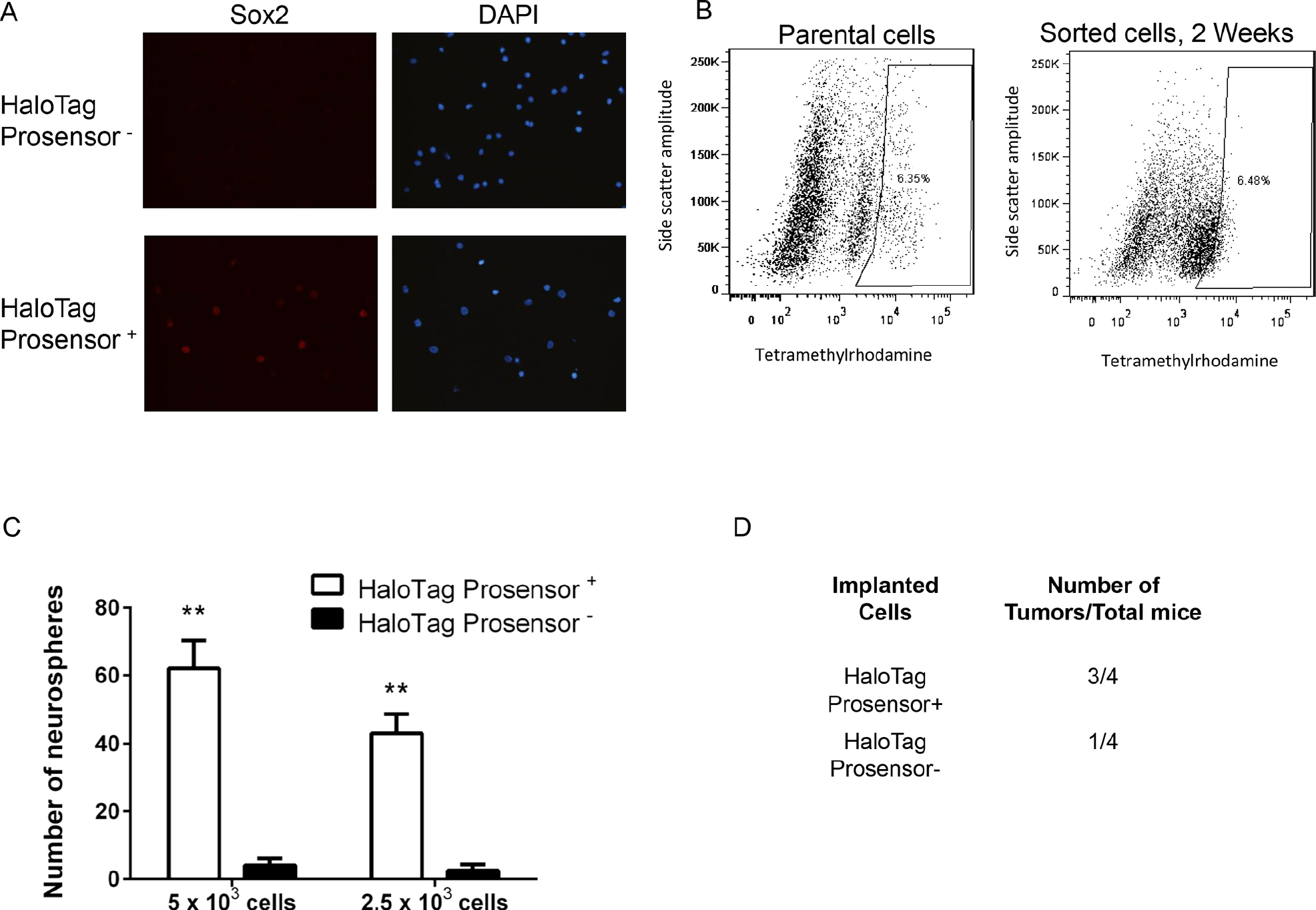
HaloTag ProSensor+ cells show properties of tumor-initiating cells. A, Representative 40× magnification images of immunofluorescence staining for Sox2 (red) in primary glioblastoma cells sorted into HaloTag ProSensor+ and ProSensor− populations. Cell nuclei were stained with DAPI (blue). B, Dot plots from fluorescence-activated cell sorting (FACS) analysis of parental HaloTag ProSensor cells labeled with HaloTag TMR ligand. The demarked HaloTag ProSensor+ cell population (box on the right of each plot) was collected and cultured in neurosphere medium for 2 weeks before repeating labeling and FACS analysis for HaloTag ProSensor+ and ProSensor− cells. Plots list the percentage of HaloTag ProSensor+ cells in each analysis. C, Cells sorted into HaloTag ProSensor+ and ProSensor− cells were seeded into nonadherent wells and cultured for 10 days before quantifying numbers of neurospheres produced by each population. The graph displays p mean values ± SEM for numbers of neurospheres from three independent experiments. **p < .01. D, Table lists numbers of mice with palpable tumors 16 weeks after implantation of HaloTag Prosensor+ or Prosensor− glioblastoma cells.
Discussion
Tumor-initiating cells are a key functional subpopulation of malignant cells in the formation of local tumors and progression to metastatic disease. 49 Through mechanisms including quiescence and expression of multidrug resistance transporters, these cells may resist conventional cancer therapies that effectively eliminate the bulk population of differentiated cancer cells. Tumor-initiating cells that survive treatment potentially account for the frequent recurrence of most solid cancers. Cancer patients with a higher percentage of tumor-initiating cells may have worse overall and progression-free survival, although this correlation has not been reproduced across all studies and types of cancer.50–52 Inconsistent correlations between the abundance of tumor-initiating cells and patient outcomes in cancer potentially reflect the inadequacies of markers used to categorize cells in individual studies and/or incomplete understanding of the functions of tumor-initiating cells in malignancy.
Our study focused on the tumor- and metastasis-initiating potential of cancer cells with low proteasome activity, a recently described functional marker that appears to enrich for tumor-initiating cells in several different solid malignancies. 13 Using two different cell line models of human breast cancer, we demonstrated that cells with low proteasome activity have significantly greater tumor-initiating potential in immunocompromised mice, confirming previous studies with breast cancer cells.13,53 For MDA-MB-231 breast cancer cells, we showed substantially increased proliferation of cells with low proteasome activity cultured in very low serum, providing a potential mechanism for enhanced growth in tumors where nutrients may be limited. In addition to greater tumor-initiating ability, we also discovered that breast cancer cells with low proteasome function produce dramatically more spontaneous and experimentally induced metastases. We observed increased metastases in both immunocompromised and immunocompetent models of breast cancer, suggesting that this cell population may be critical for metastasis in patients. Since metastases cause the death of most patients with solid tumors, therapeutically targeting cancer cells with low proteasome activity may be an effective strategy to block or treat metastatic disease.
Extending this research beyond cultured cell lines, we demonstrated that low proteasome activity identifies tumor-initiating cells in patient-derived glioblastoma cells. Similar to our study, Lagadec and colleagues showed increased formation of neurospheres from patient-derived glioblastoma cells sorted for low proteasome activity by a genetically encoded reporter construct. 30 However, we also established that low proteasome activity increases the frequency of successful tumor xenografts from patient-derived glioblastoma cells, which is the current gold standard for characterizing the tumor-initiating potential of human cancer cells. This result is essential evidence supporting proteasome function as a marker of the tumor-initiating potential of malignant cells in patients. Due to the small group size used for studies with patient-derived xenografts, we could not estimate the frequencies of tumor-initiating cells in populations with low or high proteasome activity.
Compared to the proteasome reporter based on fluorescent protein ZsGreen, the HaloTag ProSensor reporter we developed provides new flexibility for analyzing tumor-initiating cells based on low proteasome activity. The availability of HaloTag ligands with fluorescent dyes ranging from blue to far red and near-infrared permits multiplexed detection of this proteasome reporter in combination with markers for specific cell types or proteins by microscopy and flow cytometry. 54 Labeling with far red or near-infrared dyes will also improve the detection of cancer cells with low proteasome activity in animal models by intravital microscopy and/or whole animal fluorescence imaging because longer wavelengths of light transmit more efficiently through tissues and avoid endogenous autofluorescence. 55 This reporter system can also be coupled with ligands for other imaging modalities, such as positron emission tomography (PET), with improved sensitivity for whole animal studies of tumor-initiating cells during tumor progression and therapy. 56 The capability to label tumor- and metastasis-initiating cells at a defined time point will enable pulse-chase experiments to track cell divisions, localization, and the fate of these cells over time because the imaging label will remain intracellular even after degradation of the reporter protein. 57 In addition, covalent bond formation between HaloTag ligands and intact HaloTag ProSensor reporter provides a facile approach for magnetic bead isolation of cells with low proteasome activity from implanted tumors and metastases in mouse models of cancer.
The strategy used to measure proteasome activity in this study works well for preclinical studies in which cancer cell lines and even patient samples can be transduced with reporter constructs. However, we recognize that this same strategy does not translate directly to patient care. As one alternative, there are fluorogenic, cell-permeable proteasome substrates to measure proteasome activity in intact cells by flow cytometry. 58 In combination with markers for cancer cells, these substrates could be used to identify tumor-initiating cells in excised tumors based on differences in proteasome activity. Recovered cells would then be available for subsequent analyses, such as nucleic acid sequencing and implantation as patient-derived xenografts in mice for drug testing studies. These assays and analyses are feasible in research settings, but fresh, unfixed tissues are not routinely available clinically. As a second alternative, cancer cells with low proteasome activity have been shown to express lower levels of selected proteasome subunits, indicating a link between protein levels and function. 16 Differences in expression of selected components of the proteasome in cancer cells can be detected by standard immunohistochemistry of formalin-fixed, paraffin-embedded tissue sections used in clinical pathology. Similar to assays of proteasome activity, low expression of a proteasome subunit by immunohistochemistry correlated with increased risk of local recurrence and/or reduced survival in patients with breast cancer, glioblastoma, and head and neck cancer.16,59,60 Although further validation studies are needed in different cancers, this recent report highlights potential future applications of proteasome activity and/or expression as prognostic and/or predictive biomarkers in clinical oncology.
Tumor-initiating cells in solid cancers have been defined by several different markers used singly or in combination. However, these markers appear to identify distinct subpopulations of cancer cells in standard cell lines and tumors from patients. 61 Discordances among markers are likely due to several factors, including the heterogeneity of subclones within a tumor, interconversion between populations with different functions, changes in signaling within a tumor microenvironment, and more differentiated cancer cells reverting a tumor-initiating cell phenotype.62,63 Minimal overlap in subsets of cells identified with different markers may also contribute to the fact that although current approaches enrich for tumor-initiating cells, tumors form at lower frequency from cells in excluded cell populations. Cancer cells with low proteasome activity show asymmetric cell divisions and enhanced tumor-initiating potential in mice, similar to other markers. Further studies are necessary to determine to what extent the subpopulation of cancer cells with low proteasome activity overlaps with tumor-initiating cells defined by other markers in different malignancies, as well as distinct phenotypes associated with low proteasome activity.
In summary, our study establishes that low proteasome activity defines a subpopulation of cancer cells enriched for both tumor- and metastasis-initiating potential. Enhanced tumor-initiating potential of this cell population extends to xenografts of primary patient-derived cancer cells, providing an exciting new link to human cancer biology. Identifying mechanisms that control the abundance and activity of proteasomes in cancer cells and specific proteasome substrates that drive tumor progression and metastasis will advance knowledge of tumor-initiating cells and point to new treatment strategies.
Footnotes
Acknowledgment
Financial disclosure of authors: This research was supported by National Institutes of Health grants R21CA182333, 5T32EB005172-03, R01CA136553, R01CA170198, and P50CA093990.
Financial disclosure of reviewers: None reported.