
Abstract
Molecular imaging is an attractive platform for noninvasive detection and assessment of cancer. In recent years, the targeted imaging of the C–X–C chemokine receptor 4 (CXCR4), a chemokine receptor that has been associated with tumor metastasis, has become an area of intensive research. This review article focuses on positron emission tomography (PET) and aims to provide useful and critical insights into the application of PET to characterize CXCR4 expression, including the chemical, radiosynthetic, and biological requirements for PET radiotracers. This discussion is informed by a summary of the different approaches taken so far and a comparison of their clinical translation. Finally, our expert opinions as to potential future advances in the field are expressed.
CXCR4
The C–X–C chemokine receptor 4 (CXCR4, also known as fusin and CD184) is a seven-transmembrane domain G protein–coupled receptor (GPCR), the widely accepted sole ligand of which is the stromal cell–derived factor–1, the predominant isoform of which is α (SDF-1α, also known as CXCL12).1,2 Recent reports indicate, however, the existence of other ligands to CXCR4, such as macrophage migration inhibitory factor, high-mobility group protein 1, and extracellular ubiquitin.3–5 Moreover, an alternate receptor that binds SDF-1α, the C–X–C chemokine receptor 7 (CXCR7), has also been described. 6 CXCR4 is expressed throughout development and adulthood, and the CXCR4/SDF-1α axis has a fundamental role in hematopoiesis by regulating retention and homing of hematopoietic stem cells in the bone marrow.7–10 This axis also plays a crucial role in lymphocyte trafficking to sites of inflammation. 11 In the physiologic context, CXCR4 is widely expressed on most leukocytes and on a variety of other cell types, such as endothelial and epithelial cells.12,13 Besides its expression in normal tissues, CXCR4 expression has also been associated with various diseases. CXCR4 was first reported as a coreceptor for CD4+ T-cell infection of human immunodeficiency virus (HIV) type I,14,15 and more recently, a role for CXCR4 was described in the pathogenesis of rheumatoid arthritis. 16 In addition to its extensive physiologic roles and implication in immune and inflammatory disorders, CXCR4 has been found to be expressed by various human cancers.17,18
CXCR4 and Cancer
Upregulation of CXCR4 has been reported in at least 23 different epithelial, mesenchymal, and hematopoietic cancers.19–21 The percentage of CXCR4-positive tumor cells in patient samples determined by immunohistochemistry ranged from 12% in early breast cancer 22 to > 90% in ductal carcinoma in situ, 23 non–small cell lung cancer,24,25 renal carcinoma, 26 mesothelioma, 27 gastric cancer, 28 and prostate cancer. 29 In metastatic lesions of breast cancer, non–small cell lung cancer, gastric cancer, and renal cell carcinoma, the percentage of CXCR4-positive cells ranged between 63.3 and 100%.29–32 CXCR4 overexpression in tumor tissues has been correlated to poor prognosis, tumor aggressiveness, increased risk of metastasis, and a higher probability of recurrence.33–35 CXCR4/SDF-1α signaling is involved in different aspects of tumor progression. Notably, the CXCR4-based chemotaxis acts directly on tumor cell migration and invasion toward an SDF-1α gradient. High expression levels of SDF-1α have been found at the most common sites of breast cancer metastases: axillary lymph nodes, lungs, liver, and bone marrow (Figure 1).36–39 Multiple studies also report the involvement of the CXCR4 signaling in promoting angiogenesis,40–43 tumor cell proliferation and survival, 44 and resistance to chemotherapy.45–47 Therefore, due to its critical roles in cancer, CXCR4 has been designated and characterized as a potential therapeutic target, and a number of CXCR4 inhibitors have been developed, including small-molecule antagonists, 48 antibodies,49–52 peptide antagonists,53–68 and glycosaminoglycan mimetics. 69 These inhibitors have been investigated in preclinical models, and some of them have reached clinical trials 48 ; the long-term toxicity and off-target activity of these agents remain to be fully elucidated. A comprehensive description of the CXCR4 pharmacophore is outside the scope of this review and has been reported elsewhere.17,48,70

Schematic representation showing the role of the CXCR4/SDF-1α axis as a key actor in the tumor invasion and metastasis process. Molecular crosstalks between the microenvironment (including fibroblasts, lymphocytes, macrophages, neutrophils, transforming growth factor-β) and cancer cells trigger CXCR4 expression and activation, promoting a migratory and invasive phenotype and resulting in the formation of organ-specific metastasis (bone, lungs, liver, brain, and lymph nodes). The metastatic process involves a series of sequential steps, including local invasion, intravasation, extravasation, and colonization of target organs guided by an SDF-1α chemoattracting gradient.18,133
CXCR4-Targeting PET Agents
It was proposed recently that these CXCR4-targeting molecules could also provide versatile platforms for the development of imaging agents. The evaluation of CXCR4 p>expression is currently assessed by the means of tissue sample biopsies,70,71 which may not be representative of the entire primary and metastatic disease volume in the patient, 72 in spite of the high clinical value of histologic assessment in the initial tumor diagnosis. The underlying rationale is that noninvasive imaging of CXCR4, with an ability to reflect heterogeneity of protein expression, could potentially be used as a complementary diagnostic or prognostic biomarker through the detection of tumors and highly aggressive subpopulations of tumor cells, as well as a repeated measure pharmacodynamic for CXCR4-targeted therapeutic interventions. Among the various strategies developed to image CXCR4 noninvasively, this review article focuses on the development of positron emission tomography (PET) radiotracers.70,73–75 PET can significantly contribute to the clinical management of a patient by providing early functional data on disease extent, therapy response, and identification of recurrence and can also be used for therapy planning. Technological advances in PET instrumentation have enabled the transition of human imaging capabilities to the scale of small animals,76–78 and PET imaging is now recognized as a major translational “bench to bedside” molecular imaging approach due to its high sensitivity, together with the large variety and increasing portfolio of tracers for the monitoring of key biological processes in cancer. 72
The development of a CXCR4-targeting PET imaging agent, including the selection of the targeting probe, the selection of the positron-emitting isotope and its associated labeling strategy, and the validation of these agents using cell-based assays and preclinical models, provides a number of (unique and generic tracer development) challenges that are discussed below. Although several CXCR4-specific radiotracers have been developed to date and have shown feasibility in animal models, the promise of a translational agent for the clinic is yet to be met. To the best of our knowledge, only one tracer for PET, [68Ga]–CPCR4.2, has to date completed initial evaluation in humans. The result of the trial is discussed further below. Another tracer, [64Cu]–AMD3100, originally developed by Nimmagadda and colleagues, 79 is recruiting within a phase 0 trial. 80 This review article aims to provide some insights toward the establishment of the optimal translational CXCR4-specific PET agent.
Rational Design and Development of CXCR4-Targeting PET Agents
Positron Emission Tomography
PET is a noninvasive molecular and functional imaging modality that measures the in vivo biodistribution of a molecular probe labeled with a positron-emitting radionuclide. Thus, following intravenous administration, this technology provides essential insights in biological processes in intact entire (whole body) living subjects. PET scanners detect the 511-kiloelectron volt annihilation photons generated as a result of positron decay (β+ decay) by unstable proton-rich radionuclides. The detailed description of the basic principles of PET imaging is outside the scope of this review article and has been reported elsewhere.72,81,82 A wide array of compounds of biological interest (e.g., antibodies, peptides, and small molecules) have been exploited and radiolabeled with positron-emitting radionuclides for PET imaging studies of various biological processes (e.g., blood flow, metabolism, cell surface receptor expression, angiogenesis, gene expression proliferation, and apoptosis). The flexibility of labeling any molecule that targets a specific biological process or marker, as well as the ability to image the spatial distribution of the labeled molecule in vivo over time, opens up many potential uses for PET from basic and preclinical research in animal models through to clinical investigations of various human pathologies. These radiolabeled probes are administered at trace amounts—pico/femtomolar range—thus, they do not perturb normal physiology to allow in vivo studies under physiologic conditions. PET therefore enables the investigation of substrate–target interactions or physiologic processes with the advantage of not having to administer an efficacious dose of a chemical compound, as with classic pharmacologic investigations. An important characteristic of PET imaging is that it enables the distribution of the radiotracer to be measured quantitatively, assuming that appropriate corrections are made for physical factors such as γ ray attenuation and scattering in tissues. The quantitative nature of PET is largely independent of the thickness of the object and the depth of the source within the subject. As radiotracers act as substrates/ligands for normal physiologic processes, they need to undergo extensive in vitro and preclinical validation.
Development of CXCR4-Targeting PET Radiotracers
The desirable optimal features for a PET imaging agent targeting the CXCR4 receptor are discussed below and illustrated in Figure 2. It should be emphasized that, irrespective of probe type (peptide, small molecule), the ideal tracer should have a rapid and high-affinity engagement of receptor and sufficiently fast clearance to permit low uptake in background tissues and hence high contrast.

PET tracer screening cascade. Diagram describing the screening cascade and detailing the various chemical, radiochemical, biological, and clinical traits radiotracers need to present. GMP = good manufacturing practice; HPLC = high-performance liquid chromatography; IC50 = half maximal inhibitory concentration; IMP = investigational medicinal products; MS = mass spectrometry; SDF1-α = stromal cell–derived factor–1α; SOP = standard operating procedure; UV = ultraviolet.
High Binding Affinity
To achieve good contrast and to maximize tumor visualization, tracer needs to have high affinity and a slow rate of tracer–receptor complex dissociation in target tissue to preclude tracer washout. For CXCR4, a number of assays have been implemented to ensure this. Cell-based assays employing [125I]SDF-1α or fluorescently labeled compounds have been used to assess affinity. Reversal of ligand binding is usually complemented by additional studies to verify downstream biological activity, such as a cell migration, given the impact of the CXCR4–SDF-1α axis on migration. These assays underpin the initial screening cascade for novel imaging agents. Using these assays, nanomolar affinity was seen with a number of compounds developed as CXCR4 probes for PET.83,84 Affinity and docking studies in the case of the cyclopentapeptide reported by Demmer and colleagues indicated a two-site binding model with CXCR4. 83
Specificity
It is important that any tracer presents no or low affinity for other receptors, that is, the tracer must be specific to the targeted receptor. If this is not the case, then the binding of the candidate tracer to another target than initially intended will lead to false positives and/or low contrast. Routine lipid and protein kinase, as well as receptor screens, are available for ensuring high specificity, although this service has not been explored for CXCR4 antagonists. Thus, although CXCR4 imaging agents show specificity in blocking studies or when low- and high-expressing cell lines are used, selectivity against other targets remains to be determined for the imaging probes. With our increasing knowledge of chemokine–receptor interactions, it has become necessary to also consider a lack of CXCR7 interaction as part of the screening cascade, although this aspect has not been widely investigated to date. Cyclopentapeptides, tetradecapeptides, and AMD3100, like the native ligand SDF-1α, have been shown to bind CXCR7.85,86 In this specific respect, new scaffolds will be required to discriminate CXCR4 from CXCR7. Of note, the noncyclam CXCR4 inhibitor AMD11070 does not bind CXCR7, 87 and it remains to be seen if monocyclam probes bind CXCR7.
Good Penetration
Owing to their size, high-molecular-weight tracers may penetrate slowly into solid tumors; poor tumor penetration results in low absolute uptake. Although a generic issue for the development of radiotracers for PET, this issue is not deemed acute for current CXCR4 tracers as most of these are either small organic molecules or peptides. The expected high penetration of these compounds compared to antibodies (which have high binding affinity but a long residence time) should allow for the use of optimal radiohalogen labeling with fluorine 18 (which has a short half-life, high photon flux, and low energy). This ideal is, however, not obvious in the literature, and lead radiotracers (discussed below) mainly employ radiometals such as copper 64 or gallium 68 for labeling. Although gallium 68 could be considered a good compromise for the expected rapid kinetics and tissue penetration of small-molecule organic compounds and small peptides, the same argument could not be made for copper 64. This limitation is partly due to difficulties in modifying bicyclams originally exploited for PET imaging to incorporate fluorine-containing moieties 79 ; on the other hand, the ease of incorporating DOTA or NOTA moieties into peptide has opportunistically favored radiometals for labeling.
Good Contrast
Effective distinction between an overexpressing CXCR4 tumor and its background, namely muscles and blood, is essential. Conversely, nontargeted tissues should present no or low uptake. Contrast can be quantified as the signal to noise ratio. Rapid renal clearance enhances contrast in most cases but can in some instances decrease the overall intensity of tumor uptake. Thus, a balance of physicochemical properties must be achieved to ensure contrast. Broadly, common strategies to achieve this include designing tracers with low n-octanol–water distribution coefficient (logD), which are ideally charged. Conversely, tracers presenting high logD values are likely to be excreted by the hepatobiliary route and to pass the blood-brain barrier, both of which would result in nonspecific radioactivity uptake. High-molecular-weight radiotracers are also often eliminated by the hepatobiliary route. CXCR4 peptidic tracer [68Ga]–CCIC16 shows a logDpH 7.4 of −3.58 ± 0.15, 88 whereas cyclopentapeptides display a variety of distribution coefficients, ranging from 1.09 ± 0.02 ([18F]CCIC07) 89 to −2.30 ± 0.30 ([18F]CCIC30) (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). Monocyclam [64Cu]–AMD3465 displays a logP of −2.71 ± 0.37, 84 whereas bicyclam [64Cu]–AMD3100 shows a logP of 0.52 ± 0.02. 79
Appropriate Metabolism
Metabolism has two facets: on the one hand, metabolism could be desirable when it improves excretion of the tracer, but on the other hand, it might produce metabolites that bind nonspecifically, thus reducing contrast. Consequently, a tracer that presents partial metabolic instability in vivo could still prove to be efficient provided that metabolites are quickly excreted. Ideally, however, every effort should be undertaken to reduce metabolism. Unfortunately, the structure–activity relationship (SAR) around CXCR4 small molecules and peptides with respect to metabolism is less advanced, precluding substantive activity on this issue to date. For example, whereas [68Ga]–CPCR4.2 is reported to be stable in vivo, 90 a relatively similar cyclopentapeptide probe for PET, [18F]CCIC07, a 4-[18F]fluorobenzoimino PEG-functionalized cyclo(Nal1–Gly2–D-Tyr3–Orn4–Arg5) cyclopentapeptide, showed rapid biliary and renal elimination and metabolic instability (O Åberg, Pisaneschi F, Smith G, et al. unpublished work, 2012). For radiometal-labeled bicyclams such as [64Cu]–AMD3100, the possibility of transchelation and hence loss of the radiometal is a concern. Thus, in vivo stability should be a key screening variable to be empirically examined early in the development of CXCR4 probes for imaging by PET.
Careful Chemical Design
This aspect brings together all the aforementioned desirable properties. Recent disclosure of the crystal structure of CXCR4 91 permits modeling/docking studies to optimize affinity and specificity. For instance, three of the four nitrogens of small-molecule IT1t make important salt bridges or polar interactions with the receptor, whereas both cyclohexyl rings fit in small hydrophobic subpockets. These observations exhort the medicinal chemist to conserve these essential pharmacophores to maintain affinity. On the other hand, and in accordance with the SAR analysis of a series of CVX15 analogues,64–68 hexadecapeptide CVX15 principally interacts via numerous hydrogen bonds and salt bridges with the strongly negatively charged receptor binding site, whereas CVX15's β-turn points toward the extracellular milieu, admonishing us to introduce derivatizations enabling radiolabeling on the β-turn's residues.92–94 Although the spatial arrangement of the pharmacophores will mainly impact receptor binding and specificity, other characteristics, such as the overall size, charge, and logD of the molecule, will influence other pharmacokinetic behavior aspects, including excretion, metabolism, and tumor penetration. One example of this statement is the substitution of several arginine residues for citrullines in the tetradecapeptide class, which was aimed at reducing the cell toxicity associated with the high overall charge of the peptide. 65 Finally, as emphasized later in this review, the chemical design of the radiotracer will dictate which radioisotopes are available for labeling and which radiochemical reactions can be performed. This, in turn, will influence the tracer's PET and radiosynthetic properties. Therefore, the chemical design of the radiotracer is a nontrivial problem consisting of multiple variables for which a compromise between its synthetic, radiosynthetic, and pharmacokinetic features needs to be found.
High Radiochemical Purity and Specific Activity
At tracer level, it is unlikely that an antagonist could elicit a biological response and consequently adversely interfere with the biological system. However, radioactive by-products can present nonspecific binding and therefore reduce contrast. The measured variable to describe this property is radiochemical purity, which is defined as the proportion of radioactivity emanating from the desired radiotracer. In addition, nonradioactive by-products present in the injected solution and traces of the nonradioactive isotopologue can potentially compete with the tracer for receptor binding. Such nonradioactive products must be kept to a minimum and are quantified in a “specific activity” parameter, which is defined as the ratio between the activity of the sample and the amount of material in the sample (typically given in GBq.μmol−1). In the CXCR4 context, the range of specific activities varies greatly depending on the molecule labeled, the radio emitter, and the type of radiosynthesis performed. [64Cu]–AMD3100 is reported with specific activities as high as 417 GBq/μmol−1, 95 whereas the specific activities of the other tracers that allow for the visualization of tumors range between 5 and 30 GBq/μmol−1.84,93,96,97
Tumor Model
Once the above parameters have been optimized, the choice of cell lines/tumor models used for preclinical in vitro and in vivo experiments carried out to determine candidate tracer's characteristics can strongly influence the results obtained. The properties of numerous CXCR4 models were recently reviewed by Kuil and colleagues. 70 For a fair comparison of data from different studies, it is important to specify the cell model used. The in vitro characterization of the tracer, including affinity and specificity, is influenced by the cell model and the level of CXCR4 expression at the cell surface. In this regard, the two main cellular models used are CXCR4-transfected cells, which harbor a significant upregulation and expression of the receptor compared to the mock-transfected counterpart, and human cancer cells with intrinsic high expression of CXCR4.79,83,84,90,92,97–103 However, the level of CXCR4 expression at the membrane is not always reported, and for that reason, cross-comparison of data between these models has to be drawn with caution. Alternative models that recapitulate human cancers in their microenvironment, including patient-derived xenografts and genetically engineered mouse models, would potentially make a significant contribution within the experimental arsenal for the development of CXCR4-specific imaging agents and are eagerly awaited.
CXCR4-Targeting Agents
The apparent importance of CXCR4 in different pathologies encouraged efforts toward discovery of CXCR4-targeting therapeutics. A quick overview of a selection of classes of CXCR4-targeting chemicals is provided in Figure 3, but the interested reader is referred to the most recent reviews on the subject for more detail.48,104–108 The quest for CXCR4 antagonists started two decades before the determination of any crystal structures for this transmembrane receptor. 91 Early attempts at discovering potent and clinically viable CXCR4 antagonists (which were referred to as HIV-1 entry antagonists until 1994, the year when CXCR4 was discovered 109 ) were mainly focused on peptides. The discovery of tachyplesin I and polyphemusin II in the late 1990s62,63 and multiple SAR analyses based on their structures yielded a plethora of octadeca- to tetradecapeptidic CXCR4 inhibitors.64–68 Of interest, TN14003 65 and its N-4-fluorobenzoyl derivative TF14016 68 were found to be particularly active and stable in vivo and are close analogues of CVX15, which has been cocrystallized with CXCR4. 91

Selected examples of CXCR4 antagonists. The classes that constitute the major focus of this review article are boxed. Cit = citrulline; Nal = 2-naphthylalanine.
Efforts at reducing the molecular weight of these tetradecapeptides were made as smaller peptides tend to ameliorate their undesired immunoresponse. These efforts resulted in a library of cyclopentapeptides and cyclopentapeptide mimics,53–61,110,111 among which FC131 (see Figure 3) and FC122 were found to be highly potent against HIV-1 entry and CXCR4.
Numerous small molecules were also developed. The serendipitously discovered AMD3100, found as an active impurity during a research program aimed at finding an anti-HIV agent, 112 was recently approved for clinical use to mobilize hematopoietic CD34+ stem cells from the bone marrow into the circulation. In oncology, numerous clinical trials involving AMD3100 in combination with conventional chemotherapies are still ongoing. 113 In 2005, Hatse and colleagues developed the less charged monocyclam AMD3465, which proved to be 10-fold more potent than AMD3100 despite lacking the bicyclam structure that was believed to be a fundamental requisite for receptor binding. 114 Many more molecules were developed based on these structures and were thoroughly and recently reviewed by Debnath and colleagues. 48
In addition to the above, IT1t is a small molecule that has been cocrystallized with CXCR4 and has been reported to have superior binding affinity to AMD3100. 115 This molecule was developed by Novartis along with 20 other analogues published in the original article. 115 Although no other instances of antagonists with structural similarities to IT1t have been published, it can be anticipated given its favorable pharmacokinetic profile and the availability of the IT1t/CXCR4 cocrystal structure 91 that this scaffold may well attract increased attention in the future.
Radiochemical Design and Production of CXCR4-Targeting Agents
Insights into the SAR of the class of molecule or a crystal structure of the receptor (ideally cocrystallized with a representative of the tracer class of interest) could lead to determining with confidence positions on putative probes that could be modified to allow the introduction of the radioelement. The PET tracer should also be easy to synthesize in good quality. Therefore, the radiolabeling strategy should be high yielding (> 5% non–decay-corrected radiochemical yield [ndc-RCY]) and should be ideally suitable for automation to reduce the radioactive dose to the radiochemist and improve the reproducibility of radiotracer production.
The choice of the radioelement used to produce the tracer is also important as it influences the labeling step, the structure of the tracer, and some of its imaging properties (Table 1).
Comparison of the Characteristics of PET Radionuclides Used for the Labeling of CXCR4-Targeting Tracers

β = beta (electron) decay; β+ = positron emission; EC = electron capture; PET = positron emission tomography.
According to the compact notation for nuclear reactions, A+b→c+D is equivalent to A(b,c)D.
18F is usually regarded as the radionuclide of choice when it comes to positron emission. This is not only because its relatively long half-life allows for more complex labeling processes, but also because it decays almost exclusively by emission of positrons of low kinetic energy, which translates into shorter acquisition times and high resolution. 18F-Labeling can be performed via the use of [18F]fluoride (18F−) or [18F]fluoronium (18F+). [18F]F+-labeling (mainly by demetallation/fluorination) is limited by the ability to only achieve low specific activity. 116 Conditions used for labeling performed with [18F]fluoride, which can deliver higher specific activities, are often harsh and involve the use of either aliphatic or aromatic nucleophilic substitution reactions. The use of prosthetic groups has attracted much interest in recent years and enabled radiochemists to label more complex and delicate structures. On the other hand, this strategy introduces considerable structural variation, especially in small molecules. Recently, new radiolabeling methodologies have started to appear in the literature.117–121 Although a revision of these methods is outside the scope of this review, it is clear that the possibilities for 18F-labeling using these techniques are expanding and will doubtless give access to increasingly complex and efficient radiosyntheses.
Radiometals offer the advantage of a fast and tractable radiolabeling step by chelation with a precursor adorned by a multidentate chelating moiety. 122 Unfortunately, such chelating moieties are large, and their introduction in the structure of a tracer can greatly influence its affinity for the receptor. Their use is therefore limited to large tracers, such as peptides, for which such modifications are comparatively small. In addition, transchelation may be observed after administration, and it is therefore necessary to determine the exact in vivo behavior of such radiotracers. 123 Two of such radiometals are 68Ga and 64Cu. Owing to the low kinetic energy of its emitted positron, 64Cu offers good spatial resolution. Its long half-life allows scanning of subjects several hours after injection of the tracer. This provides time for the body to clear most of the nonspecifically distributed tracer from the organism, thus yielding images of high contrast. Nevertheless, only 18% of the decay is available for PET, exposing the subject to high doses of ionizing β radiation over a long period; in fact, 64Cu-ATSM, a radiospecies design for the imaging of hypoxic tumors, was recently used in preclinical evaluation for radiotherapeutic use. 124 On the other hand, 68Ga is an efficient PET radioisotope as it has 89% positron decay. Moreover, this isotope is produced in a portable generator, making it readily available for hospitals devoid of bulky and expensive cyclotrons. Its shorter half-life-means that the patient receives a lesser dose but also precludes benefiting from the improved contrast concomitant with postadministration deferment of data acquisition. Finally, 68Ga offers images of lower spatial resolution than 18F.
Depiction of CXCR4-Targeting PET Agents Developed to Date
A variety of the previously mentioned CXCR4 antagonists have been radiolabeled for PET imaging application following modification for the introduction of a radionuclide. These compounds are summarized in tabular form below and then discussed on a class-by-class basis (Table 2).
CXCR4-Targeting PET Imaging Agents to Date


Cit = citrulline; DO3A = 2-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecan-1-yl]acetyl; Nal = 3-(2-naphthyl)alanine; NO2A = 2-[4,7-bis(carboxymethyl)-1,4,7-triazacyclononan-1-yl]acetyl.
GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014.
Cyclams
Efforts at labeling AMD derivatives with 64Cu afforded [64Cu]–AMD310079,95,101 and [64Cu]–AMD3465, 84 the latter of which presented superior characteristics, such as tumor to muscle and tumor to blood ratios, which were seven- and eightfold higher at 1.5 hours postinjection, respectively. In vivo evaluation with three different cancer cell lines presenting various levels of expression of CXCR4 exhibited levels of tracer uptake varying in accordance with the expression of the receptor, namely U87.CD4.CXCR4 > HT-29 > U87. 84 These results are interesting as they suggest that the in vivo expression of CXCR4 could be quantified by PET scanning, which in turn could be related to the aggressiveness of the tumor.125–128 The observation of a high uptake of radioactivity in liver and kidneys has been assumed to result at least partly from specific interactions with an unknown target. Although uptake in the CXCR4-expressing cell model is comparatively high and suggests that [64Cu]–AMD3465 could allow for the visualization of hepatic or renal tumors expressing high levels of CXCR4, the liver and the kidneys would be exposed to high doses of ionizing radiation.
Cyclopentapeptides
Cyclopentapeptides have also been derivatized to afford tracers, either based on the structure of FC131 for labeling with p-fluorobenzaldehyde 89 or by “click” chemistry with 2-fluoroethylazide, 96 or based on the structure of FC122 for labeling with 68Ga.83,90,102 Whereas the fluorobenzaldehyde derivative [18F]CCIC07 showed no in vitro uptake and had an extremely high rate of metabolism, probably due to the presence of the polyethylene glycol linker (O Åberg, Pisaneschi F, Smith G, et al, unpublished work, 2012), the triazole derivative [18F]CCIC15 presented adequate uptake in vitro (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). Although all published work has focused on substitution of Arg3 to enable the introduction of the radioelement as this residue is believed to contribute least to the binding affinity of this class of antagonist, we have shown that replacement of FC131's D-Tyr residue can lead to improved affinity (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). The tracer [18F]CCIC30 has an IC50 value of 0.37 ± 0.12 μM, which is lower than that for FC131 (IC50 = 0.65 ± 0.22 μM) (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). This can perhaps be explained by the size of the triazole ring, which is smaller than that of the phenol unit in D-Tyr and which may therefore occupy the same pocket in CXCR4 and thus preserve all of the side-chain and backbone interactions of the unmodified cyclopentapeptide. It also cannot be excluded that one of the triazole's nitrogen participates in the binding via hydrogen bond with CXCR4's Tyr190 and the aromatic ring itself via hydrophobic contacts, mimicking D-Tyr's interactions with the receptor. [18F]CCIC30 showed a favorable in vitro profile, with twofold higher uptake in the CXCR4-positive U87.CD4.CXCR4 cells compared to isogenic CXCR4-negative U87.CD4 cells (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). Unfortunately, this did not translate in vivo, where no difference in tumor uptake was seen between the two cell lines. Moreover, although the imaging data averaged over 5 to 30 minutes postinjection highlighted the tumor, at 60 minutes postinjection, no tumor uptake was detected. Most of the radioactivity was found in the bladder (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014). This, together with evidence of metabolic stability by 60 minutes postinjection, led to the conclusion that the low tumor uptake was due to excessive renal excretion (GPC George, F Pisaneschi, E Stevens, et al, unpublished work, February 2014).
In contrast to the above, [68Ga]–CPCR4.2 permitted efficient visualization of OH-1 tumors. 90 Although the contrast was much lower than that achieved with [64Cu]–AMD3465, [68Ga]–CPCR4.2 presented limited uptake in the liver and the kidney. Along with the intrinsically superior PET characteristics of 68Ga compared to 64Cu (especially when the reconstruction algorithm considers the type of radioisotope), these excellent preclinical results encouraged a preliminary human study, which showed for the first time PET imaging of CXCR4 expression in human. 129 In a clinical setting, [68Ga]–CPCR4.2 exhibited limited uptake in nontarget tissues and the ability to visualize certain tumors, including nodal lesions, from chronic lymphocytic leukemia and lymphoma, non–small cell lung adenocarcinoma lesions, and CD30+ aggressive T-cell lymphoma [mean SUV60 values: blood 1.83, muscle 0.77, lung 0.75, liver 1.46, spleen 5.69, bone marrow 4.00, kidney 5.06, bladder 36.4. nodal lesion in chronic lymphocytic leukemia: 5.05 (. FDG). Lymphoma and lung lesions in nonsmall cell lung adenocarcinoma with CD30+ aggressive T-cell lymphoma: 9.91 and 2.70, respectively]. 130 Further clinical results are required, but [68Ga]–CPCR4.2 is already a significant milestone in the quest for a clinically useful CXCR4-specific PET radiotracer. The superior characteristics of [68Ga]–CPCR4.2 compared to [18F]CCIC30 can be attributed to the unexpectedly favorable interaction of the DOTA chelator unit. According to the authors, the addition of the DOTA moiety to FC122 led to loss of potency, but the chelation of this molecule with natGa, affording natGa–CPCR4.2, increased CXCR4 affinity by a factor of 35. 90 The authors attribute this observation to the fundamental structural differences, as well as the overall charge and charge distribution induced by the complexation of gallium.
Tetradecapeptides
Finally, tetradecapeptide TN14003 has generated the most interest from the radiochemistry community. Although the first derivatives presented unexpected red blood cell binding92,97 and/or required long (> 2.5 hours) and low-yielding (< 5% ndc-RCY) 18F-labeling procedures,94,97 it was later found that the addition of a chelating group at the peptide's N-terminus allowed red blood cell binding to be circumvented, while allowing for chelation with 64Cu and retaining acceptable affinity for CXCR4. 100 The fact that this derivatization could be tolerated was at first surprising as CVX15, a close derivative of TN14003 [CVX15 = TN14003–Gly–D-Pro–OH], was cocrystallized with CXCR4 in a pose where both its N- and C-termini were buried in CXCR4's helix bundle, filling the whole binding site. 91 This result can, however, be rationalized by considering that the shorter peptide, TN14003, allows for substantial derivatization at one of the peptide's termini. High liver and kidney uptake of the 64Cu-DO3A-N-terminus labeled TN14003, [64Cu]–DOTA–NFB, led us to engineer a revised TN14003-based CXCR4-specific PET tracer using a smaller chelating moiety to reduce affinity loss for CXCR4 when labeling with 68Ga. 88 This tracer, [68Ga]–CCIC16 (Table 3), showed improved tumor uptake in shorter incubation times and allowed for effective localization of the tumor within 1 hour postinjection in the CXCR4-overexpressing U87.CD4.CXCR4 xenograft–bearing mouse compared to published studies of [64Cu]–DOTA–NFB and [64Cu]–NOTA–NFB with the same tumor lines (Figure 4C). It was characterized by adequate contrast and promising in vivo stability and clearance, therefore showing the essential features for the identification of CXCR4-expressing tumors in a clinical setting.

Selected images from PET/CT experiments with [64Cu]–AMD3465 (A), [64Cu]–CPCR4.2 (B), and [68Ga]–CCIC16 (C). White arrowheads indicate the CXCR4-overexpressing tumors. The hollow arrowhead indicates the CXCR4-low tumor. See the original research in De Silva and colleagues and Gourni and colleagues.84,90 B = bladder; K = kidneys; L = liver.
Comparison of Radiosynthetic and Biological Properties of the Most Recent and Promising Tracers in Their Class

B = blood pool; BM = bone marrow; GB = gallbladder; IC50 = half maximal inhibitory concentration; K = kidneys; L = liver; Lg = lungs; ndc-RCY = non–decay-corrected radiochemical yield; S = spleen; T/B = tumor to blood ratio; T/M = tumor to muscle ratio; U = urine.
Information obtained from the author for correspondence.
Advantages and Limitations
In the interest of brevity, only the most recent and promising PET tracers are critically reviewed. A comparison of radiosynthesis and biological properties of three of the most recent and promising CXCR4 ligands from different structural classes—small-molecule bicyclam, cyclic pentapeptide, and 14–amino acid cyclic peptide—is considered in the following discussion (see Table 3 and Figure 4).
From a radiochemistry point of view, radiometal chelation appears favorable for CXCR4 ligands; this is mainly due to the fact that most of the tracers radio-synthesized to date are peptidic in nature. Moreover, although small molecules are not usually labeled via radiometal chelation owing to the incompatibility of the size of the chelator moiety needed relative to that of the targeting small molecule, both AMD3100 and AMD3465 were successfully radiolabeled with 64Cu. This peculiarity can be explained by the unique nature of the cyclam present in these molecules, which offers the opportunity for chelation of 64Cu while concomitantly acting as a pharmacophore for binding to CXCR4. As previously described, attempts at labeling with 18F have also been described but have tended to result in long (> T1/2) and low-yielding (< 5% ndc-RCY) syntheses, as exemplified by the efforts to label TN14003 derivatives with 2-fluoropropionate or 4-fluorobenzoate.94,97 As a side note, the use of 4-[18F]fluorobenzaldehyde or 2-[18F]fluoroethyl azide allowed for labeling in short times (< T1/2) and with acceptable efficiencies (10–20% ndc-RCY), although the obtained tracers did not show adequate biological properties.89,96 The comparatively low radiochemical yield obtained for [68Ga]–CPCR4.2 can be accounted for by the additional separation of the tracer from the remaining unreacted precursor by time-consuming semipreparative high-performance liquid chromatography (HPLC) and subsequent lyophilization. Combined with using relatively high starting activities, these protocols resulted in good specific activities, with the purpose of reducing undesired adverse effects.
Biologically speaking, the three classes of compounds explored so far all showed potential for the imaging of CXCR4 expression. [64Cu]–AMD3465 and [68Ga]–CCIC16 showed adequate in vitro uptake difference between CXCR4-expressing U87.CD4.CXCR4 and their isogenic CXCR4-low counterparts. On the other hand, Wester and colleagues chose to assess their tracer, [68Ga]–CPCR4.2, in the human small cell lung cancer cell line OH1, which endogenously expresses CXCR4. 90 No adequate control cell line is available for this cell line. In vivo, [64Cu]–AMD3465 yielded extremely high contrast, as depicted by the tumor to muscle and the tumor to blood ratios; however, the tracer also exhibited levels of uptake in the liver and the kidneys that cannot be explained by their endogenous expression of CXCR4. This tracer is therefore deemed to distribute nonspecifically to liver tissue, which would query use in liver metastases. Similar behavior was observed with its congener [64Cu]–AMD3100. Despite these concerns, [64Cu]–AMD3100 has been chosen for a small clinical trial aimed at assessing the safety and efficacy of the tracer in patients with tumors equal to or greater than 2 cm in diameter found outside the lymph nodes, bone marrow, liver, gallbladder, kidney, bladder, and brain. The study is currently recruiting participants.
These nonspecificity issues might be due to the relative simplicity of the structures of both of these tracers, and it could be expected that further modification of the molecule could improve the specificity of this type of compound, for instance, by gaining inspiration from the numerous other small molecules that have been derivatized from AMD3100. 48 Moreover, the small-molecule nature of this class could allow for efficient labeling with 18F, a radioisotope that is intrinsically more suitable for PET imaging than 64Cu; to date, no 18F-labeled small molecule–based CXCR4-specific radiotracer has been published, as emphasized by Table 2. Conversely, [68Ga]–CPCR4.2 and [68Ga]–CCIC16 showed good imaging properties, such as fast renal clearance and limited background uptake. Compared to [64Cu]–AMD3465, these two tracers exhibited much lower contrast; they were, however, still found adequate for the imaging of tumors. Similarly, uptake in metabolic organs was found to be lower with these two gallium 68–labeled tracers than with [64Cu]–AMD3465, although to different extents. The uptake of [68Ga]–CPCR4.2 in nontumor tissues was almost negligible when compared to [68Ga]–CCIC16; it can be anticipated that such low uptake might allow for the imaging of hepatic tumors. Although [68Ga]–CPCR4.2's urinary localization was not measured, it seems clear from the PET images that renal tumors cannot be imaged with this tracer. All in all, to date, [68Ga]–CPCR4.2 and [68Ga]–CCIC16 are the CXCR4-targeting PET tracers that show the most promising results across all tumor sites, whereas [64Cu]–AMD3465 had the highest sensitivity.
Perspectives and Future Directions
To date, peptidic tracers have attracted the most interesting scaffold for CXCR4-targeting radiotracers, undoubtedly because of their ease of synthesis and the consequent relative rapidity with which compounds of desirable pharmacokinetic properties can be developed. [68Ga]–CPCR4.2 is the most promising tracer of this kind, and preliminary human scans with this tracer underlie the possibility of imaging certain tumors by targeting CXCR4. We can expect to see reports of how this tracer performs in a clinical setting soon, which could enlighten us as to the suitability (sensitivity and specificity) of CXCR4 imaging for prognosis, diagnosis, and tumor phenotyping. This is particularly eagerly anticipated as although numerous factors indicate that CXCR4 is a PET target of high interest, tangible confirmation of this assumption is required. In the longer term, this could allow for patient stratification and personalized medicine with CXCR4-targeting drugs or radiotherapy. Moreover, it is possible that chelation of CPCR4.2 to radiotherapeutic metals such as 90Y or 177Lu could afford a radiotherapeutic agent, for use in a theranostic approach.
On the other hand, small molecules have so far enjoyed less attention as potential CXCR4-targeting PET agents. AMD3100 and AMD3465 are the only small molecules that have been radiolabeled for PET imaging purposes (see Table 2). However, these cyclam derivatives seem to lack specificity for CXCR4, as depicted by their high and blockable uptake in the liver and the kidneys. Given the pharmacologic attraction of CXCR4, the number of small-molecule CXCR4 antagonists being reported is rapidly increasing, and the emergence of radiolabeled analogues of these molecules is therefore to be expected. It is hoped that such new radioligands may circumvent the nonspecificity issues encountered with [64Cu]–AMD3100 and [64Cu]–AMD3465.
Other potential examples are Liotta and colleagues' 31k, 131 GlaxoSmithKline's 13, 132 and Novartis's IT1t 115 (Figure 5).
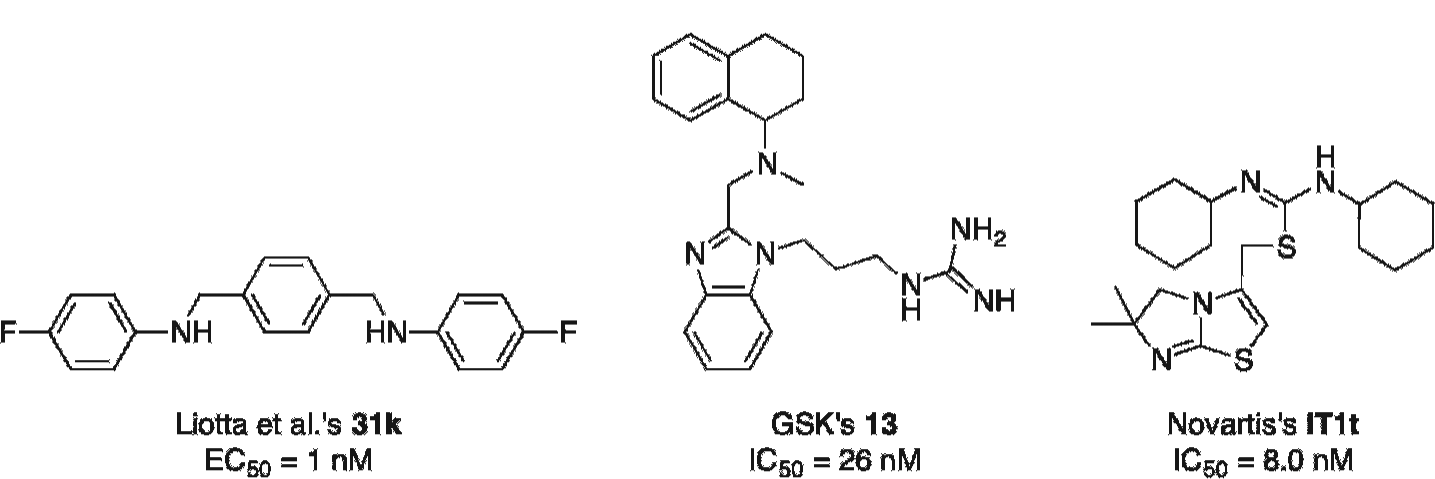
Chemical structures of three CXCR4 inhibitors that could be derivatized to afford novel PET radiotracers. EC50 = half maximal effective concentration; IC50 = half maximal inhibitory concentration.
These compounds have high affinities for CXCR4 and could be derivatized to allow for their radiolabeling with 18F. Para-xyxyl 31k 131 contains two fluorine atoms, one of which could be replaced by a nickel-based substituent to undergo a palladium-mediated fluorination with aqueous [18F]fluoride 118 and afford [18F]31k. Similarly, it can be anticipated that substituting an aromatic hydrogen of tetrahydroquinoline 13 132 for an iodine could allow for the [18F]trifluoromethylation of the resulting precursor into the corresponding [18F]trifluoromethylated 13. A careful in silico docking study and an SAR analysis will nevertheless be needed to determine which aromatic position is more adequate for the introduction of such modification.
As aforementioned, four cocrystal structures of the small molecule IT1t with CXCR4 (2.5–3.2Å, PDB: 3ODU, 3OE6, 3OE8, 3OE9) were elucidated in 2010 in a landmark study by Stevens and colleagues, along with a CVX15–CXCR4 cocrystal structure (PDB: 3OE0). 91 Taking into consideration the interesting pharmacokinetic profile of IT1t, which is characterized by high affinity and specificity for CXCR4 and in vitro stability against various cytochrome P-450 enzymes, 115 these x-ray data are expected to promote the molecular design of IT1t-based tracers.
Crystal structures are convenient as they allow for the determination of the interactions that mediate the binding of a ligand to the receptor and can help inform the de novo design of new CXCR4 ligands or the improvement of existing structures. However, the function of GPCRs is to transmit a signal through the cell membrane via a change of conformation; this type of receptor is therefore highly flexible, as illustrated by the differences between the IT1t- and CVX15-bound cocrystal structures. 91 The flexibility of CXCR4's structure admonishes the medicinal chemist to proceed with care when trying to dock molecules in silico into the crystal structure. The use of high-throughput virtual screenings should especially be avoided as this technique relies on rigid docking, whereby the receptor–ligand interactions are modeled on the “key–lock” rather than on the more accurate “hand–glove” paradigm. Although challenging, the evaluation of IT1t-based tracers is particularly attractive as it would enable the adequacy of 18F-labeled small molecules for the imaging of CXCR4 expression to be explored, an area still uncharted, as highlighted previously (see Table 2).
Conclusion
Since 1994 and the discovery of CXCR4, there has been great progress toward the development of CXCR4-targeting PET tracers. Various potent CXCR4 inhibitors have been discovered, and over the last 7 years, some of these have been engineered to allow for radiolabeling with 18F, 68Ga, or 64Cu. Although there is no clear consensus as to the precise clinical application for imaging CXCR4 to date, the potential use of such PET tracers in cancer prognosis, cancer diagnosis, tumor phenotyping, and theranostics makes research to develop such probes of great interest. 64Cu-radiolabeled AMD3465 has to date proven to be the best radiotracer with respect to absolute tumor uptake. The preliminary clinical results obtained from the evaluation of [68Ga]–CPCR4.2 are particularly encouraging regarding low liver radiotracer localization. Finally, an 18F-labeled small molecule for the noninvasive imaging of CXCR4 expression by PET has yet to be disclosed but can be expected to emerge in the near future.
Footnotes
Acknowledgments
We would like to thank Elizabeth Stevens for producing the [68Ga]–CCIC16 PET imaging data set.
Financial disclosure of authors: This work was funded by Cancer Research UK–Engineering and Physical Sciences Research Council (in association with the Medical Research Council and Department of Health, England) grant, C2536/A10337. E.O.A.'s laboratory receives core funding from the UK Medical Research Council (MC_A652_5PY80).
Financial disclosure of reviewers: None reported.