
Abstract
The potential of using endothelial progenitor cells (EPCs) in novel anticancer therapy and the repair of vascular injury has been increasingly recognized. In the present study, EPCs were labeled with N-alkyl-polyethylenimine 2 kDa (PEI2k)-stabilized superparamagnetic iron oxide (SPIO) to facilitate magnetic resonance imaging (MRI) of EPCs in a mouse lung carcinoma xenograft model. EPCs derived from human peripheral blood were labeled with alkyl-PEI2k/SPIO. The viability and activity of labeled cells were evaluated using proliferation, migration, and tubulogenesis assays. Alkyl-PEI2k/SPIO-labeled EPCs were injected intravenously (group 1) or mixed and injected together with A549 cells subcutaneously (group 2) into groups of six mice with severe combined immunodeficiency. The labeling efficiency with alkyl-PEI2k/SPIO at 7 mg Fe/mL concentration was approximately 100%. Quantitative analysis of cellular iron was 6.062 ± 0.050 pg/cell. No significant effects on EPC proliferation, migration, or tubulogenesis were seen after labeling. Seventesla micro-MRI showed the presence of schistic or linear hypointense regions at the tumor margins starting from days 7 to 8 after EPC administration. This gradually extended into the inner tumor layers in group 1. In group 2, tumor growth was accompanied by dispersion of low-signal intensity regions inside the tumor. Iron-positive cells identified by Prussian blue dye were seen at the sites identified using MRI. Human CD31-positive cells and mouse CD31-positive cells were present in both groups. Labeling EPCs with alkyl-PEI2k/SPIO allows noninvasive magnetic resonance investigation of EPC involvement in tumor neovasculature and is associated with excellent biocompatibility and MRI sensitivity.
TUMOR GROWTH, invasion, and metastasis depend on the development of a vascular network within the tumor. As the tumor volume increases beyond 2 to 3 mm3, a continual supply of blood is required to remove waste and deliver nutrients.1,2 The process of tumor neovascularization is complex.
Endothelial progenitor cells (EPCs) are precursor cells that are capable of differentiating into endothelial cells.3,4 A growing body of evidence suggests that EPCs contribute to the progression of tumor neoangiogenesis.5,6 It has been reported that the percentage of endothelial cells incorporated into perfused tumor blood vessels may be as high as 58%. 7 Recent evidence suggests that the tumor microenvironment mobilizes and recruits EPCs to tumor, where they promote angiogenesis under hypoxic condition.8,9 However, the exact molecular mechanism responsible for mobilizing EPCs is as yet unknown.
Preclinical studies using EPC transplantation to repair vascular injury and ischemic tissue10–13 or as a vector targeting angiogenesis in anticancer therapy14–17 have reported encouraging results. However, some researchers have reported that EPCs play a minimal or no role in promoting new vessel formation in tumors, indicating that their role in integrating tumor vasculature is questionable.18,19
Tumor neovasculature is usually demonstrated by histopathology or immunohistochemistry. The major limitations of these methods are that detection is not continuous, experimental animals have to be sacrificed, and follow-up is not possible. Advances in molecular imaging techniques make noninvasive and real-time analysis possible. By magnetically labeling human CD34+/AC133+ EPCs with ferumoxide-protamine sulfate (FePro), Arbab and colleagues used magnetic resonance imaging (MRI) to demonstrate the incorporation of FePro-labeled EPCs into the neovasculature of implanted flank tumors. 20
In a previous study, our research team synthesized N-alkyl-polyethylenimine 2 kDa (PEI2k)-stabilized superparamagnetic iron oxide (SPIO) nanoparticles (abbreviated as alkyl-PEI2k/SPIO), which displayed good stability and dispersibility. These particles have since been cataloged in the National Institutes of Health Molecular Imaging and Contrast Agent Database. 21 The main characteristics of alkyl-PEI2k/SPIO can be categorized as follows: (1) it leads to higher MRI sensitivity at the same iron concentration or achieves similar MRI contrast and better biocompatibility with smaller dosages and (2) it has both imaging and drug delivery functions with dual functional carriers. 22 Liu and colleagues used alkyl-PEI2k/SPIO to label mesenchymal stem cells (MSCs) and injected the labeled MSCs dispersed in a collagen hydrogel subcutaneously into the flanks of BALB/c mice. 23 MRI showed that the hypointensity of the labeled MSC implant remained visible for at least 19 days. Another study also used alkyl-PEI2k/SPIO to label tumor cells and tracked the metastasis of tumor cells in lymphatics by cellular MRI. 24 Based on these findings, bylabeling with self-assembled alkyl-PEI2k/SPIO and tracking with high-resolution MRI, it should be possible to noninvasively investigate the involvement of EPCs in tumor neovasculature. These results should further clarify the role of EPCs in tumor angiogenesis and provide possibilities for monitoring EPCs as a drug vector for antitumor angiogenesis therapy.
In the present study, we first introduced alkyl-PEI2k/SPIO into human peripheral blood-derived EPCs. We evaluated labeling efficacy and investigated the biological effects on peripheral blood EPCs after in vitro labeling. To investigate patterns of EPC migration in tumor, labeled EPCs were administered to a mouse lung carcinoma xenograft model in two different ways. EPC labeling in vivo was tracked using 7.0 T MRI. MRI findings were subsequently confirmed using immunohistochemistry.
Materials and Methods
Twenty fresh peripheral blood samples (20 mL/sample) were collected via cubital venous access from 10 healthy volunteers (two samples/volunteer). Samples were kept in tubes prepared with heparin sodium to prevent coagulation. The samples were used within 2 hours.
The study protocol was approved by the Institutional Ethics Review Committee of the West China Hospital (Sichuan University). All subjects provided written informed consent.
Isolation and Culture of EPCs
Blood samples were diluted in phosphate-buffered saline (PBS; 1:1) and layered carefully onto 2:1 density-gradient medium (Histopaque-10771, Sigma-Aldrich, St. Louis, MO) in 15 mL conical centrifuge tubes (Corning, NY). The samples were centrifuged at 400g for 30 minutes at 20°C in a Legend Mach 1.6R Centrifuge (Thermo Fisher, Waltham, MA). After centrifugation, the opaque mononuclear cell layer was collected, washed twice with PBS (1:2), and resuspended. The cells were seeded in 75 cm2 plastic cell culture flasks (Corning, USA), which had been coated with human fibronectin (25 μg/mL, Prospec, Israel) 3 hours previously to induce cell attachment. The flasks were incubated with endothelial growth medium (EGM)-2 MV Single Quots (Lonza, Allendale, NJ) at 37°C in a CO2 incubator (Thermo Fisher), and the concentration of fetal bovine serum (FBS) was adjusted to 10%. The medium was changed every 3 days; unattached cells were removed on day 4. EPCs were digested with 0.25% trypsin-0.1% ethylenediaminetetraacetic acid (EDTA) and passaged while growing into a cobblestone-like monolayer that reached confluence. Morphologic changes were monitored using an inverted microscope (Eclipse TE2000-U, Nikon, Japan).
Functional and Flow Cytometric Characterization of EPCs
As a functional property of endothelial cells, the ability of EPCs to uptake acetylated low-density lipoprotein (ac-LDL) and to bind to Ulex europaeus agglutinin-I (UEA-I) was determined as follows. In one well of a six-well culture plate, 1 × 10 4 EPCs were incubated at 37°C with 2 mL of EGM-2 supplemented with 10 μg/mL of 1,1′-dioctadecyl-3,3,3′,3′- tetramethyl-indocarbocyanine perchlorate (DiI)-ac-LDL (Molecular Probes, the Netherlands). The cells were protected from light for 4 hours and then washed twice with PBS and fixed with 4% paraformaldehyde for 20 minutes. The fixed EPCs were incubated at 37°C for 1 hour with 10 μg/mL fluorescein isothiocyanate (FITC)-UEA-I (Sigma-Aldrich). After being washed twice with PBS, the cellular nuclei were counterstained with 2% 4′-6-diamidino- 2-phenylindole (DAPI) (Sigma-Aldrich) for 5 minutes. The cells were washed three times with PBS, coverslipped, and photographed with a fluorescent microscope (Nikon). All experiments were performed in triplicate, and EPCs were derived from three different blood samples.
Phenotype markers of EPCs were analyzed by flow cytometry. EPCs were collected and washed twice with PBS. The cell concentration was adjusted to 2 × 10 6 /mL with PBS. The washed EPCs (100 μL) were incubated with 20 μL of either FITC mouse antihuman CD31 (BD Pharmingen, USA), phycoerythrin (PE) mouse antihuman CD34 (BD Pharmingen), or Alexa Fluor 647 mouse antihuman CD309 (vascular endothelial growth factor receptor 2 [VEGFR-2], BD Pharmingen) at room temperature for 30 minutes. FITC mouse IgG1, PE mouse IgG1, and Alexa Fluor 647 mouse IgG1 (all from BD Pharmingen) were used as isotype controls. They were then washed with PBS, centrifuged, and resuspended in 500 μL PBS. Flow cytometry was performed using a Cytomics FC500 flow cytometer and CXP software (Beckman Coulter, Miami, FL).
Alkyl-PEI2k/SPIO Labeling of EPCs
Alkyl-PEI2k/SPIO was synthesized using a previously described procedure.23,25,26 Briefly, amphiphilic polyethylenimine was obtained by alkylation of branched PEI2k with 1-iodododecane in ethanol. SPIO nanoparticles were synthesized using iron (III) acetylacetonate as precursors in benzyl ether at 300°C for 1 hour. The product was purified through ethanol wash and resuspended in hexane. Alkyl-PEI2k/SPIO was synthesized by transferring SPIO into the aqueous phase with the help of amphiphilic polyethylenimine. According to previous studies23,24 and our preliminary experiments, EPCs were labeled with 7 μg/mL alkyl-PEI2k/SPIO in EGM-2 at 37°C for 12 hours. The labeled cells were washed three times with PBS to remove any excess iron particles before use.
Prussian Blue Staining and Cellular Iron Quantification of Labeled EPCs
The uptake of SPIO was verified by Prussian blue staining. SPIO-labeled and unlabeled EPCs were fixed with 4% paraformaldehyde for 20 minutes and incubated for 30 minutes with fresh Perls reagent (A: 10% potassium ferrocyanide; B: 10% hydrochloric acid). The cells were washed gently, counterstained with 1% Nuclear Fast Red for 3 minutes, and photographed using a microscope (Nikon Eclipse 80i). Labeling efficiency was accessed in five random fields at × 200 magnification.
The cellular iron content was determined by atomic absorption spectroscopy, and 2 ×10 6 SPIO-labeled EPCs were collected and resuspended with PBS; 150 μL of 37% HCl was added to digest the labeled EPCs. The digested solution was diluted to 7 mL with distilled water. A standard curve was plotted using a standard GSB iron solution (NCS, China). The content of iron in the solution was measured by an atomic absorption spectrometer (Analyst 800, PerkinElmer, Waltham, MA). Three copies of labeled EPCs were performed, and the experimental data represent the mean ± standard deviations of three independent experiments.
Cell Proliferation Assay
Cell proliferation was determined by CCK-8 assay (Dojindo, Japan). SPIO-labeled or unlabeled EPC suspension (100 μL of 1 × 10 3 cells/well) was inoculated onto a 96-well plate. CCK-8 solution (10 μL/well) was added to 10 wells of labeled or unlabeled EPCs and incubated for 4 hours on days 2 to 7. The absorbance of each well was measured with a microplate reader (Model 680, Bio-Rad, Hercules, CA) at 490 nm, and a growth curve was plotted.
Migration and Tubulogenesis Assay
Two sets of experiments (labeled EPCs group and unlabeled EPCs group) were performed to evaluate the migration of EPCs after labeling. Six hanging cell transwell inserts (8 μm pore, BD Pharmingen) were placed in six wells of a 24-well plate. SPIO-labeled or unlabeled EPC suspension (three samples of each group, 500 μL/sample, 2 × 10 4 cells/mL) was plated in the upper well of the transwell inserts, and the collected supernatant of medium from the incubated A549 lung cancer cells was plated on the bottom well of the inserts. After 24 hours' incubation at 37°C, the EPCs on the upper side of the membrane were removed and those that had migrated through the membrane and become attached on the lower side were washed with PBS, fixed with 4% paraformaldehyde for 20 minutes, and stained with hematoxylin-eosin (HE). The number of migrated EPCs was observed and counted in five random fields per well at × 200; magnification using an inverted microscope (Nikon Eclipse TE2000-U).
In vitro tube-forming capacity was evaluated in EPCs labeled with SPIO-PEI2k. Six wells of a 96-well plate were plated with Matrigel basement membrane matrix (BD Pharmingen) and incubated at 37°C in a CO2 incubator for 3 hours. SPIO-labeled or unlabeled EPC suspensions (labeled EPCs group and unlabeled EPCs group were set, three samples of each group, 100 μL/sample, 2 × 10 6 cells/mL) were then plated on the top of each Matrigel well in cell suspension containing EGM-2 supplemented with 10% FBS. The plates were incubated at 37°C for 6 hours. Prussian blue staining was performed as described above. The number of tubule structures was observed and counted in five random fields per well at X200 magnification under an inverted microscope (Nikon Eclipse TE2000-U).
Animal Xenograft Model
Animal experiments were performed according to a protocol approved by the Animal Ethics Review Committee of Sichuan University (Animal Care and Use Committee at Sichuan University). Eighteen male mice with severe combined immunodeficiency (SCID) (Beijing HFK Bioscience, China), 6 to 8 weeks of age, were divided randomly into three groups. All mice were raised under specific pathogen-free conditions at the Animal Center of West China Hospital, Sichuan University.
Human lung cancer A549 cells (from the Laboratory of Signal Transduction and Molecular Targeting Therapy, West China Hospital, Sichuan University) were cultured at 37°C in 75 cm2 plastic cell culture flasks (Corning) containing RPMI-1640 medium supplemented with 10% FBS. The cells were passaged when they reached 80 to 90% confluence.
Seven days after subcutaneous inoculation of 1 × 10 7 A549 lung cancer cells in 100 μL PBS into the right inguen, 5 × 10 5 labeled EPCs in 100 μL PBS were injected intravenously (group 1, n = 6) or were coinjected subcutaneously with 1 × 10 7 A549 cells (group 2, n = 6). Mice receiving 1 × 10 7 A549 lung cancer cells in 100 μL PBS were inoculated into the right inguen without EPCs acting as controls (group 3, n = 6).
In Vivo MRI
MRI was performed using a 7 T MRI system (BioSpec 70/30, Bruker, Germany) with a 40 mm volume coil. Images were acquired using a T2-weighted turbo rapid acquisition with relaxation enhancement (RARE) sequence (repetition time [TR]/echo time [TE] = 3,000/33 ms, number of excitations [NEX] = 1, matrix = 256 × 256, field of view [FOV] = 3 cm2, slice thickness = 1 mm) and a T2*- weighted gradient echo sequence (TR/TE = 500/5 ms, NEX = 8, matrix = 256 × 256, FOV = 3 cm2, slice thickness = 1 mm). Serial in vivo MRI was performed on days 7, 14, 21, and 28. Furthermore, daily MRI was performed after EPC administration in group 1 and up to the presence of iron-positive hypointensity.
During all in vivo MRI studies, the mice were anesthetized with 1.5 to 2% isoflurane in oxygen using an anesthetic machine (Matrx VIP 3000, Midmark, Dayton, OH), referring to a reported study 27 and a previous study in our laboratory. 24 The mice's respiration and heart rate were controlled at 50 to 80 breaths, 360 to 400 beats per minute, respectively, and their temperature was maintained at 37°C with a water bath.
Prussian Blue Staining and Immunohistochemistry
Mice were euthanized by cervical dislocation after MRI on day 28. Tumors were removed and divided in half. Assays were undertaken for 5 μm sections from frozen sections (frozen in liquid nitrogen) and 5 μm sections from paraffin blocks (fixed with 4% paraformaldehyde embedded in paraffin). The paraffin-embedded tumor sections were stained with HE to display the stroma and vessels in tumor. Prussian blue staining was used to identity iron-positive cells by staining consecutive paraffin block sections with Perls reagent for 30 minutes.
Immunofluorescence staining was performed to analyze the expression of endothelial CD31 cell markers. Consecutive slides from frozen sections were fixed with 4% paraformaldehyde at room temperature for 10 minutes and washed three times with PBS. They were then incubated at 4°C overnight with antihuman CD31 antibody (1/100 dilution, Thermo Scientific, USA) and antimouse CD31 antibody (1/100 dilution, BD Pharmingen). After thorough washing, the cells were incubated in the dark at 37°C for 1 hour with secondary antibody FITC-conjugated goat antimouse IgG (1/500 dilution; ZSGB-BIO, China) and tetramethylrhodamine isothiocyanate (TRITC)-conjugated goat antirat IgG (1/200 dilution; ZSGB-BIO). Sections were counterstained with 2% DAPI for 5 minutes, washed five times with PBS, and coverslipped. The cells were viewed using a light microscope (Nikon Eclipse 80i) or a fluorescent microscope (DM4000B, Leica, Germany).
Statistical Analysis
Data were expressed as means ± standard deviations. Differences between group means were assessed by Student t-test using SPSS version 13.0 software (SPSS Inc., Chicago, IL). Values of p < .05 were considered statistically significant.
Results
EPC Characteristics
Mononuclear cells began to attach onto the fibronectin-coated plates 48 hours after culturing. Small colony clusters were formed with round cells centrally and spindle-shaped cells peripherally from day 7 onwards. The cells enlarged and became gradually elongated. After a further 10 days, some were arranged in tubes or cords. The characteristic cobblestone morphology of endothelial cells developed as EPCs reached confluence at about 20 days (Figure 1, A and B).
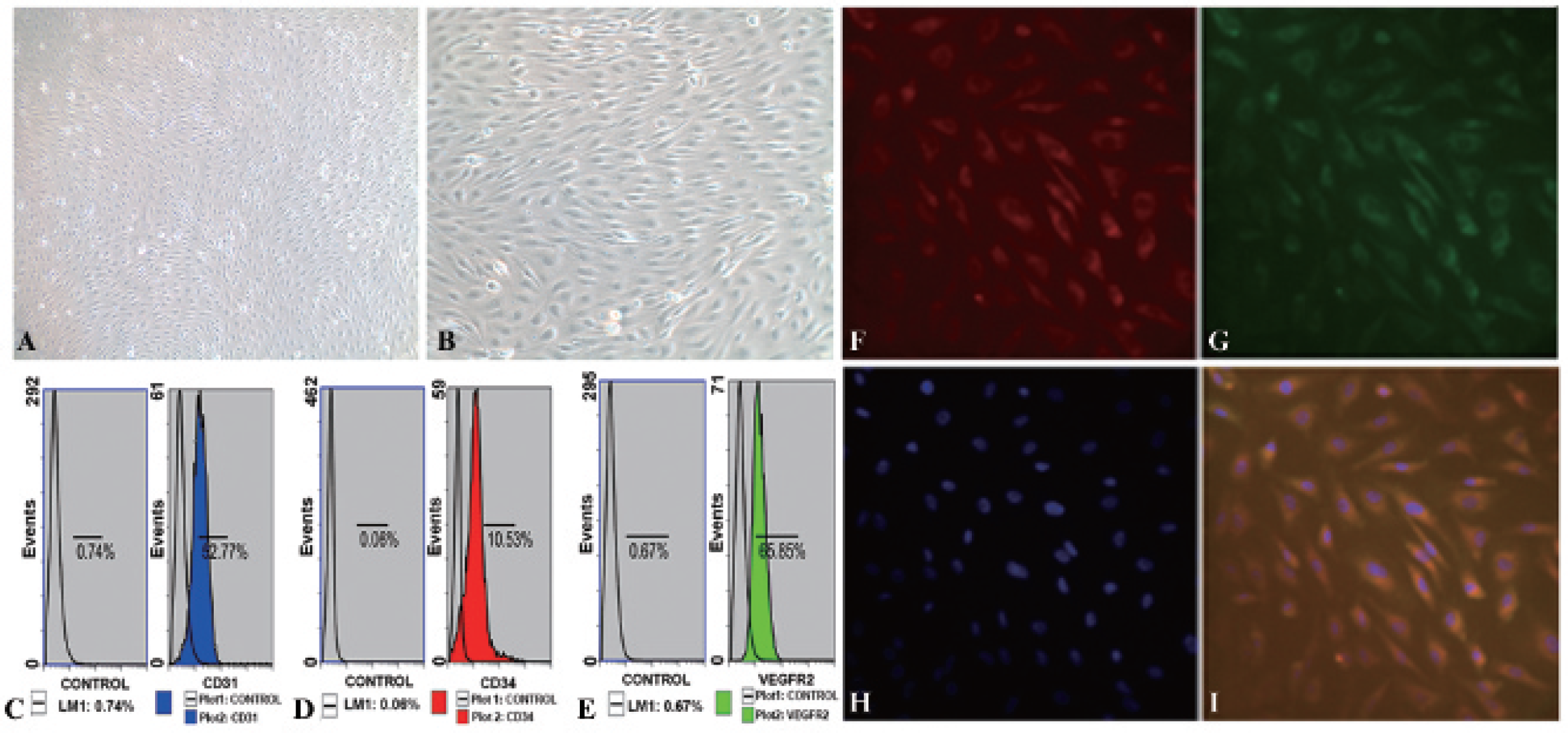
Morphologic, functional, and flow cytometric characterization of endothelial progenitor cells (EPCs) from human peripheral blood. Mononuclear cells were collected from human peripheral blood by centrifugation and incubated with 10% FBS/EGM-2 MV. The characteristic “cobblestone” morphology of endothelial cells was seen on reaching confluence at about 20 days (original magnification: A, X100; B, X200). C to E, Flow cytometric analyses of the expression of endothelial markers CD31, CD34, and VEGFR-2. Cell immunofluorescence demonstrated the uptake of DiI-ac-LDL by EPCs (red fluorescence) and binding to FITC-UEA-I (green fluorescence). The double-stained cells showed yellow fluorescence when images were merged. The nuclei were stained blue by DAPI (F: Dil-ac-LDL; G: FITC-UEA-I; H: DAPI; I: merged figures F-H; × 400 original magnification).
The expression of EPC biomarkers was examined by flow cytometry. The percentage of cells that were positive for CD31, CD34, and VEGFR-2 was 52.8%, 10.5%, and 65.9%, respectively (Figure 1, C-E). Immunofluorescence experiments demonstrated that human peripheral blood-derived EPCs were able to uptake DiI-ac-LDL (red fluorescence) and bind with FITC-UEA-I (green fluorescence). Yellow fluorescence was seen in merged images (Figure 1, F–I).
Labeling Efficiency and Biocompatibility in EPCs after Labeling with Alkyl-PEI2k/SPIO
Iron labeling was confirmed by Prussian blue staining. Manual counting using a microscope indicated that labeling efficiency for EPCs with alkyl-PEI2k/SPIO at 7 μg Fe/mL was approximately 100%. No blue iron particles were seen in unlabeled EPCs (Figure 2, A and B). Quantitative analysis with atomic absorption spectroscopy showed that the cellular iron content of EPCs was 6.062 ± 0.050 pg/cell.

Endothelial progenitor cells (EPCs) labeled with magnetic alkyl-PEI2k/SPIO at low concentrations showed excellent labeling efficacy and biocompatibility. Prussian blue staining showed that labeling efficiency for EPCs with alkyl-PEI2k/SPIO at 7 μg Fe/mL was approximately 100% (A, X400 original magnification). No blue iron particles were seen in unlabeled EPCs (B, X400 original magnification). Migration assay showed that cells in the labeled EPCs group (D, X200 original magnification; average number of migrated cells is 80.6 ± 8.0) migrated through transwell membranes at the same rate as the unlabeled EPCs group (C, X200 original magnification; average number of migrated cells is 77.6 ± 4.6, p = .39). Tubulogenesis assay showed that cells in the labeled EPCs group (F, X200 original magnification; average number of tubes is 9.4 ± 1.5) formed comparable numbers of tubes with the unlabeled EPCs group (E, X200 original magnification; average number of tubes is 9.0 ± 1.0, p = .615). Histograms G (migration assay) and H (tubulogenesis assay) showed no differences in migration and tubulogenesis capacity between labeled and unlabeled EPCs, respectively. Cell proliferation was determined using CCK-8 assay. Growth curves indicated no statistically significant differences between labeled and unlabeled EPCs (I). Alkyl-PEI2k/SPIO had no effect on EPC proliferation.
Proliferation of labeled EPCs was monitored using a CCK-8 assay. Growth curves showed no statistical differences (p > .05) between labeled and unlabeled EPCs (Figure 2I). Alkyl-PEI2k/SPIO had no effect on EPC proliferation.
Effect on EPC Migration and Tubulogenesis after Labeling with Alkyl-PEI2k/SPIO
EPCs migrated through the transwell membrane following stimulation by the supernatant of A549 lung cancer cells. Cells in the labeled EPCs group migrated at the the same rate as cells in the unlabeled EPCs group (p = .39, Figure 2, C, D, and G).
A tubulogenesis assay was performed with labeled or unlabeled EPCs. After 6 hours' culture on Matrigel, tube-like structures were formed by both labeled and unlabeled EPCs. Quantitative analysis showed that there was no difference in the number of tube-like structures produced in 10% FBS by labeled and unlabeled EPCs (p = .615; Figure 2, E, F, and H).
In Vivo MRI
Lung carcinoma xenograft models were established by subcutaneous inoculation. Two mice (one of group 1 and one of group 2) were excluded from these experiments because of failure to obtain tumors.
In group 1, there was no evidence of iron-induced hypointensity before injection of labeled EPCs. Schistic or linear hypointense regions were observed at the periphery of tumors on T2-weighted and T2*-weighted images starting from days 7 to 8 after intravenous administration of labeled EPCs. During follow-up, these regions had gradually extended to the inner layers of the tumor. In group 2, a focal central region of low signal intensity was initially observed. This gradually dispersed from the center and migrated to the periphery as tumors continued to grow. MRI demonstrated that iron-induced hypointensity was more pronounced on T2*-weighted imaging than on T2-weighted imaging. The hypointense regions remained visible by day 28 in both groups 1 and 2, whereas they were more pronounced in group 2 than in group 1. Iron-induced low signal intensity was not observed in group 3 during MRI (Figure 3).

In an in vivo MRI study, 1 × 10 7 A549 lung cancer cells were inoculated subcutaneously into the right inguen of SCID mice to establish carcinoma xenografts (circle); 5 × 10 5 labeled endothelial progenitor cells (EPCs) were injected intravenously 7 days after injection of A549 cells (group 1) or coinjected with A549 cells subcutaneously into mice (group 2), whereas xenografts without EPCs acted as controls (group 3). Serial in vivo MRI was performed for up to 28 days at a 7 T MRI system. Group 1: schistic or linear hypointense regions were observed at the periphery of tumors starting from 14 days after injection of A549 cells. This gradually extended into the inner layers (arrow). Group 2: a central focal region of low signal intensity was initially observed at the beginning and gradually dispersed from the center and migrated to the periphery as the tumors increased in size (arrow). Group 3: no definite iron-induced low signal intensity was observed in tumors during MRI. It was confirmed by Prussian blue staining (see Figure 4D).
HE staining showed that the stroma thickened significantly, and abundant microvessels were seen throughout the tumor (Figure 4A). Prussian blue staining confirmed that positive cells were present at tumor sites in groups 1 and 2 (Figure 4, B, C, E, and F). No Prussian blue-positive cells were detected in group 3 (Figure 4D).

Hematoxylin-eosin (HE) and Prussian blue staining. Mice were euthanized at day 28 after MRI, and tumors were sectioned according to the hypointensity in MRI. HE staining (A) showed significantly thickened stroma and abundant microvessels throughout the tumors. Prussian blue staining (B-F) confirmed that positive cells (arrows) were at the margin (B and E, group 1) or in inner (C and F, group 2) of tumors corresponding to MRI. However, no iron-positive cells were found in group 3 (D). Original magnification: B to D, × 200; E to F, × 400.
To further determine whether the labeled EPCs were incorporated into tumor neovascular structures, the expression of endothelial differentiation marker CD31 was determined by immunohistochemistry analysis. Two antibodies (antihuman and mouse CD31 antibody) were used to distinguish human-derived vessels from mouse-derived vessels. Fluorescent microscopic imaging showed that human EPCs incorporated into tumor neovasculature and displayed a strip and sinusoid structure. Different from antihuman CD31 green fluorescence, mouse-derived vessels displayed red fluorescence (Figure 5, A-D). However, no human-derived vessels were found in mice receiving lung cancer cells without EPCs (Figure 5, E–G).

Immunofluorescence staining showed endothelial markers with anti human and mouse CD31 antibodies. After administration of human endothelial progenitor cells (EPCs) to mice xenografts, tumors were sectioned and immunofluorescence staining was performed with antihuman and mouse CD31 antibodies. Fluorescent microscopic imaging showed that human EPCs incorporated into tumor neovasculature, human-derived vessels (arrows) showed a strip and sinusoid structure (green fluorescence), and mouse-derived vessels (arrowheads) displayed red fluorescence. However, no human-derived vessels were found in mice receiving lung cancer cells without EPCs (group 3). The merged images were provided (D and G), and the nuclei were stained blue by DAPI. Original magnification: A to D, × 400; E to G, × 200.
Discussion
EPCs are derived mainly from peripheral blood, bone marrow, and umbilical cord blood. 4 Fewer EPCs are initially obtained from peripheral blood, but they are of higher purity that those derived from marrow. This is because mononuclear cells isolated from bone marrow mix with other cell types, resulting in multilineage differentiation potential.28,29 The use of umbilical cord blood-derived EPCs for therapeutic angiogenesis is limited because of immunologic graft-versus-host diseases induced by the host's immune defense mechanisms. 30 For these reasons, human peripheral blood-derived EPCs were chosen in the present study. The lung adenocarcinoma cell line (A549) used in our experiments is a tumor with a rich blood supply that was suitable for investigating neovascularization. 31
Mononuclear cells from fresh human peripheral blood were obtained by density gradient centrifugation and cultured with EGM-2 MV Single Quots. Our previous experience indicated that mononuclear cells differentiate to EPCs well when the concentration of FBS is adjusted to 10%. The identification of EPCs mainly depends on morphologic and functional characterization and phenotypic analysis. 32 In our study, peripheral blood-derived EPCs showed the characteristic cobblestone morphology and were able to uptake DiI-ac-LDL and bind with FITC-UEA-I. Flow cytometry demonstrated the expression of EPC biomarkers such as CD31, CD34, and VEGFR-2. The cell surface phenotype CD34, CD133, and VEGFR-2 have gained widespread use as a means to measure putative circulating EPCs.32–34 Lin and colleagues and Duda and colleagues further revealed that increasing expression of CD31 and decreasing expression of CD133 and CD34 indicated the differentiation of early EPCs into late EPCs.35,36 Thus, we specifically analyzed CD34 expression in the present study, low expression of CD34 indicated that EPCs were differentiating toward late EPCs.
Noninvasive imaging techniques provide a means of monitoring cell delivery and biological processes in vivo. Numerous nanomaterials have been produced for biomedical applications, some of which have been used in clinical practice.37–42 Current Food and Drug Administration-approved labeling agents include Feridex (Advanced Magnetics, USA) and Resovist (Bayer Schering Pharma, Germany). 43 In previous reports, the concentration of Fe using these agents to label cells in vitro was in the range of 20 to 50 μg/mL.
In the present study, the magnetic material alkyl-PEI2k/SPIO, which we used to label EPCs, showed excellent biocompatibility and magnetic resonance sensitivity. The nanoparticles were 54.7 ± 9.5 nm in diameter, and the T2 relaxivity of alkyl-PEI2k/SPIO was 345 Fe mM−1 S−1. 23 A controlled clustering structure has been designed, which allowed us to wrap multiple SPIO species. When multiple SPIO species gather together, T2 relaxivity time is greatly improved compared to single SPIO.22,23 Labeling dosages of iron in our study were much lower compared to previous reports, resulting in excellent labeling efficacy and MRI effects. When EPCs were labeled using 7 μg Fe/mL, the labeling efficiency was approximately 100% confirmed by Prussian blue staining. The cellular iron content was 6.062 ± 0.050 pg/cell, which is sufficient for cellular MRI imaging (> 2 pg/cell, as previously reported). 44
Safety and biocompatibility are both critical concerns for applications of nanomaterials. The transfection agent polyethylenimine 25 kDa (PEI25k) is a highly effective gene with cytotoxic properties causing cell death, apoptosis, or inhibition of cell differentiation. Low-molecular-weight polyethylenimines such as PEI2k showed comparable transfection efficiency to PEI25k, but with much lower cytotoxicity. 45 In our study, CCK-8 assay results indicated that alkyl-PEI2k/SPIO did not alter EPC proliferation. Labeled EPCs also had the ability to uptake ac-LDL and bind with UEA-I, indicating that alkyl-PEI2k/SPIO did not affect EPC differentiation potential. The labeled EPCs had comparable in vitro migration and tubulogenesis capacity to unlabeled EPCs and were capable of incorporation into tumor neovasculature in vivo. In our previous studies, alkyl-PEI2k/SPIO was also used to label MSCs in vitro and metastatic tumor cells (human colorectal cancer LoVo cells) in lymphatics in vivo, without affecting their viability.23,24
The mechanism of EPC migration and incorporation into tumor neovasculature is complex, and the time of EPC migration to the tumor site is determined by multiple factors, such as tumor location, tumor size, and the circulatory level of cytokines. In addition, different detecting or imaging equipment may lead to different results due to its sensitivity and resolution.4,6 Fluorescence studies have previously shown that carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled EPCs migrate to intracranial gliomas 7 days after intravenous introduction of labeled EPCs. 46 Arbab and colleagues tracked FePro-labeled EPC incorporation into the subcutaneously implanted tumor by two sets of experiments. 20 Magnetically labeled EPCs could be detected at the tumor periphery as early as 3 days after EPC administration in preformed tumors (3 days after implantation) and within 5 to 7 days in concurrently implanted tumors by ex vivo MRI, whereas hypointense regions were observed on in vivo and ex vivo MRI when tumors grew to 1 cm at 12 to 14 days in both groups. Their results suggest an early incorporation of EPCs into tumor neovasculature in mice when the tumor was approximately 0.2 cm in size. In our study, we observed the migration of labeled EPCs injected intravenously 7 days after implanted tumors (group 1) or coimplanted subcutaneously with tumor cells (group 2). In group 1, iron-positive hypointensity was observed at the periphery of tumors 7 or 8 days after intravenous administration of EPCs to mice with xeno-grafted lung carcinoma. The time presenting magnetically labeled EPCs on in vivo MRI was similar to that in the studies reported above. Moreover, our study observed that magnetically labeled EPCs, injected either in the circulation (group 1) or in the center of the tumor (group 2), migrate to the periphery of tumors. These findings, confirmed by Prussian blue and immunofluorescent staining, provided evidence of the migratory capacity of EPCs. Nevertheless, as circulating EPCs travel with blood in vivo and are uncontrollable, it is difficult to monitor them in real time at an MRI scanner. We restricted the MRI range to tumor regions in the present study, and the distribution and delivery of labeled EPCs in circulation during the initial 7 days were unclear. In another study, by tracking the FePro-labeled AC133+ cells coimplanted in the flank of nude mice with tumor cells at a MRI imaging scanner, Arbab and colleagues further investigated the expression of angiogenic factors produced by tumor or surrounding tissue responsible for the migration and homing of EPCs to sites of active angiogenesis in tumor. 47 They found that hypoxia-inducible factor 1α (HIF-1β)-dependent stromal cell-derived factor 1 (SDF-1) might be the factor for AC133+ cells homing to these angiogenic sites, whereas the role of VEGF is unclear.
One concern is whether the Prussian blue cells are solely labeled EPCs or other cells, such as macrophages, which can phagocytose dead labeled EPCs or irons released from labeled EPCs. To our knowledge, host macrophages could phagocytose dead labeled EPCs or irons released from labeled EPCs, although they would be cleared by the host in the liver or spleen quickly and dissolved into elemental or chelated iron. Thus, the Prussian blue-positive cells are solely labeled EPCs that present hypointense regions on MRIs. This was well demonstrated in Arbab and colleagues' two studies.20,47 In the present study, after in vivo MRI on 28 days, tumors were removed and divided in half for Prussian blue staining and immunofluorescence staining corresponding to hypointense regions on MRIs. As a consequence, the hypointensity, Prussian blue-positive cells, and antihuman CD31-positive cells (green fluorescence) matched each other and presented at identical sites of tumors.
One drawback of using in vivo MRI to track magnetically labeled cells is the loss of signal at times corresponding to either biodegradation or dilution of iron following cell division. This imaging modality is therefore not suitable for long-term monitoring of transplanted cells. Additionally, the present study did not determine the efflux and associated organ distribution of exogenous iron. These topics require further investigation.
In summary, the present study demonstrates that self-assembled alkyl-PEI2k/SPIO can be used to label human peripheral blood EPCs with high efficacy. No significant effects on EPC viability and differentiation were found after labeling. The labeled EPCs could be tracked using a 7.0 T magnetic resonance scanner until they had become incorporated into the tumor neovasculature of a lung carcinoma xenograft model in mice. A reliable cell labeling and tracking strategy will enable EPCs to be monitored as a drug vector for antitumor angiogenesis therapy.
Footnotes
Acknowledgments
Financial disclosure of authors: The study was supported financially by the National Basic Research Program of China (No. 2011CB935800), the National Natural Science Foundation of China (81130027, 81071155, 81271572), and the Natural Science Foundation of Shanghai (10JC1418100, 10411952800).
Financial disclosure of reviewers: None reported.