
Abstract
18F positron emission tomography (PET) has a number of attributes that make it clinically attractive, including nearly 100% positron efficiency, very high specific radioactivity, and a short half-life of ≈ 110 minutes. However, the short half-life of 18F and the poor nucleophilicity of fluoride introduce challenges for the incorporation of 18F into complex molecules. Recently, the tetrazine-trans-cyclooctene ligation was introduced as a novel 18F labeling method that proceeds with fast reaction rates without catalysis. Herein we report an efficient method for 18F labeling of free cysteines of peptides and proteins based on sequential ligation with a bifunctional tetrazinyl-maleimide and an 18F-labeled trans-cyclooctene. The newly developed method was tested for site-specific labeling of both c(RGDyC) peptide and vascular endothelial growth factor (VEGF)-SH protein. Starting with 4 mCi of 18F-trans-cyclooctene and only 10 μg of tetrazine-RGD (80–100 μM) or 15 μg of tetrazine-VEGF (6.0 μM), 18F-labeled RGD peptide and VEGF protein could be obtained within 5 minutes in 95% yield and 75% yield, respectively. The obtained tracers were then evaluated in mice. In conclusion, a highly efficient method has been developed for site-specific 18F labeling of cysteine-containing peptides and proteins. The special characteristics of the tetrazine–trans-cyclooctene ligation provide unprecedented opportunities to synthesize 18F-labeled probes with high specific activity for PET applications.
HIGH-MOLECULAR-WEIGHT biologic macromolecules, such as single-stranded oligonucleotides, 1 peptides, 2 and proteins, 3 are being considered with increasing frequency for use as radiopharmaceuticals, and applications of radiolabeled macromolecules are rapidly gaining importance in nuclear medicine. With a half-life of 109.8 minutes, low β+-energy (0.64 MeV), and ease of production, 18F represents the ideal radionuclide for routine positron emission tomography (PET). Its low positron energy results in short positron linear range in tissue, which could lead to higher resolution in PET imaging. Furthermore, the half-life of 18F is long enough to allow syntheses, transportation, and imaging procedures to be extended over hours, while the patient is subjected to a limited amount of radiation exposure. However, the synthesis of 18F-labeled proteins with high specific activity is challenging. 18F-containing prosthetic groups are often required. Moreover, due to the slow rates of conventional conjugation chemistry, a large excess of protein is often required to obtain high radiochemical yield. Fast, efficient labeling methods in 18F radiochemistry are therefore necessary for developing imaging agents in nuclear medicine and life science.
Previously, we and others developed a novel bioconjugation that involves reaction between tetrazines and trans-cyclooctenes (TCOs).4,5 The fast rates of these reactions enable high reactivity at low micromolar concentrations within minutes and without an excess of either reactant.4,6,7 With this foundation, we developed an extremely efficient method for synthesizing 18F-labeled probes with high specific activity based on tetrazines and an 18F-labeled trans-cyclooctene (18F-TCO).8,9 We also used the tetrazine ligation for the first time in a microPET imaging study using an αvβ3 integrin targeted RGD probe that was constructed through this efficient cycloaddition reaction. 10 Recently, this kind of tetrazine/TCO reaction was successfully applied for a pretargeted imaging strategy.6,11 These studies demonstrated how the fast rates and efficient reactivity of the tetrazine ligation distinguish it from most existing 18F labeling methods.
In our previous study, the tetrazine functionality was introduced through lysine acylation. A concern for unselective protein modification of lysine residues is possible interference with biologic activity as the modification of one or more lysines located at or near the active site can reduce the binding affinity. As cysteines are much less abundant than lysine, aspartic acid and glutamic acid residues, thiol-reactive agents can be used to modify peptides and proteins with higher selectivity than amine and carboxylate-reactive reagents. To further explore the scope of the efficient tetrazine ligation, herein we report a new method for 18F labeling of free cysteine–containing bioligands. The newly developed method was tested for site-specific labeling of c(RGDyC) and vascular endothelial growth factor (VEGF)-SH protein.
Materials and Methods
General
All chemicals obtained commercially were of analytic grade and used without further purification. No-carrier-added 18F was produced via the 18O(p,n)18F reaction by the bombardment of an isotopically enriched [18O]water target (95% enrichment; Isonics, Golden, CO) with 11 MeV protons using a Siemens RDS-112 negative ion cyclotron (Siemens Medical Solutions USA, Inc, Knoxville, TN). Reversed-phase extraction C18 Sep-Pak cartridges were obtained from Waters (Milford, MA) and were pretreated with ethanol and water immediately before use. The syringe filter and polyethersulfone membranes (pore size 0.22 μm; diameter 13 mm) were obtained from Nalge Nunc International (Rochester, NY). c(RGDyC) was obtained from Peptides International (Louisville, KY). The protein VEGF-SH was provided by Dr. Xiaoyuan Chen at the National Institutes of Health. The semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a Vydac protein and peptide column (218TP510; 5 μm, 250 × 4.6 mm) was performed on a Dionex 680 chromatography system (Thermo Fisher, Sunnyvale, CA) with a UVD 170U absorbance detector and model 105S single-channel radiation detector (Carroll & Ramsey Associates, Berkeley, CA). The recorded data were processed using Chromeleon version 7.1 software (Thermo Fisher). With a flow rate of 1.0 mL/min, the mobile phase was changed from 95% solvent A (0.1% trifluoroacetic acid [TFA] in water) and 5% B (0.1% TFA in acetonitrile [MeCN]) (0–2 minutes) to 5% solvent A and 95% solvent B at 22 minutes. Ultraviolet (UV) absorbance was monitored at 218 nm, and identification of the peptides was confirmed based on the UV spectrum using a photodiode array detector. MicroPET scans were performed on a microPET R4 rodent model scanner (Siemens Medical Solutions USA, Inc). The scanner has a computer-controlled bed and 10.8 cm transaxial and 8 cm axial fields of view (FOV). It has no septa and operates exclusively in the three-dimensional list mode. Animals were placed near the center of the FOV of the scanner.
Chemistry
Synthesis of N1-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-N5-(6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)glutaramide [Tetrazinyl Maleimide]
A flame-dried flask was charged with 2,5-dioxopyrrolidine-1-yl 5-oxo-5-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)amino)pentanoate (7) 0.030 g (0.065 mmol) and N-(2-aminoethyl)maleimide trifluoroacetate salt 0.020 g (0.078 mmol). The flask was evacuated and then refilled with nitrogen. Anhydrous N,N-dimethylformamide (1 mL) was added, and the mixture was stirred. Diisopropylethylamine (0.032 mL, 0.195 mmol) was added dropwise to the mixture. The reaction was stirred for 24 hours at room temperature. The reaction was condensed in vacuo and then loaded onto a silica gel column and run using a gradient of 0 to 10% MeOH in CH2Cl2 as the eluent to give 0.015 g (0.031 mmol, 48%) of the title compound as a dark red solid. 1H NMR (400 MHz, dimethyl sulfoxide [DMSO]-d6) δ 10.56 (s, 1H), 9.06 (s, 1H), 8.94 (d, J = 4.3 Hz, 1H), 8.65 to 8.58 (m, 2H), 8.45 to 8.41 (m, 1H), 8.16 (t, J = 7.8 Hz, 1H), 7.99 to 7.93 (m, 1H), 7.76 to 7.70 (m, 1H), 7.01 (s, 2H) 3.49 to 3.43 (m, 2H), 3.24 to 3.17 (m, 2H), 2.42 (t, J = 7.22 Hz, 2H), 2.09 (t, J = 7.5 Hz, 2H), 1.86 to 1.78 (m, 2H); 13C NMR (100 MHz, DMSO) 5 172.7 (C), 172.5 (C), 171.6 (C), 163.5 (C), 163.2 (C), 151.1 (CH), 150.7 (C), 144.2 (C), 141.7 (C), 139.0 (C) 138.3 (CH), 135.0 (CH), 127.1 (CH), 126.6 (CH), 125.4 (CH), 124.7 (CH), 37.7 (CH2), 37.3 (CH2), 36.1 (CH2), 34.9 (CH2), 21.1 (CH2); HRMS-ESI m/z. [M + H] calculated for C23H22N9O4, 488.1795; found 488.1796.
Synthesis of 19F-TTM-RGD
The conjugation was modeled after a reported procedure. 12 Thus, the tetrazinyl-maleimide (TM; 200 μg, 0.41 μmol) in 100 μL DMSO and c(RGDyC) (200 μg, 0.33 μmol) in 500 μL phosphate buffer (50 mM, pH 6.5–7.0) were mixed together at room temperature. After the mixture was stirred for 5 hours, the tetrazinyl-maleimide-RGD conjugate (TM-RGD) was purified by semipreparative HPLC. The collected fractions were combined and lyophilized to afford a pink powder. TM-RGD was obtained in 85% yield.
19F-TCO was synthesized according to our previous report. 8 TM-RGD (100 μg, 92 nmol, in 100 μL DMSO) and 19F-TCO (200 μg, 0.33 μmol, in 100 μL DMSO) were mixed together at room temperature. After the mixture was stirred for 5 minutes, the conjugate was purified by semipreparative HPLC. The collected fractions were combined and lyophilized to afford the final product as a white powder. 19F-TTM-RGD was obtained in 92% yield with a 13.2-minute retention time on analytical HPLC. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (m/z): [M + H]+ calculatedd for C57H73FN15O13S, 1226.5; found 1226.4.
Synthesis of 18F-TTM-RGD via Cycloaddition of 18F-TCO and TM-RGD
18F-TCO (148 MBq, 4 mCi, in 50 μL ethanol) was added to the TM-RGD solution (10 μg, in 50 μL DMSO) followed by 5-minute incubation at 40°C. The conjugate of 18F-TCO and TM-RGD, referred to as 18F-TTM-RGD, was purified by semipreparative HPLC. The collected fractions were combined, and the solvent was removed by rotary evaporation under reduced pressure. 18F-TTM-RGD was reconstituted in 1 μL phosphate-buffered saline (PBS) and passed through a 0. 22 μm syringe filter for in vivo animal experiments.
Synthesis of 18F-TTM-VEGF
The TM (200 μg, 0.41 μmol, in 100 μL DMSO) and VEGF-SH (100 μg, 5.5 nmol, in 500 μL PBS) were mixed together at room temperature for 5 hours. The conjugate was purified by size-exclusion PD-10 column and concentrated by a Centricon filter (Millipore, Bedford, MA). The final concentration of TM-VEGF conjugate was determined based on UV absorbance at 280 nm using unconjugated VEGF of known concentrations as standards. The final concentration was then adjusted to 50 μg/mL. For 18F labeling, 18F-TCO (148 MBq, 4 mCi, in 50 μL ethanol) was added to the TM-VEGF (15 μg) in water and incubated for 5 minutes at 40°C. The conjugate (shortened as 18F-TTM-VEGF) was purified by PD-10 column using 1× PBS as the eluent. The 3.0 to 4.0 mL fraction containing VEGF proteins was collected for in vivo animal experiments.
Cell Culture
Human glioblastoma cell line U87MG was obtained from the American Type Culture Collection (Manassas, VA) and was cultured in Dulbecco's Modified Eagle's Medium containing high glucose (Gibco, Carlsbad, CA), which was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were expanded in tissue culture dishes and kept in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every other day. A confluent monolayer was detached with 0.05% trypsin–ethylenediaminetetraacetic acid (EDTA) and 0.01 M PBS (pH 7.4) and dissociated into a single-cell suspension for further cell culture.
MicroPET Imaging
Animal procedures were performed according to a protocol approved by the University of Southern California Institutional Animal Care and Use Committee. For static microPET scans, the mice bearing U87MG xenografts were injected with 3.7 MBq (100 μCi) of 18F-TTM-RGD via the tail vein (n = 3 for each group). At 0.5, 1, and 2 hours postinjection, the mice were anesthetized with isoflurane (5% for induction and 2% for maintenance in 100% O2) using a knockdown box. With the help of a laser beam attached to the scanner, the mice were placed in the prone position and near the center of the FOV of the scanner. The 3-minute static scans were then obtained. Images were reconstructed using a two-dimensional ordered-subsets expectation maximization (OSEM) algorithm. Regions of interest (ROI; five pixels for coronal and transaxial slices) were drawn over the tumor on decay-corrected whole-body coronal images. The maximum counts per pixel per minute were obtained from the ROI and converted to counts per milliliter per minute using a calibration constant. With the assumption of a tissue density of 1 g/mL, the ROI were converted to counts per gram per minute. Image ROI-derived %ID/g values were determined by dividing counts per gram per minute with injected dose. Similarly, 3.7 MBq (100 μCi) of 18F-TTM-VEGF was injected to healthy nude mice to study its distribution with microPET. Static scans were performed at 0.5 and 3 hours postinjection.
Results
Chemistry and Radiochemistry
The synthesis of the bifunctional TM is shown in Figure 1, and the syntheses of RGD conjugates are shown in Figure 2A. The conjugation of the bifunctional TM with c(RGDyC) in phosphate buffer with pH 6.5 to 7.0 afforded the TM-RGD conjugate in 85% yield. The cycloaddition reaction of TM-RGD and nonradioactive 19F-TCO provided the standard for the F-labeled product. The pink color of TM-RGD disappeared immediately on the addition of 19F-TCO, indicating that the tetrazine had completely reacted on mixing. The identity of the 19F-TTM-RGD was confirmed by MALDI-TOF-MS. 18F-TCO was produced using the protocol developed in our laboratories. 8 The radiolabeling yield for 18F-TTM-RGD was counted nearly quantitatively from 18F-TCO (Figure 3A). The radiochemical purity of 18F-TTM-RGD was more than 95% as determined by radioHPLC. The specific activity of 18F-TTM-RGD was estimated to be approximately 3 to 6 Ci/μmol. Following a similar protocol, 18F-TTM-VEGF was obtained in high yield (75% after purification) from VEGF-SH.

Synthesis of N1-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-N5-(6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl) glutaramide.

The different synthesis routes for 18/19F-TTM-RGD. A, Synthesis of TM-RGD followed by 18F labeling with 18F-TCO. B, Synthesis of 18F-TTM followed by c(RGDyC) conjugation.
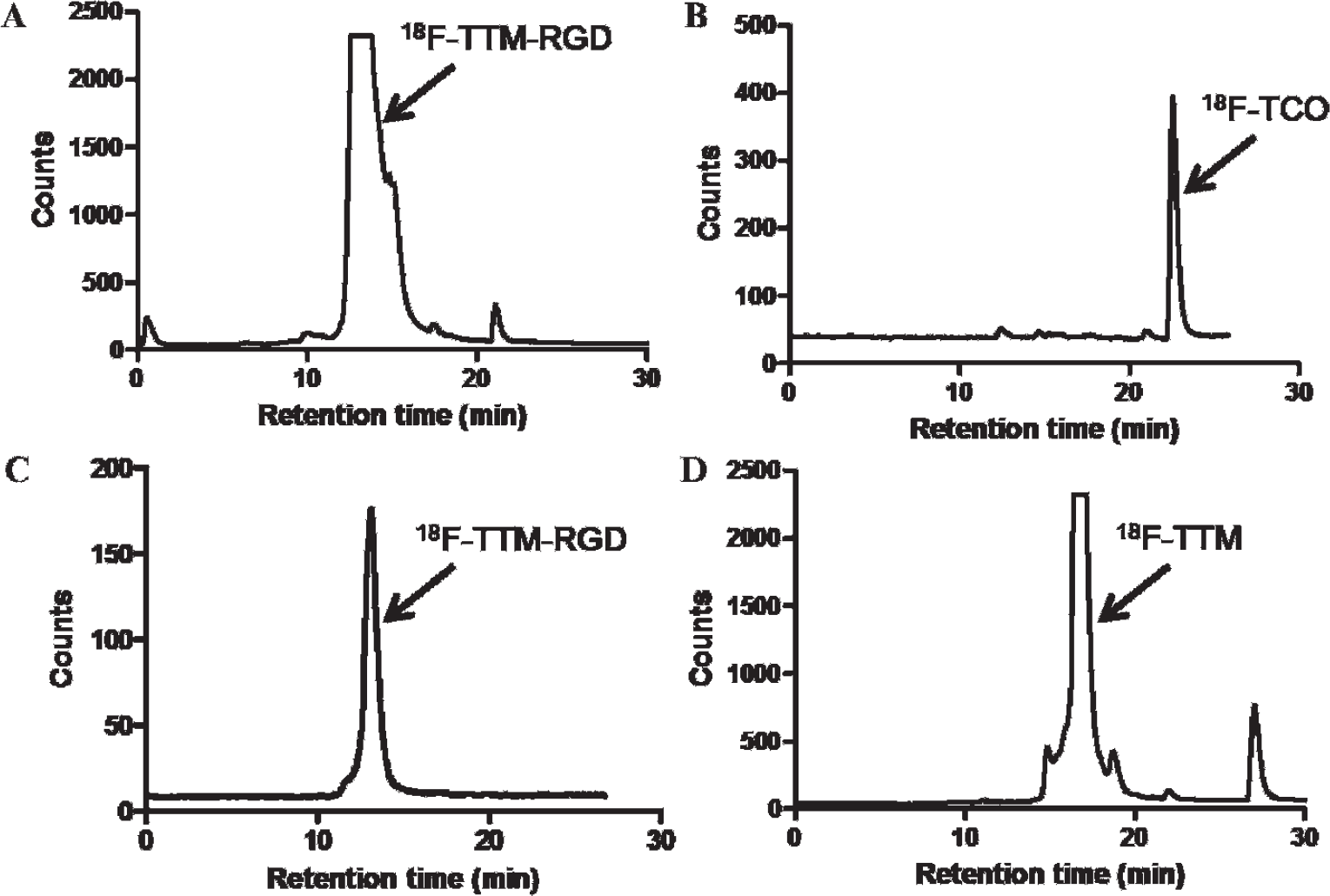
The radioHPLC profiles of 18F-TTM-RGD purification and quality control (QC). A, Purification of the crude mixture of 18F-TCO and TM-RGD. B, The QC of 18F-TCO. C, The QC of 18F-TTM-RGD. D, The purification radioHPLC profile for the crude mixture of 18F-TCO and tetrazinemaleimide.
To compare the efficiency of the tetrazine ligation with direct conjugate addition of a sulfhydryl-containing peptide to a maleimide, we performed the reaction between 18F-TCO and the TM directly, which produced the cycloadduct 18F-TTM (see Figure 1B). The conjugate 18F-TTM-RGD could also be efficiently obtained through the reaction of 18F-TTM and c(RGDyC); however, a relatively long reaction time (20 minutes) and a higher concentration of c(RGDyC) (1.8 mM) were needed. By contrast, the approach outlined in Figure 1A requires fewer manipulations of 18F-labeled intermediates and is complete within 5 minutes at room temperature at a concentration that is 18 times lower (0.1 mM).
MicroPET Imaging
Representative coronal microPET images of U87MG tumor–bearing mice (n = 3/group) at different times after intravenous injection of approximately 3.7 MBq (100 μCi) of 18F-TTM-RGD are shown in Figure 4A. The tumors were clearly visible, with good contrast at all time points. The mice showed high abdominal activity accumulation. Prominent uptake of 18F-TTM-RGD was observed in the kidneys and urinary bladder at early time points, indicating that this radiotracer is excreted through the renal system as well. Quantification of tumor and major organ activity accumulation in the microPET scans was achieved by measuring the ROI that encompass the entire organ on the coronal images. The tumor and major organ uptake of 18F-TTM-RGD is depicted in Figure 5A. The tumor uptake of 18F-TTM-RGD in U87MG tumor was 1.98 ± 0.33, 1.80 ± 0.15, and 1.27 ± 0.33 %ID/g at 0.5, 1, and 2 hours postinjection, respectively. As can be seen from Figure 5A, the uptake of the 18F-TTM-RGD decreased rapidly with time in the muscle, which afforded a better contrast at late time points (2 hours postinjection).

A, Decay-corrected whole-body coronal microPET images of athymic female nude mice bearing U87MG tumor from a static scan at 0.5, 1, and 2 hours after injection of 18F-TTM-RGD. Tumors are indicated by arrows. B, Decay-corrected whole-body coronal microPET images of normal athymic female nude mice from a static scan at 0.5 and 3 hours after injection of 18F-TTM-VEGF. C, Two-dimensional projection of the microPET images of athymic female nude mice after injection of 18F-TTM-VEGF.

MicroPET quantification of tumor and major organs after injection of (A) 18F-TTM-RGD and (B) 18F-TTM-VEGF, respectively.
The biodistribution of 18F-TTM-VEGF was evaluated in normal Sprague-Dawley nude mice. Representative coronal microPET images and the two-dimensional projections at 0.5 and 3 hours postinjection of about 3.7 MBq (100 μCi) 18F-TTM-VEGF are shown in Figure 4, B and C. The activity was mainly accumulated in kidneys, which correlated well with a previous report. 13 MicroPET quantification was performed by measuring the ROI, and the result is shown in Figure 5B. The kidney uptake of 18F-TTM-VEGF was 23.20 ± 2.15 and 16.51 ± 1.52 %ID/g at 0.5 and 3 hours postinjection, respectively. Low muscle uptakes were observed (2.98 ± 0.66 and 1.59 ± 0.57 %ID/g at 0.5 and 3 hours postinjection, respectively).
Discussion
The development of 18F-labeled PET probes is rapidly gaining importance in nuclear medicine. 18F is generally incorporated into the bioligands through three types of functional groups: amino groups,14–16 carboxylic acid groups, 17 and sulfhydryl groups. As cysteines are much less abundant than lysines, aspartic acid and glutamic acid residues, thiol-reactive agents have been used to site-selectively modify peptides and proteins.18,19 Previously, several thiol-reactive 18F-synthons have been reported, all of which bear a maleimide group for conjugate addition of thiols under mild conditions.12,20–22 These labeling methods use mild reaction conditions and give high radiolabeling yields. However, the preparation of 18F-fluorinated probes by conventional methods requires a relatively large amount of bioligand (100–1,000 μg) in the bioconjugation step. For peptides or proteins with high cost and low availability, it is desirable to minimize the amount of wasted ligand. Furthermore, for large peptides and for protein labeling, the unlabeled ligands cannot necessarily be efficiently separated from the 18F-labeled ligands. As unlabeled ligands are competitive inhibitors of the 18F-labeled probes, the unlabeled ligands can dramatically decrease the sensitivity of radiolabeling. Thus, it is a premium consideration to decrease the equivalency of labeling precursor while maintaining fast and efficient reactivity.
In this study, we developed a bifunctional TM that is capable of selectively introducing tetrazine functionality to bioligands through the sulfhydryl group, and subsequent bioconjugation with 18F-TCO. To evaluate the new labeling method, two ligands, c(RGDyC) and VEGF-SH, were selected for labeling reaction.
The conjugation between TM and c(RGDyC) or VEGF-SH is efficient at room temperature. After purification, the radiolabeling of TM-RGD and TM-VEGF was performed with low loadings of bioligands. Starting from 18F-TCO, the synthesis of 18F-TTM-RGD was achieved in nearly quantitative yield. The total radiosynthesis time was about 90 minutes from the end of bombardment. For 18F-TTM-VEGF, the 75% labeling yield was calculated by collecting and measuring the radioactivity from intermediate fractions (3–4 mL) that eluted from a PD-10 size-exclusion column (later fractions were excluded from the yield calculation).
Previously, several thiol-reactive 18F-tagging molecules have been described,12,20–22 all of which bear a maleimide group allowing for thiol-specific Michael addition reaction. The total synthesis time of those 18F tags ranges from 100 to 150 minutes, with 10 to 20% non–decay-corrected radiochemical yields. As 18F-TCO can be prepared with a total synthesis time of 50 minutes and in 71% radiochemical yield, the present method represents an efficient alternative for the introduction of 18F labels to the sulfhydryl functions of biologic molecules.
In the microPET study, 18F-TTM-RGD was found to accumulate in U87MG tumor, which is consistent with our previous PET RGD studies.23–25 The tumor uptake of 18F-TTM-RGD in U87MG tumor was 1.98 ± 0.33, 1.80 ± 0.15, and 1.27 ± 0.33 %ID/g at 0.5, 1, and 2 hours postinjection, respectively. Compared with the N-succinimidyl-4-[18F]fluorobenzoate (18F-SFB)-labeled RGD (18F-FB-RGD), 25 the tumor uptakes were slightly higher. However, the uptakes of normal organs, such as kidneys and liver, are also higher than those of 4-[18F]fluorobenzoate-labeled RGD (18F-FB-RGD). For example, the liver uptake was 0.39 ± 0.05 %ID/g for 18F-FB-RGD and 2.35 ± 0. 24 %ID/g for 18F-TTM-RGD at 2 hours postinjection. The higher uptake in liver might be attributed to the increased lipophilicity of 18F-TTM-RGD compared to the RGD peptide itself. Future studies will involve modification with hydrophilic polyethylene glycol (PEG) linkers to decrease the liver uptake.
In addition to RGD peptides, we also tested our method for protein labeling. The interaction between VEGF and its receptor (VEGFR) has been extensively studied for angiogenesis-related pathways, which are very important for tumor growth.26–29 Previously, VEGF protein–based tracers have been developed for PET, single-photon emission computed tomography (SPECT), and optical imaging of tumor angiogenesis, all of which showed high renal uptake due to the VEGFR1 expression in kidneys.30–34 In the present study, we evaluated the 18F-TTM-VEGF in normal mice, and the biodistribution pattern correlated well with previous reports. The kidney uptake was 23.20 ± 2.15 and 16.51 ± 1.52 %ID/g at 0.5 and 3 hours postinjection, respectively (Figure 5B). The high kidney uptake is consistent with the literature.13,27,35,36 These results indicate that the radiolabeling was successful and the labeling moiety had a minimal effect on VEGF binding to its receptor. In conclusion, a highly efficient method has been developed for site-specific 18F labeling of cysteine-containing peptides and proteins. The special characteristics of the tetrazine-TCO ligation provide unprecedented opportunities to synthesize 18F-labeled probes with high specific activity for PET applications.
Conclusions
Despite the great potential of using 18F-labeled proteins for both diagnoses and therapeutic monitoring, their application has been limited because it is very challenging to synthesize 18F-labeled proteins in adequate yield with high specific activity. In this study, we developed an efficient method for the construction of 18F-labeled proteins with high specific activity based on the tetrazine-TCO ligation. We demonstrated that TM can conjugate with peptides and proteins bearing a free cysteine residue and that the resulting bioconjugates can be efficiently 18F-labeled through 18F-TCO ligation. The major advantage of this strategy lies in the fact that this cycloaddition reaction is highly efficient and enables very low bioligand loading. This TM reagent should be useful for the labeling of a variety of peptides, proteins, or oligonucleotides containing free sulfhydryl groups. Future studies will be directed toward the creation of even more reactive bioconjugation partners and improving the selectivity for tumor uptake relative to kidney uptake. This advance in 18F labeling methodology should enable the discovery of critical imaging probes for cancer diagnosis and treatment monitoring in both basic and clinical research.
Footnotes
Acknowledgment
Financial disclosure of authors: This work was supported by National Institutes of Health grant number P20 RR017716 from the Centers of Biomedical Research Excellence Program of the National Center for Research Resources, grant number P30CA014089 from the National Cancer Institute, and research grant DE-SC0002353 from the Department of Energy and by the University of Southern California Department of Radiology. Spectra were obtained with instrumentation supported by National Science Foundation CRIF:MU grants CHE 0840401 and CHE-0541775.
Financial disclosure of reviewers: None reported.