
Abstract
Molecular imaging allows clinicians to visualize disease-specific molecules, thereby providing relevant information in the diagnosis and treatment of patients. With advances in genomics and proteomics and underlying mechanisms of disease pathology, the number of targets identified has significantly outpaced the number of developed molecular imaging probes. There has been a concerted effort to bridge this gap with multidisciplinary efforts in chemistry, proteomics, physics, material science, and biology—all essential to progress in molecular imaging probe development. In this review, we discuss target selection, screening techniques, and probe optimization with the aim of developing clinically relevant molecularly targeted imaging agents.
Development of Molecular Imaging Agents
Molecular imaging relies on appropriate targets or biomarkers and the ability to generate probes that are selective and specific for those biomarkers. With the genomics and proteomics eras as well as increased understanding of the etiology of disease, targets are being rapidly identified, but the number of imaging agents to those targets has not kept pace. Bridging this gap has many far-reaching implications for the future directions of molecular imaging and radiologic sciences, including the early detection of disease (eg, atherosclerosis, cancer, diabetes) by the identification of early molecular signatures, accurate disease staging and treatment stratification, and the evaluation of various treatments and therapeutics (ie, efficacy and dosing).
In general, there are three aspects of probe development, although sometimes these can be combined. The first is to identify a biologic target for the condition of interest. The second is to generate a compound that binds to this target. The third is to convert the binding compound to a probe and to optimize in vitro and in vivo characteristics such as affinity, pharmacokinetics, toxicity, and elimination, with validation. In this review, we discuss target selection, screening techniques for identifying the targeting moiety, and probe optimization with the aim of developing clinically relevant molecularly targeted imaging agents.
Targets
The outpouring of information about aberrant molecular pathways and the proliferation of tools (such as genechips and other microarrays) have led to a great deal of information about the etiology of human disease. It is becoming easier and easier to select a list of proteins that are relevant to any biologic problem from a literature search. Unfortunately, not every protein is useful for imaging. A good target has several characteristics:
It is expressed differently in diseased tissue than surrounding normal tissue—preferentially not at all in the normal range, with high levels in the diseased tissue.
The target is highly expressed, although the exact density of receptors required varies with the sensitivity of the imaging modality. For example, in prostate cancer, the bombesin receptor is expressed on the order of 10,000 copies/cell. 1 This target has proven adequate for single-photon emission tomography (SPECT) using 99mTc.2,3 However, similar probes proved inadequate to image the same condition by magnetic resonance imaging (MRI) using iron oxide nanoparticles for contrast (L. Josephson and F. Reynolds, unpublished data, 2004).
It has to be accessible. The endothelium lining of blood vessels forms a tangible barrier. Therefore, unless the target is an endothelial cell, a probe must be able to extravasate out of the blood vessels and into the tissue of interest. In many diseases, such as cancer or atherosclerosis, the endothelial barrier is compromised and the lymphatics are impaired, leading to the enhanced permeability and retention effect. Many nontargeted and targeted imaging agents take advantage of this circumstance, allowing delivery and accumulation to the diseased area. This is especially important for nanoparticles large enough to stay in the circulation: the fenestrations in normal vessels exclude extravasation into those tissues, but the enlarged pores from the disease conditions allow selective perfusion in those areas. Similarly, the plasma membrane is another barrier that must be overcome if the desired target is a cytoplasmic or nuclear protein (Figure 1). Currently, there are few effective mechanisms to breach the plasma membrane and ensure homogeneous distribution throughout the cellular compartments; therefore, almost all probes bind to extracellular or membrane-associated targets.
To increase the sensitivity and contrast, it is preferable for a cell surface target to be internalized on binding and quickly recycled to the cell surface. This pumps the targeted agent into the cell to gain a measure of amplification relative to the bloodstream concentration. 4
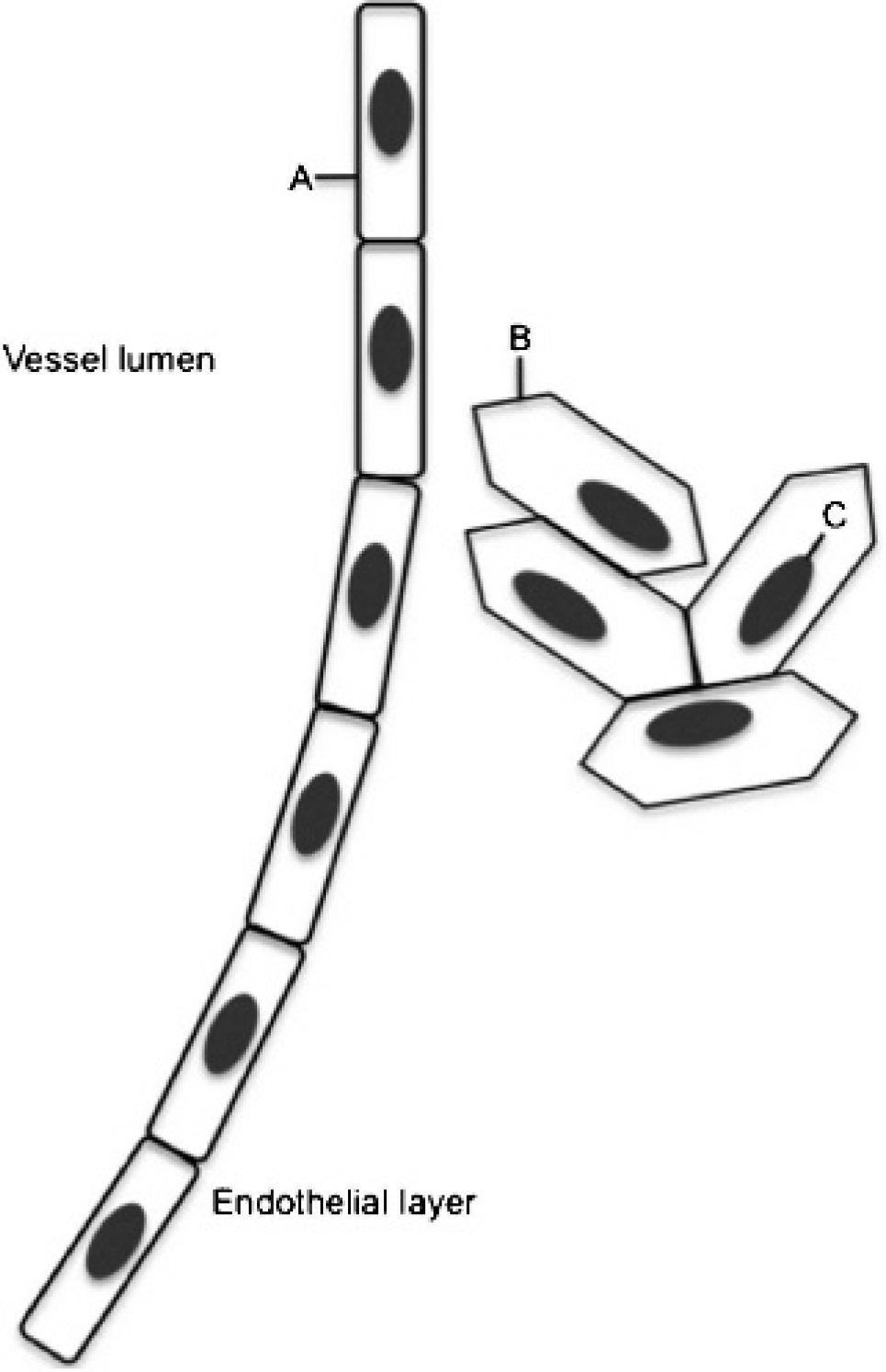
Physical barriers to target. A, The target is on the surface of the endothelium and accessible from the bloodstream. B, The target is at the cell surface and is accessible from the bloodstream only if the endothelial layer is compromised. C, The target is inside the cell. Any probe binding to this target must cross both the endothelium and the cell membrane.
Although genomics and proteomics have given investigators a wealth of potential targets, there is information to be gained by performing a no a priori knowledge screen using one of the combinatorial techniques described below.5–8 For example, a novel marker of pancreatic cancer has been identified using phage display and diseased tissue. 5 In this example, the protein plectin1 is on the cell surface for pancreatic cancer, whereas in normal cells, it is strictly cytoplasmic, taking advantage of the plasma membrane barrier to allow exquisite selectivity between normal and diseased tissue. With this type of screen, it is not always trivial to determine the exact receptor the probe binds to, but it is essential in this case to determine what other tissues, if any, express the mystery protein on their cell surface.
Targeting Moiety Identification
Peptides
Peptides have many characteristics that make them useful as imaging agents. They have a short blood lifetime, are nonimmunogenic, are relatively inexpensive to synthesize using standard conditions on commonly available automated machines, and are easy to chemically modify. Although using a synthetic protein from a known protein–protein interaction to generate an imaging agent is feasible, these tend to have long blood lifetimes if they do not trigger an immune response, are expensive, and may lose their binding ability when modified. Truncating a known protein binding partner into a peptide, however, can resolve these problems while retaining much of the affinity of the original protein–protein interaction.9–11 These peptide fragments can be transformed into an imaging agent,2,3,12 but a project such as this is an immense amount of work requiring biologic details, including structure, that are presently unknown for many targets.
Generating libraries of possible targeting compounds offers a way to circumvent the limitations of systematic design. With the 20 natural amino acids forming the entire palette from which proteins are made, the diversity of a library of peptides containing every combination of 7 amino acids (1.3 × 109 sequences) will usually have a few members that bind with a target with desired affinity. Libraries can be synthesized chemically, which allows the simple addition of nonnatural amino acids and access to derivatives, or biologically, on phage, yeast, or bacteria, allowing the researcher to harness the power of biologic enzyme systems to speed up and simplify the screening process. Both methods can generate either linear or disulfide-constrained libraries. Owing to entropic effects, disulfide-constrained libraries tend to generate fewer leads with better affinities compared to linear peptide libraries, 13 but chemically modifying the peptides afterward can be challenging.
As synthesizing and screening 109 peptides one by one is unfeasible with present technology, many methods have been developed to synthesize, screen, and deconvolute peptide libraries, including pin synthesis,14,15 library encoding, 16 various positionally encoded methods,17,18 and affinity chromatography methods. 19 The most comprehensively developed chemically synthesized system appears to be the one bead, one compound methodology.20–24 This system uses a pool-and-split technique to generate random peptides on 100 µm polystyrene beads. The beads are split into 19 separate pools; each pool has a different amino acid added to it using standard chemistry (cystine is not used outside defined locations to avoid random crosslinking). All the separate reactions are mixed together and again split into 19 reactions (Figure 2). This is continued until the peptides are long enough to provide the desired diversity. The peptides on any one bead are identical, but for all but the shortest peptides, no two beads will have the same sequence. 22
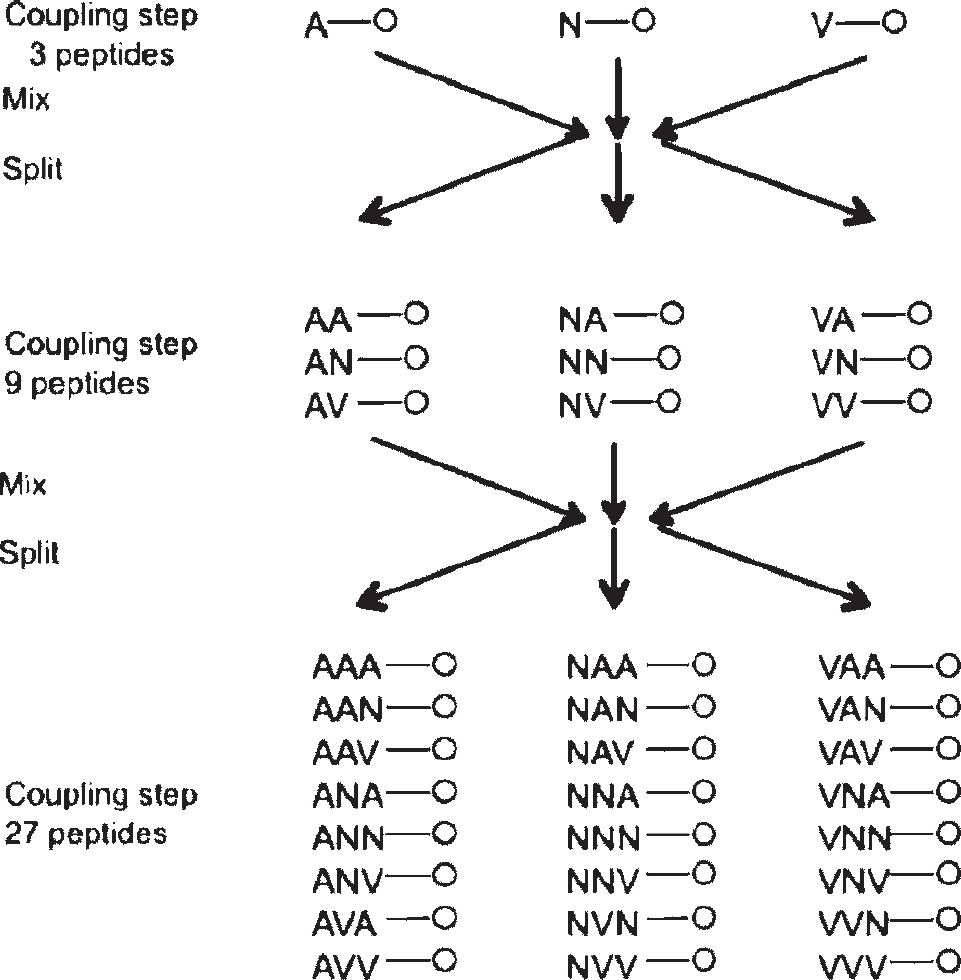
Pool-and-split library synthesis scheme.
Screening of proteins for binding peptide sequences consists of mixing the biotin-labeled protein with the beads and then adding streptavidin–alkaline phosphatase.22,24 The chromogenic substrate bromo-chloro-indolyl phosphate is added, and the beads that turn visibly blue are collected using microdisection tools. The bound receptor and streptavidin are removed, and the beads are reprobed with streptavidin–alkaline phosphatase to remove the peptides that bind to streptavidin rather than the intended protein. Each bead is individually sequenced by automated Sanger degradation to recover the peptide sequences.22,24
Recovering sequences that bind to cells uses a slightly more complex protocol.20,24 Trypsinized cells are mixed with the on-bead library and allowed to sit for 48 hours. If the cells have surface markers that bind to a peptide, they grow on the bead containing that sequence, which can be identified under a microscope. It is possible to use a negative cell line by labeling those cells with calcein AM, a green fluorescent dye. To restrict the screen to a specific protein on the cell membrane, a third screen is run on the beads that bound using an antibody to the desired protein to block binding.20,24
The one bead, one compound method allows rapid screening using common equipment. There is great flexibility in the library structure; standard peptide synthesis permits
Phage Display
Researchers have harnessed the power of biologic methods to make and screen libraries. Peptide sequences have been encoded on a plasmid attached to the peptide 25 with the library expressed in Escherichia coli, peptides attached to bacterial polysomes,26,27 displayed on yeast28–30 or attached to bacteria and on bacteriophage.
Phage display has been the workhorse technique with thousands of publications for screening peptides with desired binding characteristics. It has been used to screen for peptides that bind to protein, such as streptavidin,31–38 small molecules, 39 cells (eg, prostate cancer,40–45), inorganic compounds (eg, platinum metal46–50), and even substrates such as paint chips (titanium dioxide 51 ). It has been used in vivo in animals6,52–57 and humans 58 and can be engineered to display nonnatural amino acids,59,60 although with more difficulty than chemical synthesis. Phage display has not been able to find peptide sequences that bind to specific nucleotide sequences, however, as the screens yield sequences enriched in postive amino acids that bind indiscriminately to the ribose or deoxyribose backbone (see, for example, Tan and Frankel 61 —hits are essentially polyarginine).
Although there are a very large number of phage screening libraries for peptide binding, including different bacteriophage,62,63 and expression of various valency levels on different coat proteins, 13 most of the literature uses one of the M13 bacteriophage libraries commercialized by New England Biolabs (Ipswich, MA), or freely provided by George Smith from the University of Missouri. These are filamentous phage with the peptide library expressed on the five PIII proteins on one end of the viron, one peptide per protein. The various libraries differ in the length of the peptides of the library and disulfide constraint. Longer peptide sequences are best for selection against targets where the binding will be relatively weak but repeating, such as inorganic crystals. The tradeoff is in peptide diversity; the typical phage display protocol uses around 1010 viron particles, enough to completely cover a 7–amino acid library, but the commercial 12–amino acid library has 1015 possible combinations. Most proteins bind to other proteins through relatively small areas 64 (hot spots), which tend to be areas that phage peptide sequences also bind to.64,65 Constrained libraries have peptides that are forced into a limited number of configurations, an example of which is the disulfide-constrained libraries. Another way to confer confirmation restraints involves histidine complexation to metal ions. 66 None of these libraries are long enough to form stable tertiary structures.
The protocol for panning for clones that bind to a target, although it is subject to the creativity of the researcher and can be modified to emphasize different factors of importance, is rather basic. 13 Phage are incubated on the target, often immobilized on a plate or some other geometry. The nonbinding phage are washed off, and the bound phage are eluted, titered, and amplified. This is repeated for two to five rounds, after which clones are picked and sequenced. Many protocols include a subtraction step at the first panning to remove phage that bind to similar but undesired targets in addition to the selection.39,44
Elution of the phage from the target has been an area of great creativity. The M13 bacteriophage is very stable, allowing researchers to use harsh, nonspecific elution conditions to weaken the peptide-target binding, such as high salt concentrations, denaturing conditions, pH extremes, or even proteases. 13 Phage that bind to specific sites on a target can be eluted by displacement with the natural ligand (see, for example, Giebel and colleagues 31 ) or an antibody to the target.67,68 If the phage-target binding is extremely strong, it may be necessary to amplify the phage without elution.69,70
An important nuance allows the differentiation between phage that are internalized into a cell, presumably by binding to a receptor that is internalized, and phage that are confined to the surface. Once all the phage bound to the cell are eluted, cell lysis will free all the clones that were able to enter the cell membrane. 71 As the cell is constantly sampling the media by pinocytosis and turning over receptors, the time the cells are exposed to phage is an important parameter for selectivity; ideally, this would be similar to the turnover time of the receptor targeted. 71 Unfortunately, this information is difficult to discover, especially for a screen against an unknown target.
In vivo phage display screening is slightly different from in vitro. The phage is usually injected intravenously and allowed to circulate long enough for blood clearance (about 15 minutes to an hour 55 ). The exposure of the phage to all the surfaces of the endothelium removes undesired clones and can function as a subtraction step. 4 Phage are recovered by excising the tissue of interest, homogenizing it, and adding it to bacteria for amplification. As with the in vivo screens, this is repeated as needed, usually for two to four rounds.
The biodistribution of unlabeled phage gives one of the limits of in vivo screening. In mice, the virons tend to accumulate in the spleen, liver, lung, and, to a lesser extent, kidney, with some variation with mouse breed. 55 This can cause problems in isolating phage from these organs owing to the high number of nonspecific clones. For example, an in vivo screen for lung binding peptides yielded three clones that bound 143 that were isolated in five rounds of panning. 72 The second limitation is the ability of the phage to reach the desired tissue. Intravenously injected phage are limited in their ability to exit the endothelium unless it is compromised (as in cancer or inflammation). 71 An in vivo screen against cardiomyocytes using an intravenous injection, for example, will be challenging as the phage may have difficulty gaining access to the cardiomyocytes.
Although phage display is the main method in the literature for discovering peptide sequences that bind to a target, there are a number of papers where bacterial display is used. This technique consists of a random peptide sequence engineered to be expressed on an extracellular protein of the bacterium. The first and most common modification is the peptide sequence placed into a disulfide-constrained loop of E. coli thioredoxin (trxA), which is inserted into an E. coli flagellum protein (fliC),73,74 commercialized by Invitrogen (Carlsbad, CA) under the name FliTrx, although it does not presently appear to be available. Other proteins used are FhuA, 75 OmpA,76–78 and FimH. 79 Bacteria other than E. coli have also been used. 80 The differences between the different platforms are not obvious from the literature with the exception of OmpA, 77 which allows for linear peptide sequences free at either the C-terminus or the N-terminus. However, unlike phage display, this technology does not appear to be as well developed. The main application is panning against immobilized38,73,74,81, and dissolved proteins,76,78 with some work involving cell binding41,75,77,83 and metal ions.79,84,85
The advantages of bacteria versus phage display are a greater potential diversity (approximately 10 times that of phage 76 ), simpler laboratory workup and culture, 38 and the ability to use antibiotics with resistance genes to limit contamination with wild-type bacteria. 38 The disadvantages are that bacteria have many extraneous proteins that can bind to the target 73 and the inability to do in vivo selection owing to immune response. The commercial bacteria display system appears to have quality control issues, with some researchers finding that the peptide insert is a different length than expected.38,79 The authors of one article comparing phage to bacteria display stated that bacteria display is inferior to phage display 38 ; however, their data suggest that part of the issue may be unfamiliarity with the technology.
Screening is very similar to phage display. Bound bacteria are eluted by vortexing to tear them from their flagella, titered using absorbance, and expanded. If the screen was on cells, they can be lysed with pure water to collect the bacteria 75 or just vortexed, as with immobilized proteins.41,83 Internalization into cells can be selected for by eliminating all noninternalizing clones with gentamicin. 75 The peptide expressed by the binding bacteria is determined by sequencing the appropriate part of the genome. An alternative panning protocol is to attach a biotin onto a target protein and conduct the binding in solution, using phycoerythrin-streptavidin and fluorescence-activated cell sorting (FACS) to isolate the binding bacteria.76,77
Aptamers
Ribonucleic acid (RNA) and single-stranded deoxyribonucleic acid (DNA), owing to their specific interactions, will form complex secondary and tertiary structures that are not seen with peptide libraries. 86 The advantages of using aptamers are that it is easy to generate extremely large starting libraries of 1015 different compounds, achieving greater diversity than with peptides,87–91 and the techniques (polymerase chain reaction [PCR], translation of RNA to DNA and back) are commonly used in many laboratories. RNA also has very good pharmacokinetics for imaging, with a blood half-life of around 2 to 3 minutes.92,93 The primary elimination mechanism is blood degradation by endonucleases followed by renal excretion, both of which can be modified for longer lifetimes.
Researchers at multiple laboratories described the aptamer library generation and screening process at about the same time, one screening RNA oligomers to find compounds that bind to selected dyes 87 and another using the same technique to find RNA ligands that bind to an RNA binding protein 94 (naming the process systematic evolution of ligands by exponential enrichment [SELEX]). A third article used similar technology to generate catalytic sequences that would cleave a DNA substrate. 95 Since then thousands of studies have examined these aptamers for binding proteins,88,90,93,96–98 small molecules,87,90,99,100 and even chemical weapons.101,102 Several of these compounds have progressed to human clinical trials,98,103–105 with one compound approved by the Food and Drug Administration (FDA) to treat macular degeneration.96,106 The affinities can be in the picomolar range, leading to the nickname chemical antibodies.
The initial library is a chemically synthesized random RNA chain between 15 and 100 nucleotides long
107
flanked by fixed primer sequence handles for reverse transcription and PCR amplification. The primer sequences do not influence binding
107
and are excised in the final probe. A number of chemically modified nonnatural nucleotide bases are accepted by RNA enzymes that can be used for additional diversity,89,90,108 but they do not appear to enjoy wide use. For in vivo applications, such as imaging agents, the polymer chain needs to be stabilized against nucleases. An elegant method of doing this is to select against the mirror image of the target, such as a peptide or protein made of
Screening is very similar to phage panning, with two exceptions. Aptamers do not show amplification bias and the washing step is less effective, leading to many more panning iterations, usually 10 to 20 or more. The protein or small molecule of interest is typically attached to a resin and packed into a column, similar to affinity chromatography, but can also be presented on nitrocellulose membrane. Temperature is mentioned in a few articles, with one demonstrating that an aptamer selected for high affinity at one temperature may lose that affinity when the temperature is changed. 93 The literature of screening against cells is limited, but the basic approach is identical to phage display.105,111 Internalization can be selected for using trypsin to cleave off the targeted proteins and their attached aptamers. 112 In vivo selection has been reported once at this time, 113 using a 2′-fluoro stabilized RNA library to isolate aptamers that bind to a mouse model of colon cancer metastases.
Small Molecules
There is a long history of screening small molecules to discover drug leads but a much shorter publication record for imaging. Assays have been developed to look at binding,114,115 including fluorescence polarization, surface plasmon resonance, and calorimetry, but these techniques are not high throughput. The two solutions in the literature to avoiding expensive compound by compound screening are to immobilize the compounds on a slide or to use a tag on the small molecule to simplify the screen.
The success of gene chips and other microarray technology has led to a number of articles attempting to extend the idea into other areas, including small molecules. Both attachment114,116–127 and detection114,122,124,128 are facets that have been extensively published. An array of small molecules is bonded to a solid surface, with the identity of each small molecule positionally encoded. Protein, cell lysates, homogenized tissue, or other material to be tested is washed over the microarray, and adherence of proteins is measured. The detection is typically fluorescence or biotin on the protein, but antibodies 122 and surface plasmon resonance 128 are also used.
A common approach to identifying small molecules that can be used for imaging is to make libraries of compounds that already have the imaging moiety attached.7,129,130 In one example, magnetic nanoparticles used for MRI contrast were labeled with a fluorescent tag and a small molecule. Binding of the small molecule to a cellular or protein target is monitored directly by either the fluorescent tag or an antibody to it. The advantage of this technique is that a hit is the agent; attachment of a binding moiety to the nanoparticle is already completed. The disadvantage is that each small molecule is tested individually, limiting the diversity of the library. The literature studies referenced here used an MRI probe. Some other imaging techniques would be very expensive to do this way, such as 18 F labeling for positron emission tomography (PET).
Other groups have used a polynucleotide tag to label each small molecule, giving them a unique identifier that can be amplified by PCR. The basic idea has been in use for almost two decades, 131 where a peptide sequence (or any other polyamide) was encoded in the attached DNA sequence. The basic protocol is similar to phage panning. The mix of small molecules is exposed to either a cell or an immobilized protein, and the nonbinding samples are washed away. The samples that bind may be repanned, often with PCR amplification and library resynthesis. Another protocol is to mix the DNA-labeled small molecules with the protein(s) in solution, remove unbound material by size exclusion, and detect the DNA attached to the small molecule by complement binding on a gene chip. 132 Other variations have the DNA sequence encoding the reactions to make the small molecule in a split-and-pool type combinatorial synthesis, either adding a section of DNA when the reactions are run 133 or synthesizing the DNA first using combinatorial methods and using it to direct the synthesis.134,135 Very large libraries can be constructed by these methods, and as the DNA indicates the reactions, biologic amplification techniques can be used. A final variant has the DNA enclosed in a phage,136,137 a variant that will protect the tag during the various synthetic and screening steps.
One final note on small molecule screening: most small molecules can be modified only at certain locations on their scaffold, or the modification will interfere with binding. 138 If a small molecule is attached to a plate at the position required for binding, it will not bind its target, even if there is high affinity in solution. Adding complexity, some sites will be permissive, but with size or charge limitations. 139
Antibodies
Antibodies were the first compounds developed that could bind specifically to a known protein target. It is possible to find commercially available antibodies that bind to almost any desired disease state, even discriminating between single molecules such as phosphates on specific amino acid residues. Furthermore, hybridoma and other technologies have facilitated the generation of antibodies on a large scale. There is a large repertoire of reliable chemical modifications based on N-hydroxysuccinimide (NHS) ester chemistry to link them to chelators, peptides, or nanoparticles for imaging. As can be imagined, there is an extensive literature using antibodies to target imaging agents. However, antibodies are expensive, have very long blood lifetimes, 140 and can elicit an immune response if made in a different species, 141 and labeling can affect the affinity, 142 although this must be determined empirically as it varies from antibody clone to antibody clone. Attempts to use antibody fragments give better pharmacokinetics, but this often yields reduced binding and tumor uptake. 140 There are several reviews on using antibodies as imaging agents,143,144 with a cohort of antibody based agents clinically approved.145–147
Optimization and Development
Optimization and development consist of determining the binding partner of the targeting compound (if not already known), optimizing the affinity, converting it into an imaging agent, and improving its in vivo characteristics. Selectivity is optimized in the screen that gave the lead if the screens were designed properly. Optimization is too broad a subject to cover as one section of a review article, so only the most common problems and solutions will be given.
A good imaging agent will have a blood lifetime long enough for the compound to bind but short enough to allow imaging soon after administration, can reach the target (either penetrate the endothelial wall or bind to a protein on the endothelium), will bind strongly to the target and not accumulate anywhere that will interfere with imaging, and will be completely nontoxic in the dosages used. Ideally, there will be an amplification mechanism, such as receptor-mediated internalization or signal amplification.148,149 These are different design end points than those used in developing drugs. Drugs, by design, will perturb a process in the body, whereas imaging agents should have minimal physiologic effect. Off-target binding in drugs is not desired but may be an acceptable tradeoff, whereas off-target binding of imaging agents causes false-positive results and high background, rendering the compound difficult to use. Side effects in a therapeutic agent may be tolerated, depending on the condition being treated. The same side effects in an imaging agent are intolerable as some fraction of the people dosed with the compound will be healthy. Ideally, drugs will remain in the bloodstream for long periods of time to minimize dosing requirements, but good imaging agents are cleared quickly to reduce background. A drug may be administered for years for some conditions, whereas an imaging agent will be dosed at most a few times, so chronic effects are examined differently. Although both drugs and imaging agents optimize pharmacokinetics and pharmacodynamics, the goalposts are very different.
If the exact protein target of the identified compound is unknown, it should be identified before further work is done to help determine off-target binding and possible side effects. Unfortunately, this is a difficult task. The most common method with peptides and aptamers is affinity chromatography.113,150 Although this method often works well, if the target protein is membrane stabilized, isolating the protein will denature it, abrogating the binding. Some researchers have used bioinformatics tools, such as Basic Local Alignment Search Tool,56,58,151–153 to identify what protein the peptide is mimicking, but there are serious limitations to that approach. First, the peptides identified are not likely to match exactly the protein that binds to the same site they are using. Often only a few amino acids in the sequence are essential for binding; the rest are essentially random, leading to poor matches. The second limitation is that a bioinformatics approach assumes that the protein that binds will do so along one linear section of its amino acid sequence. Often the binding site will consist of a point where a protein is folded, bringing areas that are distant in the primary structure of the protein in close proximity. 65 A peptide mimicking a protein with multiple folds will not show sequence alignment. Therefore, it is often necessary to use protein–protein binding techniques, such as yeast two hybrid, to solve this problem.
Once the binding partner is known, the affinity is determined and improved if needed. All things being equal, the higher the affinity of the compound the better as this allows lower doses, which can help reduce off-target binding. In addition, it may be necessary to trade off affinity to optimize another parameter, such as specificity or selectivity during optimization. The easiest way to improve the affinity of an aptamer, peptide, or small molecule consists of determining the moieties that are essential for binding, building a new library with the essential components unchanged, and repeating the screening in a process called affinity maturation.65,154–156 For peptides, a series are synthesized, each one differing from the original targeting peptide by the mutation of one position for an alanine. The affinity of each peptide is determined, and the peptides with significantly poorer affinity indicate the essential residues. The target is then rescreened with a new library, holding these amino acids constant. A similar process is done with aptamers, but owing to their length and the lack of an innocuous nucleotide, it is much more expensive and time consuming. A simpler method is to make several libraries with different sections randomized and rescreen. 89 It is possible to do this work as part of the initial screen in both phage display and aptamer screening by randomly mutating the compounds between selections.95,154 Small-molecule affinity is a more difficult process involving chemical synthesis. The idea is to systematically modify the molecule and look at structure-activity relationships to develop heuristics about how features such as physical size, hydrophobicity, and charge at different portions of the molecule affect the affinity. Changing any of the structures can change features such as specificity and toxicity, which is why this process is done early in the drug development timeline. This is an entire field of chemistry in itself—as much art as science. Different experienced groups starting with the same lead molecule often arrive at very different results.
If it is impossible to improve the affinity sufficiently by these techniques, avidity effects can compensate on the final agent. If an agent with multiple copies of a binding sequence has one that binds to a receptor, the remaining copies will be positioned to bind, leading to an artificially improved affinity. In effect, the three-dimensional tracking problem has been reduced to two dimensions. 157 This will not make a millimolar binding affinity into a working agent but can make a borderline affinity workable.
The next step is to modify the binding moiety to make it more resistant to enzymes that will cleave it. Both peptides and aptamers are usually degraded in the blood by endogenous enzymes and removed by the kidneys too quickly to be effective imaging agents. The ways to prevent this for aptamers, that is, 2’ substitutions and 3’ capping, were mentioned in the screening section as these modifications will affect binding. These can be added at this stage, but a mutation experiment is needed to see which residues are sensitive to the substitution.
158
It is much less work to include these substitutions in the original screen. For peptides, capping the N-terminius
159
and substituting
Developing the agent with the optimized binding moiety is the next step and is dependent on the imaging modality used. The targeting ligand is attached to a contrast segment, and the agent is optimized. The contrast segment varies with imaging modality. For SPECT, this is usually a chelator binding 99mTc or 111 In. 161 For PET, it is usually 18 F. 162 MRI uses chelated gadolinium for T1 imaging or magnetic nanoparticles for T2 contrast. 163 X-ray and computed tomographic techniques use heavy atoms (usually iodine but occasionally gold) to provide contrast, either attached directly or as a nanoparticle. Optical methods use fluorescent dyes. Although this is not a complete list of contrast species for these imaging techniques, they are the ones most commonly used clinically. Each imaging moiety has its own associated chemistry and requirements for functional groups on the targeting ligand. Most if not all of these imaging moities can be used with any of the targeting compounds mentioned in this review. With few exceptions, the preference of the researcher and the achievable affinity and selectivity, rather than the exact chemical species (aptamer, peptide, small molecule, or antibody) used, will be definitive. An example of a rare poor choice would be using a short half-life isotope such as 18 F with a long circulating nanoparticle. When the imaging agent has cleared from the blood to a level that gives low background, the radioactivity would have decayed as well.
Once the first iteration of the probe is made, it should be tested in cell culture to see if the affinity and specificity are still adequate and in an animal model to look at target or background, off-target binding, toxicity, and optimal dosing or timecourse. If any of these parameters are insufficient, the probe must be modified.
Blood lifetime is governed by probe degradation, elimination, and uptake by the immune system. Degradation in the blood should have been addressed before the synthesis of the probe. Elimination is caused by phagocytosis by immune cells, renal clearance, and various pathways of liver clearance. For volatile compounds, lung clearance will also be significant, 164 but this is unusual for molecular imaging probes. All of these elimination mechanisms will act on anything injected into the bloodstream. Owing to a number of poorly understood active transport phenomena, it is difficult to tell a priori exactly how a compound will be removed and where the agent will collect nonspecifically. However, there are heuristics about how compounds are eliminated that can be used to skew the dominant mechanism. Nanoparticles and compounds of similar size are usually predominantly opsonized and removed by phagocytes. Mobile phagocytes, such as macrophages and neutrophils, will bring the probe to the lymph nodes, whereas stationary phagocytes, such as Kupffer cells of the liver, will simply degrade them where they are injested. Reducing opsonization with polymer coatings will greatly slow this process down, but by removing a major elimination mechanism, the blood lifetime will be increased, often considerably. 165 Hydrophilic compounds smaller than ≈6 nm in diameter can be filtered by the kidneys; for compounds below 4 nm in diameter, this removal is very rapid. Hydrophobic compounds bind to serum albumin in the blood and will be cleared by the liver through the bile duct to the intestines or by partitioning into the blood plasma for renal excretion. 164 It is common for hydrophobic compounds to be oxidized or otherwise rendered more hydrophilic by the cytochrome P-450 liver enzymes followed by renal or hepatobillary excretion. 164 These heuristics can be used as a basis for modifying a probe to influence the blood lifetime and redirect it away from areas to be imaged. For instance, a small hydrophilic probe may be eliminated renally too quickly to be a good agent. To increase blood lifetime, the probe can be made more hydrophobic to increase binding to albumin (decreasing renal clearance and increasing hepatic clearance, and incidentally often improving the affinity and reducing specificity) or by increasing the size of the compound by adding either a polyethylene glycol (PEG) chain or a protein (slowing but not necessarily eliminating renal clearance).92,105
Conclusions
Much of the technology to generate an imaging agent is new and still developing. Advances in molecularly targeted probe development will likely increase the ability to diagnose patients and perform preclinical and basic research.
Footnotes
Acknowledgments
We would like to thank Frederick Epstein for critical reading of the manuscript.
Financial disclosure of authors: K.A.K. is funded by the Johnson & Johnson Corporate Office of Science and Technology, the Wallace H. Coulter Foundation, an American Association for Cancer Research PanCAN career development award, the Department of Defense Congressionally Directed Medical Research Program, National Institute of Biomedical Imaging and Bioengineering grant EB010023, and National Cancer Institute grant RO1 CA137071.
Financial disclosure of reviewers: None reported.