
Abstract
Molecular imaging has moved to the forefront of drug development and biomedical research. The identification of appropriate imaging targets has become the touchstone for the accurate diagnosis and prognosis of human cancer. Particularly, cell surface- or membrane-bound proteins are attractive imaging targets for their aberrant expression, easily accessible location, and unique biochemical functions in tumor cells. Previously, we published a literature mining of potential targets for our in-house enzyme-mediated cancer imaging and therapy technology. Here we present a simple and integrated bioinformatics analysis approach that assembles a public cancer microarray database with a pathway knowledge base for ascertaining and prioritizing upregulated genes encoding cell surface- or membrane-bound proteins, which could serve imaging targets. As examples, we obtained lists of potential hits for six common and lethal human tumors in the prostate, breast, lung, colon, ovary, and pancreas. As control tests, a number of well-known cancer imaging targets were detected and confirmed by our study. Further, by consulting gene-disease and protein-disease databases, we suggest a number of significantly upregulated genes as promising imaging targets, including cell surface-associated mucin-1, prostate-specific membrane antigen, hepsin, urokinase plasminogen activator receptor, and folate receptors. By integrating pathway analysis, we are able to organize and map “focused” interaction networks derived from significantly dysregulated entity pairs to reflect important cellular functions in disease processes. We provide herein an example of identifying a tumor cell growth and proliferation subnetwork for prostate cancer. This systematic mining approach can be broadly applied to identify imaging or therapeutic targets for other human diseases.
IN THE PAST DECADE, various imaging technologies, including positron emission tomography (PET), single-photon emission computed tomography (SPECT), computed tomography (CT), magnetic resonance imaging (MRI), and optical imaging, have evolved rapidly to facilitate the noninvasive diagnosis, prognosis, and guided treatment of human cancer.1,2 The aberrant expression and unique biochemical function of cell surface- or membrane-bound proteins in tumor cells make them desirable targets for molecular imaging.1–4 A well-established imaging strategy is to design radiolabeled or optical-based ligand probes that home in on the specific cell surface- or membrane-bound targets with high affinity. This imaging strategy might be advantageous for several reasons. First, the imaging probes could provide an image intensity correlating with the amount of targets actively present on tumors given the easily accessible locations. 1 Second, low-molecular-weight imaging probes employed in this strategy, such as radiolabeled small molecules and peptide derivatives, generally enjoy ease of synthesis, rapid biodistribution, and rapid clearance. 4 Third, the ligand probes are not required to penetrate the cell membrane, a formidable obstacle. Furthermore, the functional information obtained from imaging these targets can be used to better understand the pathology of human cancer for diagnostic and therapeutic applications.
The strategy of using enzyme-activated imaging probes shows great promise for cancer diagnosis and drug development.1,2 As an example, we developed an enzyme-mediated cancer imaging and therapy (EMCIT) technology, 5 which aims to hydrolyze and precipitate water-soluble radioactive prodrugs within the extracellular spaces of solid human tumors for imaging or therapy. To identify EMCIT-suitable targets, we reported a rapid literature mining approach for searching PubMed abstracts and retrieving entities (using the LSGraph program, no longer available) by keyword and Gene Ontology (GO) term filtering. 6 The application of this approach led to the identification of a number of cancer-related hydrolases, including prostatic acid phosphatase (PAP), prostate-specific antigen (PSA), and sulfatase-1 (SULF1). Although useful, this literature mining approach cannot convey quantitative expression information and does not allow for biologically meaningful ranking and prioritization of the identified targets. The emergence of high-throughput technologies such as microarray has greatly changed the course of target discovery for diagnosis and therapeutic intervention. 7 For example, to identify molecular signatures potentially important in human cancer, scientists would compare an overwhelmingly large number of gene expression profiles of a particular cancer type and of its corresponding normal tissue, and then a handful of genes with particular characteristics are filtered and selected for further experimental validation. However, the sifting of thousands of differentially expressed entities appears to be difficult for researchers who do not possess statistical or computational proficiency. 8
In recent years, a variety of databases warehousing high-throughput microarray data have been developed to facilitate the discoveries of useful biologic targets or insights. For instance, Gene Expression Omnibus (GEO; <http://www.ncbi.nlm.nih.gov/geo/>) has emerged as one of the major public repositories for a wide variety of microarray data. Numerous interesting diagnostic or prognostic signatures have been identified through the analysis of stored GEO microarray data with the use of implemented clustering and visualization tools. 9 Similarly, Oncomine is one of the principal public cancer microarray platforms incorporating 264 independent microarray data sets, totaling more than 18,000 microarray experiments, which span 35 human cancer types. 10 Recently, we successfully identified lists of putative bloodborne biomarkers for six common human cancers through a combined analysis strategy in Oncomine and a curated pathway knowledge base. 11
Here we describe an integrated bioinformatics approach to analyzing Oncomine data for the discovery of potential targets for cancer imaging. Our procedure was developed in the way that cancer microarray data can be analyzed in the context of other biologic data, such as gene-gene/protein-protein interaction networks. To identify suitable targets for molecular imaging, the method is based on (1) analyzing in six common cancers all gene signatures upregulated relative to their normal tissues in the Oncomine database; (2) further filtering of the list of upregulated genes by a combination of relevant GO AmiGO (<http://amigo.geneontology.org/cgi-bin/amigo/go.cgi>) terms, implying extracellularity and membrane-bound after critical evaluation of all GO vocabularies and a stringent corrected Q value (false discovery rate) cutoff (Q ≤ 0.05) 12 ; (3) enlarging and enriching the entities by related functions and biomolecular interaction networks, which can be achieved by importing the list into the Ingenuity knowledge base (Ingenuity Pathway Analysis [IPA] software, Ingenuity Systems, Redwood City, CA; <http://www.ingenuity.com/products/pathways_analysis.html>) or manually importing the list into biomolecular interaction databases (see Materials and Methods); (4) resolving duplicates and genes with inappropriate locations, such as “cytoplasm” or “nucleus,” from the list and keeping those that belong to cell surface- or membrane-bound; and (5) ranking of the entities in the final list based on the absolute Student t value (abs[t]), normalized previously across various studies in Oncomine. This bioinformatics approach can be employed to derive suitable imaging targets appearing in the most common and lethal types of human prostate, breast, lung, colon, ovarian, and pancreatic cancers.
For prostate cancer, as an example, we retrieved 749 upregulated genes implying extracellularity and membrane-bound, from a total of 181,361 measured genes of different studies, of which 13,353 are upregulated in prostate cancer relative to normal prostate (Table 1). These 749 signatures were then imported to the Ingenuity knowledge base and enlarged by related functions and biomolecular interaction networks embedded in curated interaction databases, including BIND (Biomolecular Interaction Network Database; <http://bond.unleashedinformatics.com/>), BIOGRID (Biological General Repository for Interaction Datasets; <http://www.thebiogrid.org/>), DIP (Database of Interacting Proteins; <http://dip.doe-mbi.ucla.edu/>), INTACT (protein interaction database; <http://www.ebi.ac.uk/intact/site/index.jsf>), HiMAP (Human Interactome Map; <http://www.himap.org>), MINT (Molecular Interaction Database; <http://mint.bio.uniroma2.it/mint/>), and MIPS (Database for Genomes and Protein Sequences Databases; <http://mips.gsf.de>) (see Materials and Methods), resulting in 1,404 entities from which 374 entities were identified as cell surface- or membrane-bound proteins. Further, these 374 entities were clustered into 33 hydrolases, including enzymes, phosphatases, and peptidases; 32 G protein-coupled receptors; 39 ion channels; 23 kinases; 53 transmembrane receptors; 65 transporters; and 129 other proteins accordingly (see Table 1). By manually reviewing the identified entities at the gene-disease knowledge base GeneCards (<http://www.genecards.org/>) and knowledge base iHOP (information hyperlinked over proteins) (<http://www.ihop-net.org/UniPub/iHOP/>), we established a number of potential hits for cancer imaging, all confirmed by the previous literature to be promising imaging targets for human cancer. Finally, we suggest that this integrated bioinformatics approach could be broadly applied in discovering and prioritizing valuable imaging and therapeutic targets for other human diseases.
Number of Potential Hits Identified in Six Common and Lethal Tumors by Integrated Bioinformatics Analysis of Genomic Profiles*

IPA = Ingenuity Pathway Analysis.
Completed by 02/02/09.
Sum of measured genes in all data sets filtered by <<cancer vs normal>>.
Gene Ontology keywords include <<extracellular space>>, <<extracellular region>>, <<cell surface>>, <<plasma membrane>>, and <<integral to membrane>>.
Materials and Methods
The focus of our analysis approach is to retrieve and filter genes significantly upregulated in cancer compared to normal tissues, encoding cell surface- or membrane-bound proteins, to a manageable gene list. The choice of microarray platform or database, statistical cutoff criteria, and controlled annotations (GO terms) in the mining strategy is variable, depending on the particular interest of the user. Our strategy is based on the combination of Oncomine, an advanced, publicly accessible, cancer microarray platform; the Ingenuity Pathways knowledge base; biomolecular interaction databases; and other knowledge bases.
Data Collection and Filtration
Oncomine 3.0 is a cancer microarray database incorporating 392 independent microarray studies, totaling more than 28,880 microarray experiments, which span 41 cancer types. Oncomine is unique in that it provides differential expression analyses comparing most major types of cancer to respective normal tissues. More importantly, it is integrated with the GO annotations filter, which permits users to identify genes with particular biologic processes, molecular functions, and cellular locations. Thus, in comparison with other cancer microarray data sources, Oncomine is more biologist friendly for in-depth data analysis on a per-gene basis. For each of the six cancer types, all upregulated genes comparing cancer versus normal tissue samples were collected irrespective of the different microarray platforms used in these studies. The suitability and significance of the entities as molecular imaging targets were assessed later by literature review.
These cancer gene profiles were then filtered by their subcellular locations. In this study, we wanted to identify cell surface- or membrane-bound targets suitable for cancer imaging. Consequently, these targets should not be secreted or shed into circulation. To search and identify these entities, we filtered the upregulated genes associated with the following GO terms: <<extracellular space>>, <<extracellular region>>, <<cell surface>>, <<plasma membrane>>, and <<integral to membrane>>. Each GO annotation term was conceived and consulted in the GO database to identify all relevant hits. Other users can apply suitable GO controlled terms as filters to identify new targets for a particular problem.
The overexpressed genes identified by GO terms were collected and further filtered by the Q (false discovery rate) cutoff value, which has been widely used as a sensible measure of the balance between the number of true positives and false positives in DNA microarray experiments. Q value controls the proportion of positive calls that are false positives. To account for multiple hypothesis testing in the Oncomine database, Q values are calculated as follows: Q = Np/R, where p is the p value, N is the total number of genes analyzed, and R is the sorted rank of the p value. 10 Because a typical microarray data set contains thousands of genes, Q value can be used as a much more sensible cutoff than p value to evaluate the upregulated genes in cancer microarray data sets. We chose a stringent Q value cutoff of 0.05, and only those upregulated genes with a Q value less than 0.05 were kept in the final list.
Finally, to select genes with an indication of significant overexpression across all data sets, we used the abs(t) value to rank and prioritize the overexpressed hydrolytic enzymes. In Oncomine, each gene is assessed for differential expression with the Student t-test, calculated as t =<y1> – <y2>/δ, where y1 and y2 are the mean expression values in two conditions and δ is the common variance of the two distributions normalized across different studies. Therefore, the t value should be a good measure of the change in expression level in cancer tissue relative to normal tissue. For each gene, only the highest abs(t) value from multiple data sets was kept in the list to avoid the inherent noise in the different platforms.
Gene Mapping
The National Center for Biotechnology Information's Entrez gene identifier was chosen as the target identifier in the analysis. We used the Ingenuity knowledge base to assign an identifier to each entity in the list. The Ingenuity knowledge base is acquired by manual curation of full texts of peer-reviewed scientific publications. It covers information on more than 500,000 mammalian genes or proteins, molecular concepts, and millions of their pathway interactions. If no tag or accession ID was available, the entity was mapped using gene symbol or gene synonyms. Then the duplicated entities were resolved from the list. The lists were then imported to the network analysis of the Ingenuity package, permitting interpretation of the data set in the context of biologic processes, pathways, and molecular networks.
Enrichment Analysis
The derived lists of entities were enlarged and enriched by related functions and biomolecular interaction networks embedded in seven publicly accessible interaction databases, including BIND, BIOGRID, DIP, INTACT, HiMAP, MINT, and MIPS, implemented within the Ingenuity knowledge base. Researchers may enlarge the entities by Ingenuity or manually import and enlarge the entities by these protein interaction databases. Researchers could also choose to enlarge the entities by other interaction networks according to their particular interests. For instance, researchers could choose to enlarge the entities by protein-protein domain interactions embedded at SCOPPI (Structural Classification of Protein-Protein Interfaces; <http://www.scoppi.org/>).
Literature Review of the Candidate Entities
Further analysis and assessment of the resulting hits were performed retrospectively using GeneCards and iHOP, two curated databases that find links and cited articles to genes/proteins and identify the particular gene product if the gene name or synonym is known. The entities obtained were checked by carefully reading the associated literature references or original publications. The accuracy of the findings is assessed using control entities, selected as candidate molecules by other studies or well-known and clinically useful imaging targets.
Results and Discussion
The bioinformatics analysis approach, assembling Oncomine cancer microarray database and biomolecular interaction networks implemented within the IPA knowledge base, was employed to identify cell surface- or membrane-bound targets for six common tumor tissues (prostate, breast, lung, colon, ovary, and pancreas) (see Table 1 and Figure 1). These specific tumor tissue names were applied in the profile search of Oncomine to yield all of the available microarray data sets for each cancer tissue type. With the <<filter>> module in Oncomine set to the analysis type <<cancer vs normal>>, the cancer microarray data sets are limited to 4 to 15 microarray data sets per cancer type. These microarray data sets contain between 3,064 and 19,645 upregulated gene expression profiles for the cancer relative to its normal tissue. The next step is the filtering of upregulated genes by the GO cellular component terms <<extracellular space>>, <<extracellular region>>, <<cell surface>>, <<plasma membrane>>, and <<integral to membrane>>. The combination of these GO location terms defines the extracellular compartments of cancerous cells, including those elements bound or integral to cell membranes. Further sorting by a very stringent Q (false discovery rate) value cutoff of 0.05 reduces the number of upregulated genes to between 211 (colon) and 2,782 (ovary) for the six cancer types.

Working scheme of an integrated bioinformatics analysis approach to identify potential cancer imaging targets.
The import of retrieved genes from Oncomine to the Ingenuity knowledge base facilitates the interpretation of the data sets in the context of biologic networks or pathways. Approximately 140 to 810 entities were mapped within the literature-curated molecular network after removing duplicates and cytoplasmic proteins. Next, about 352 to 1,421 entities were found by enlarging and enriching the entities by related functions and biomolecular interaction networks embedded in seven interaction databases (as described in the Materials and Methods). Further, the filtering of subcellular locations led to the identification of about 72 to 398 cell surface- or membrane-bound entities (see Supplementary File 1). These cell surface- or membrane-bound targets were then clustered into hydrolases, G protein-coupled receptors, ion channels, kinases, transmembrane receptors, transporters, and other proteins according to biologic functions and ranked according to the abs(t) values. Further review of these prioritized hits in gene-disease and protein-disease databases and reading of the articles cited for the hits enabled us to select those targets suitable for cancer imaging.
Identification of Well-Known Imaging Targets
As positive controls, we list below a few well-known imaging targets, detected by our work, that have also been well established or are already being used as clinical imaging targets.
Somatostatin Receptors
Somatostatin is a cyclic 14-amino acid peptide that acts at many sites to regulate and inhibit the release of many hormones and other secretory proteins. Somatostatin receptor (SSTR) is a family of cell surface G protein-coupled receptors (including SSTR1, SSTR2, SSTR3, and SSTR5) with seven transmembrane segments that mediate the biologic effect of somatostatin, and the upregulation of SSTRs is associated with many tumors. 13 We detected SSTR2, STTR3, and STTR5 as highly overexpressed entities for breast cancer with the gene expression pattern SSTR5>SSTR2>SSTR3. On the other hand, Kumar and colleagues determined the upregulation pattern of the SSTRs in breast cancer as SSTR1>SSTR2>SSTR3>SSTR4>SSTR5 through reverse transcriptase-polymerase chain reaction (RT-PCR) and immunocytochemistry techniques. 14 This discrepancy implies that the expression pattern derived from genomic profiles is not necessarily consistent with that observed at the protein level. Indeed, nonmetabolized radiolabeled peptide probes targeting at SSTRs have been intensively explored for clinical cancer imaging. For example, a 64Cu-labeled somatostatin analogue comprising 8-amino acid residues (octreotide) was designed for imaging of various cancers expressing SSTRs with high affinity and efficient renal clearance 15 ; a variety of 111In/68Ga/90Y/177Lu-labelled SST-analogues have been prepared for the imaging and therapy of SSTR-rich human tumors 16 ; and near-infrared fluorescent dye-labeled peptides have been investigated as sensitive tracer agents for clinical imaging of human tumors overproducing SSTRs. 17 Therefore, our study has confirmed the important role of SSTRs in clinical cancer imaging and diagnosis.
Erythroblastic Leukemia Viral Oncogene Homologue 2 (ERBB2/HER2).
ERBB2, commonly referred to as HER2/neu by clinicians, encodes a cell surface-bound tyrosine kinase receptor that is involved in the signal transduction pathway leading to cell growth and differentiation through the binding of specific factors. Amplification or overexpression of this gene has been reported in numerous cancers, including breast and ovarian tumors. Herceptin, a humanized antibody with a high binding affinity to HER2, has been formally approved by the Food and Drug Administration (FDA) to treat metastatic breast cancer. Our study, consistent with prior experimental findings, has detected ERBB2 as an upregulated gene in prostate, breast, ovarian, and pancreatic tumors. Because of its frequent overexpression in human cancer, HER2 has attracted intensive research attention for daily clinical imaging. For instance, a 124I-radiolabeled antibody has been developed as a radiotracer for the imaging of HER2-positive tumors with PET 18 ; a nanoshell bioconjugated contrast agent targeting at HER2 receptor has been developed as an imaging probe for breast cancer with high resolution 19 ; and herceptin-dye conjugates have been prepared as photoacoustic CT probes for HER2 expression in breast cancer. 20
Integrin αvβ3
Another well-known target for molecular imaging that was also identified by our study is ITGAV, which encodes integrin αvβ3, a membrane-bound protein composed of an alpha chain and a beta chain interacting with extracellular matrix ligands. It is known that integrin αvβ3 plays a key role in the signaling transduction pathway and angiogenesis of many tumors. In our study, we confirmed the upregulation of ITGAV in prostate and lung cancer. In fact, multimodality approaches have been applied to image integrin αvβ3 expression in vivo for the noninvasive early detection of cancer. As examples, a paramagnetic contrast agent targeting at endothelial integrin αvβ3 has been developed to detect tumor angiogenesis using MRI, 21 an 18 F-labeled tracer selective for integrin αvβ3 with high uptake has been developed to diagnose cancer using PET, 22 and contrast-enhanced ultrasonography with microbubbles targeting at integrin αvβ3 has been employed to image tumor angiogenesis. 23
Epidermal Growth Factor Receptor
Epidermal growth factor receptor (EGFR) encodes a cell surface tyrosine kinase receptor that binds to epidermal growth factor and regulates cellular growth and proliferation. The upregulation of EGFR has been associated with a number of cancers, including lung cancer, colon cancer, and glioblastoma. The identification of EGFR as an oncogene has led to active development of anticancer therapeutics against EGFR, including FDA-approved gefitinib for lung cancer 24 and cetuximab for colon cancer. 25 Further, EGFR has been selected as a target for noninvasive imaging of EGFR expression and signaling activity for patients under chemotherapy. For example, a 124I-labeled small molecular tracer has been developed for noninvasive PET imaging of EGFR kinase activity 26 ; a bioluminescent imaging approach has been developed to study the biologic regulation of EGFR activity in vivo. 27 Again, our microarray data mining confirmed the upregulation of EGFR in prostate, breast, and lung cancer. For colon and ovarian cancer, we detected EGFR as a potential hit among the enlarged entities through multiple protein-protein interaction networks, including MAPK, Akt, and JNK pathways.
Identification of Promising Imaging Targets
One advantage of our mining strategy is the prioritization (according to absolute t values) of top-ranked overexpressed genes with biologic evidence implicating their significant role in cancer. Previously, little attention has been paid to their potential as imaging targets, probably because of the challenge in validating a large pool of candidate genes. These top-ranked genes are valuable because they are quantitatively more overexpressed than the other genes and thus increase the values as targets for cancer imaging. Those scientists interested in discovering diagnostic or therapeutic targets could further analyze and validate these candidate targets to make them clinically useful. We want to remind readers that the results from genomic profiling studies are not sufficient on their own as they do not confirm whether the product of the gene is (1) present or truly differentiated at the protein level, (2) localized at the desired location (outer cell membrane), and (3) normally functioning (for instance, a number of receptors are present but nonfunctioning in the ligand-receptor interaction). Therefore, the targets derived from genomic profiling studies need to be validated at the protein level through experimental techniques such as RT-PCR or immunohistochemistry. Below we assess the potential of five of the top-ranked genes identified in our analysis as clinically useful imaging targets as these have already been evaluated and confirmed experimentally.
Mucin-1, Cell Surface Associated (MUC1)
MUC1 encodes a membrane-bound, glycosylated phosphoprotein anchored to the apical surface of many epithelia. Remarkably, we identified MUC1 as the most significantly upregulated gene in ovarian cancer among the 391 upregulated genes encoding cell surface- or membrane-bound proteins. In addition, MUC1 is also upregulated in breast and lung cancer. This is consistent with prior experimental findings that MUC1 is highly overexpressed in human tumors, including breast, lung, and ovarian 28 (see Supplementary File 1). Further, the upregulation and underglycosylation of MUC1 on the surface of tumor cells can lead to the exposure of the core peptides of its extracellular domain and oligosaccharides. 29 By pathway analysis, we found MUC1 interacting with at least 60 molecules that are involved in the ERK, Src, and nuclear factor κB signaling pathways. All of these unique biochemical features have made MUC1 a very attractive target for cancer therapy and diagnosis. Recently, Salouti and colleagues described the development of a 99mTc-labeled monoclonal antibody targeting at MUC1 for in vivo imaging of breast cancer with high affinity and high sensitivity. 30 This is in agreement with our prediction that MUC1 can be used as a promising target for clinical imaging of human cancer, considering its significant upregulation and accessible location on the membrane of cancerous cells.
Folate Hydrolase, Prostate-Specific Membrane Antigen (FOLH1/PSMA)
FOLH1, also referred to as PSMA, encodes a membrane-bound glycoprotein acting as a glutamate carboxypeptidase that cleaves N-acetyl-
Hepsin, Transmembrane Protease, Serine 1 (HPN)
HPN encodes a cell surface serine protease, hepsin, which plays an essential role in the cell growth and maintenance of normal cell morphology. 33 Hepsin can function as an activator for other extracellular proteases or directly degrade the extracellular matrix of surrounding cells by cleaving after basic amino acid residues such as Arg and Lys. 34 Strikingly, we identified HPN as the most overexpressed gene encoding cell surface- or membrane-bound proteins in prostate tumor (Table 2), consistent with prior experimental findings that HPN was collectively upregulated among 500 patients bearing prostate cancer. 34 More importantly, hepsin is overproduced in different stages of prostate tumors, ranging from precursor lesions of prostate cancer to hormone-refractory metastatic tumors. All of these properties identify hepsin as a potential imaging target for the diagnosis and prognosis of prostate cancer. Recently, Kelly and colleagues developed hepsin-targeting imaging probes by conjugating multiple peptides with fluorescent nanoparticles, which bind with hepsin with high affinity. 35 Similarly, we envision that peptide-conjugated, water-soluble, and radioactive prodrugs targeting at hepsin could be developed for noninvasive diagnosis of prostate cancer based on our EMCIT concept. 5
Top 10 Ranked Cell Surface- or Membrane-Bound Entities Identified from Genomic Profiles for Six Tumor Types

Plasminogen Activator, Urokinase Receptor (PLAUR/uPAR)
uPAR, encoded by PLAUR, is a glycolipid-anchored receptor for urokinase-type plasminogen activator (uPA). The binding of uPAR by uPA activates the Ras/extracellular signal-regulated kinase pathway, which leads to cell proliferation, migration, and invasion. 36 Therefore, the dysregulation of the uPA/uPAR system has been linked to tumor growth and metastasis. 37 PLAUR is also highly upregulated in breast and pancreatic cancer (see Supplementary File 1). Thus, it might be possible to develop imaging methods for quantifying uPA/uPAR expression for cancer diagnosis and prognosis. Indeed, an early study successfully employed peptide-conjugated fluorescent probes to map tumor-associated uPA activity. 37 Recently, Li and colleagues developed a linear 64Cu-labeled peptide targeting at uPAR for noninvasive in vivo PET imaging. 38 These experimental findings confirm that the computational approach described in our studies is valid and is capable of identifying molecular targets that can be useful in the detection of malignant tumor lesions.
Folate Receptor 1, Adult (FOLR1/FBP)
FOLR1, a member of the folate receptor family encoding folate binding protein (FBP), has drawn our attention because we detected it as the third most overexpressed gene (see Table 2) in ovarian cancer. Moreover, it was uniquely overexpressed in ovarian cancer through a comparison study across six human tumor types. In line with our predictions, prior clinical studies have demonstrated the overproduction of the folate receptor at the protein level (overproduced in > 90% of ovarian carcinomas); there is limited expression in normal tissues. 39 Additionally, the overproduced level of the folate receptor is closely correlated with the aggressiveness of ovarian cancer from stage 1 to stage 4 disease. 39 Given all of these properties, the folate receptor has emerged as an attractive imaging target for the early detection and prognosis of ovarian cancer. Indeed, numerous radiolabeled folate receptor-targeting agents (66–68Ga, 111In, 99mTc, and 64Cu labeled) have been developed for clinical radionuclide imaging in recent years.40 Notably, most of these radiopharmaceuticals have achieved very good tumor to background tissue contrast, probably owing to their unique and significant upregulation in ovarian cancer. Therefore, the future of folate receptor-based imaging seems very promising for the detection of ovarian cancer with high specificity and affinity.
Identification of “Focused” Interaction Networks for Cancer Imaging
Another attribute of our integrated analysis approach is the ability to identify “focused” protein interaction networks underlying derived entities. One great challenge of molecular imaging is to visualize and unravel key pathways that are unique for a specific disease process, such as human cancer.1,2 Moreover, genes or proteins with potential as therapeutic end points are more likely to function as a cooperative group or network in human cancer. Given such cooperative pathways or subnetworks, researchers could formulate imaging strategies and devise high-affinity specific molecular probes to study and better understand complex biologic processes such as the initiation and progression of human cancer. Fueled by the increasing amount of data repositories for gene-gene and protein-protein interactions, it has become feasible to identify or construct relevant interaction subnetworks from high-throughput data. As an example, we constructed a tumor cell growth and proliferation subnetwork underlying prostate cancer using the retrieved upregulated candidate entities as seeds.
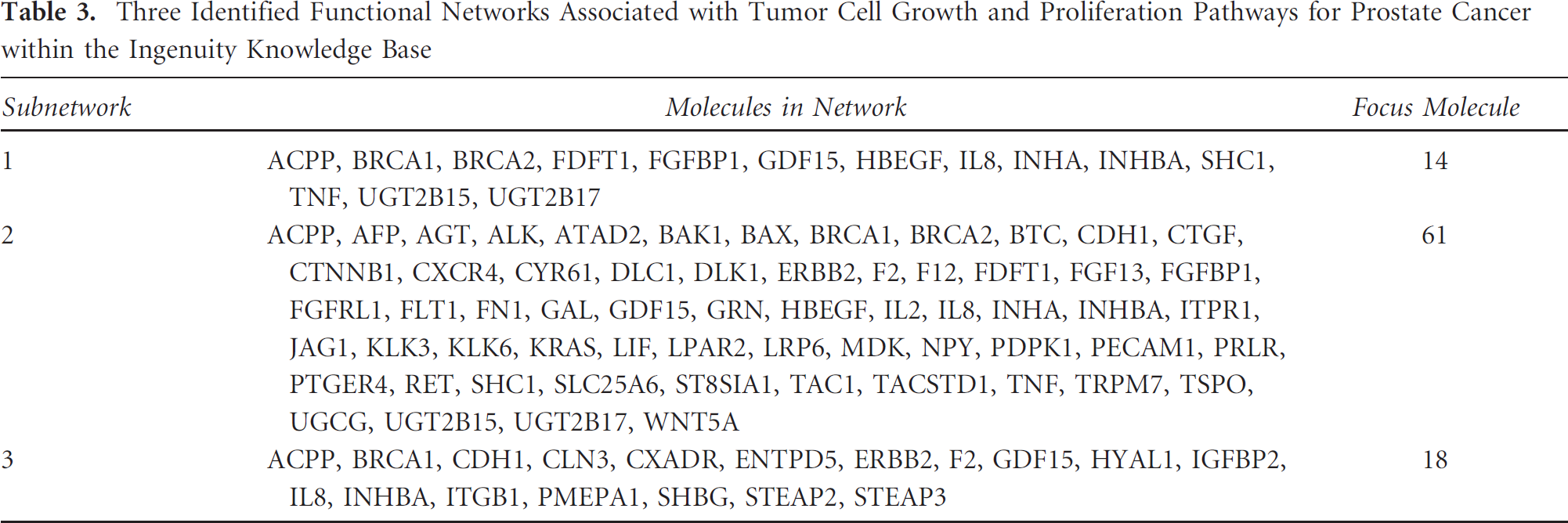
Monitoring the rate of tumor cell growth and proliferation by molecular imaging is an effective way to detect tumor aggressiveness and tumor response to therapy. Consequently, genes or gene products that regulate tumor cell growth and proliferation pathways could be potential imaging targets for prognosis. Using prostate cancer as an example, we identified three functional subnetworks (Table 3) consisting of numbers of entities (called focus molecules) that are closely associated with tumor cell growth and proliferation via the network analysis. We hypothesized that these tumor-relevant molecules could be combined to define a broad genetic roadmap that can lead to prostate tumor progression and metastasis. Subsequently, we clustered and uploaded the identified focus molecules to analyze, organize, and present the landscape of their interrelationships in graphic form within the Ingenuity package (Figure 2). In this landscape, entities are represented as nodes, and the biologic relationships between two nodes are represented as a line (direct protein-protein interactions) or a dashed line (regulations of bindings, inhibitions, proteolysis, phosphorylation, or modifications), also called an edge. Particularly, on exploring the links in this subnetwork, we found IL8, KLK3, TNF, IL2, ACPP, AGT, ITGB1, CXCR4, FN1, ERBB2, SHC1, BAX, and CTNNB1 to be 13 upregulated entities to the core of this subnetwork, acting as a hub by interacting with surrounding focus molecules (see Figure 2). These hub entities with high connectivity (a large number of interactions with other entities) in the networks are highly influential and might be preferred diagnostic or therapeutic targets. 41 Strikingly, we found direct experimental evidence to support the contention that many of these hub entities are responsible for the progression or metastasis of prostate tumor. For example, IL8, which encodes a chemokine protein secreted by prostate cells, may be responsible for the androgen-independent growth of advanced prostate cancers through interactions with G protein-coupled receptors in signaling pathways,42,43 and CXCR4, which is located on the cell surface with seven transmembrane regions, can regulate the MAPK kinase pathway and eventually lead to the progression and invasion of prostate tumor. 44 Consequently, we extracted a cooperative and focused interaction subnetwork consisting of these hub entities (see Figure 2) and speculated that imaging strategies could be formulated to visualize these entities or their interactions as prognostic indicators for prostate cancer. For instance, clinical imaging researchers could devise suitable radiopharmaceuticals homing at these hub entities to monitor their concordant overproduction, which might be a strong indication of tumor invasion. Although the detailed mechanisms of pathogenesis for these hub entities remain unclear, they may all pave the way to the progression or malignancy of prostate tumor. Nevertheless, further careful experimental validations are required to confirm their usefulness in clinical cancer imaging because protein-protein interactions are often measured in vitro or artificial systems and may not occur in vivo. 8

Identification of a focused protein-protein interaction network that may lead to prostate tumor progression and metastasis. Gene products are represented as nodes and biologic relationships (direct and indirect) are shown as lines (protein-protein interactions) and dashed lines (regulations of bindings, inhibitions, proteolysis, phosphorylation, or modifications). The hub entities in this subnetwork are highlighted in orange, including IL8, KLK3, TNF, IL2, AGT, ACPP, ITGB1, CXCR4, FN1, ERBB2, SHC1, BAX, and CTNNB1. These hub entities were extracted to form a focused interaction subnetwork, as represented on the right of the figure. The subcellular locations are also indicated on the right as extracellular space, plasma membrane, and cytoplasm and nucleus.
Three Identified Functional Networks Associated with Tumor Cell Growth and Proliferation Pathways for Prostate Cancer within the Ingenuity Knowledge Base
Conclusions
We have described herein a rapid, integrated, and biologist-friendly approach to mine potential targets for cancer imaging, based on the assembling of a public cancer microarray database and curated knowledge base. For six common and lethal human tumor types, the application of this analysis approach led to the identification of upregulated genes encoding putative cell surface- or membrane-bound proteins as candidate targets for noninvasive cancer imaging. Future research could be geared to designing “smart” radiopharmaceuticals that possess high affinities and specificities to these entities once their upregulation has been confirmed at the protein level through experimental techniques. Regardless, the integrated bioinformatics analysis of genomic profiles offers the advantage of prioritizing and ranking candidate genes for overexpression, which is crucial for the design of imaging probes to accurately detect cancer. Furthermore, the retrieved entities have been analyzed in the context of protein-protein interactions to construct focused interaction subnetworks useful for cancer imaging and extracting deeper biologic insights for human cancer. Such derived subnetworks associated with tumor cell growth and proliferation for cancer prognosis are very useful and warrant further investigation. Consequently, we strongly believe that the integrated analysis approach described above can be applied broadly to discover targets that are useful in diagnosis or therapy and to advance our understanding of the underlying biologic mechanisms of many human diseases.
Footnotes
Acknowledgments
Financial disclosure of authors: This work was supported in part by US Department of Defense grant W81XWH-06-1-0043, Radioimaging and Radiotherapy of Prostate Cancer (to A.I.K.). Work in the Y. Yang laboratory was supported by Start-up Fund for Excellent Young Overseas Fellows no. 3016–893318 at Dalian University of Technology.
Financial disclosure of reviewers: None reported.
Notes