
Abstract
Inhibiting the proteolytic activity of the 26S proteasome has been shown to have selective apoptotic effects on cancer cells and to be clinically efficacious in certain malignancies. There is an unmet medical need for additional proteasome inhibitors, and their development will be facilitated by surrogate markers of proteasome function. Toward this end, ectopic fusion of the destruction domain from ornithine decarboxylase (ODC) to reporter proteins is often used for assessing proteasome function. For luciferase-based reporters, we hypothesized that the oxygen-dependent destruction domain (ODD) from hypoxia-inducible factor 1α (HIF-1α) may provide improved sensitivity over luciferase-ODC, owing to its extremely rapid turnover by the proteasome (HIF-1α has a half-life of less than 5 minutes). In the current study, we show that ODD-luciferase affords a greater dynamic range and faster kinetics than luciferase-ODC in sensing proteasome inhibition in vitro. Importantly, ODD-luciferase also serves as an effective in vivo marker of proteasome function in xenograft tumor models, with inhibition being detected by noninvasive imaging within 3 hours of bortezomib administration. These data establish ODD-luciferase as a surrogate marker of proteasome function that can be used both in vitro and in vivo for the development of novel proteasome inhibitors.
THE 26S PROTEASOME is a highly selective enzyme complex that uses adenosine triphosphate to degrade polyubiquitylated proteins. It consists of a barrel-shaped 20S core particle that harbors proteolytic activity and is capped on either side by a 19S regulatory particle. A highly regulated ubiquitin conjugation system ensures that only proteins destined for degradation are targeted to the proteasome, and the majority of cellular proteins, including those involved in deoxyribonucleic acid (DNA) replication and cell cycle machinery, are eventually turned over in this manner.1, 2 Studies have shown that blocking proteasome activity can disrupt a wide variety of cellular functions and induces apoptosis in certain cell types.3, 4 Malignant cells appear to be more susceptible to the apoptotic effects of proteasome inhibition in comparison with normal cells, in part owing to greater metabolic demands5, 6 and in part through heightened dependence on signaling pathways that are regulated by the proteasome. 7
Three main classes of compounds, peptide aldehydes, nonpeptide natural compounds, and peptide boronates, have been found to inhibit the proteasome. 8 To date, bortezomib, a peptide boronate, is the only proteasome inhibitor approved for use by the US Food and Drug Administration. First approved in 2003 to treat refractory or relapsed multiple myeloma, bortezomib has since been approved for use as an initial treatment for multiple myeloma and for the treatment of mantle cell lymphoma. 9 Research has indicated that multiple myeloma cells rely on nuclear factor κB (NF-κB) activity for their growth and survival, and bortezomib is thought to be particularly effective in treating this disease owing to its ability to block proteasome-mediated degradation of IκB, a key inhibitor of NF-κB. 6 Bortezomib's success has reinforced the concept that targeting the proteasome may be an effective therapeutic strategy for treating other types of cancers, and clinical trials are under way to determine its utility in treating various hematologic malignancies and solid tumors. 10 Despite its success, however, resistance to and toxicity from bortezomib have been observed in patients who are on long-term treatment.6, 10 Many different molecular mechanisms, including upregulation of growth and antiapoptotic factors, may contribute to the development of resistance.
Novel proteasome inhibitors may be efficacious in treating multiple myeloma cases where bortezomib is no longer well tolerated or resistance has developed or in treating cancers for which bortezomib is not effective. Techniques that allow the rapid and sensitive detection of proteasome inhibition are vital to high-throughput screening efforts for such new compounds. Ornithine decarboxylase (ODC), which catalyzes the rate-limiting step in polyamine synthesis, contains a well-characterized proteasome destruction signal in its carboxyl-terminus. 11 When fused to green fluorescent protein (GFP) or luciferase, a 37– to 53–amino acid sequence from ODC accelerates proteasome-mediated degradation of the reporter.12, 13 In the presence of a proteasome inhibitor, destruction of the ODC-fused reporter diminishes and thus fluorescence or bioluminescence intensity increases, providing a measure of proteasome blockade. The ODC domain, attached to a fluorescent marker protein, is thus commonly used as a sensor of proteasome activity in cells.
GFP is a very stable protein, with a half-life of over 24 hours. Destabilization by fusion of GFP with the ODC degradation domain (GFP-ODC) results in significant reduction in stability (the half-life of ODC is approximately 2 hours) and improved dynamic range for assessing proteasome inhibition in vitro. However, for the purposes of in vivo imaging, fluorescence imaging of GFP has significant limitations, including interference by autofluorescence from endogenous chromophores in animal tissues, absorbance of both excitation and emission by hemoglobin, and a long half-life, which dampens the rapidity of change in reporter activity.14, 15 In contrast, a significant portion of the bioluminescence emission spectrum of firefly luciferase is red-shifted beyond the hemoglobin absorbance spectrum and endogenous luminescence is low in animal tissues, thus facilitating in vivo imaging of deep tissues.14, 15 However, since the half-life of native firefly luciferase is approximately 3 hours, 16 fusion to the ODC degradation domain (LucODC) may not adequately shorten protein half-life and may limit dynamic range. In an effort to produce greater destabilization, we explored the utility of using the oxygen-dependent degradation domain (ODD) of the α-subunit of hypoxia-inducible factor 1α (HIF-1α) to destabilize luciferase. Under normal growth conditions, the half-life of HIF-1α is less than 5 minutes and is among the shortest of all known proteins. Under normal oxygen tension, two residues within the ODD are prolyl-hydroxylated, leading to binding of the von Hippel-Lindau (pVHL) protein, polyubiquitylation, and subsequent proteasome-mediated degradation. 17 Under hypoxic conditions, hydroxylation of the ODD is inhibited, pVHL no longer binds, and the level of HIF-1α protein increases exponentially. 18 Previous studies have shown that fusing the HIF-1α ODD to Gal4, GFP, or luciferase rapidly accelerates their destruction under normal oxygenation, 19 22 making ODD-fusion proteins powerful sensors of hypoxia. We hypothesized that the rapid proteasome-mediated degradation of ODD-luciferase could be exploited to efficiently screen proteasome inhibitors in vitro and could be used as a pharmacodynamic readout of proteasome inhibition in vivo.
Methods
Plasmids and Cell Lines
An insert encoding firefly luciferase, followed by a picornovirus ribosomal slippage peptide (F2A), mCherry fluorescent protein, another picornovirus ribosomal slippage peptide (T2A), and, lastly, puromycin-N-acetyltransferase, was codon-optimized for expression in human cells and synthesized as a minigene (GeneArt, Regensburg, Germany). This insert was subcloned into the FUW lentiviral plasmid 23 to create FUW-Luc-mCherry. To create ODD-luciferase, DNA encoding residues 527 to 645 of human HIF-1α was cloned as an amino-terminal fusion to the firefly luciferase gene within FUW-Luc-mCherry. To create luciferase-ODC, DNA encoding residues 410 to 461 of mouse ODC was cloned directly downstream and in frame with the carboxy-terminus of firefly luciferase. All lentiviruses were packaged by standard cotransfection with plasmids encoding helper functions in 293T cells. 23 HCT116 colon cancer cells and U87 glioma cells (American Type Culture Collection, Manassas, VA) were infected with high-titer lentiviruses and were subsequently selected with 2 μg/mL of puromycin to establish stably transduced cells. All chemicals were from Sigma (St. Louis, MO), except as otherwise indicated.
In Vitro Luciferase Assays and Immunoblotting
Cells were seeded in 96-well plates, and the indicated drug treatments (eg, bortezomib and MG132) were performed in triplicate wells.
Xenografts and In Vivo Imaging
Five million HCT116 cells that had been stably transduced with FUW-Luc-mCherry or FUW-ODDLuc-mCherry were subcutaneously injected into the flanks of NCr nude mice. On day 6 postinjection, mice with palpable tumors (approximately 30–50 mm3) were imaged for mCherry expression (IVIS Spectrum, Caliper Life Sciences, Hopkinton, MA) to ensure tumor establishment.
Results
In Vitro Assessment of Proteasome Inhibition
We created a tripartide lentiviral expression vector in which firefly luciferase (wild-type luciferase [wtLuc]), the mCherry fluorescent protein, and the puromycin resistance gene were produced from a single messenger ribonucleic acid through intervening picornovirus ribosomal slippage sequences. 24 The ODD domain, consisting of amino acids 527 to 645 of human HIF-1α, was fused to the N-terminus of luciferase to create ODDLuc. The degradation domain consisting of the terminal 52 amino acids of mouse ODC was fused to the C-terminus of luciferase to create LucODC. The coding sequences were cloned in the FUW plasmid, 23 which directs high-level expression using the ubiquitin C promoter, creating FUW-wtLuc-mCherry, FUW-ODDLuc-mCherry, and FUW-LucODC-mCherry lentiviral vectors (Figure 1A). Given that the destabilizing effects of ODD- and ODC-fusion are restricted to the luciferase polypeptide to which they were fused, cells transduced with all three lentiviruses produce high-level and equal expression of both mCherry and puromycin.

Destabilization of luciferase by ODD and ODC fusion. A, Schematic of luciferase reporters. The ubiquitin C (UbC) promoter drives expression of all coding sequences. For each vector, the production of three separate polypeptides is accomplished by intervening picornovirus ribosomal slippage peptides (F2A and T2A). B, Absolute (raw) luciferase activity of wtLuc, ODDLuc, and LucODC in stably transduced HCT116 cells under basal growth conditions. C, Half-life of ODDLuc, LucODC, and wtLuc was determined in HCT116 cells stably transduced with each reporter. Cells were treated with 20 μM cycloheximide for the indicated amount of time. In vitro dose-response curve for the effects of (D) bortezomib or (E) desferrioxamine on the activity of each luciferase reporter in HCT116 cells (10 hours). Significance was calculated by the Student t-test: **first data point at which p < .005. All data are represented as mean ± SD of triplicates. Graphs are representative of at least three independent experiments. F, HCT116 wtLuc, ODDLuc, and LucODC cells were treated for 10 hours with 0, 4, 8, or 16 nM bortezomib, as indicated. Cell lysates were subjected to SDS-PAGE followed by immunoblotting for luciferase protein.
We used these plasmids to package high-titer lentivirus and infected HCT116 colon cancer and U87 glioma cells. Stably transduced cells were produced by selection with puromycin. At baseline, the absolute luciferase activities of LucODC and ODDLuc were significantly lower than that of wtLuc, indicating that the ODC and ODD degradation domains were working to destabilize the luciferase protein (Figure 1B). To determine the half-lives of the different luciferase alleles, we used cycloheximide to inhibit protein synthesis in HCT116 cells and measured luminescence intensity over time. As previously reported, 16 the half-life of wtLuc was approximately 180 minutes (Figure 1C). The LucODC fusion had a reduced half-life of approximately 90 minutes (see Figure 1C), consistent with the half-life of native ODC being ≈2 hours. In contrast, fusion of luciferase to the HIF-1α ODD resulted in the shortest half-life of all three reporters at < 30 minutes (see Figure 1C). Similar results were obtained in U87 cells (data not shown).
To determine the utility of each reporter for measuring proteasome activity, we assessed luciferase activity after 10 hours of treatment across a range of bortezomib concentrations. The activity of wtLuc was minimally affected by bortezomib, whereas a statistically significant (p < .005) increase in both ODDLuc and LucODC activity occurred at all concentrations > 4 nM (Figure 1D). In response to higher concentrations of bortezomib, ODDLuc readings increased by ≈14-fold, whereas LucODC activity increased only by ≈2-fold. The EC50 of bortezomib on ODDLuc activity was approximately 7 nM. To verify that the change in ODDLuc activity represented a change in its stability, we used desferrioxamine to inhibit the prolyl hydroxylases responsible for targeting ODD to pVHL and the ubiquitin-proteasome degradation pathway. 17 Desferrioxamine also caused a statistically significant (p < .005), dose-dependent increase in the activity of the ODDLuc reporter, whereas there was no effect on LucODC or wtLuc (Figure 1E). We used immunoblotting for luciferase protein to confirm that the observed increases in reporter activity were correlated with actual increases in luciferase protein. Cells were treated with bortezomib for 10 hours, and lysates were subjected to SDS-PAGE. Immunoblotting with an antiluciferase antibody verified that bortezomib treatment resulted in an accumulation of the ODDLuc and ODCLuc proteins (Figure 1F). Together, these results demonstrate that ODD-fusion destabilizes luciferase significantly more than fusion to ODC, resulting in improved dynamic range for assessment of proteasome inhibitors.
To characterize the kinetics with which ODDLuc serves as a biomarker for proteasome inhibition, we performed a time course experiment with a dose of bortezomib (64 nM) that afforded maximal luciferase signal at 10 hours (see Figure 1D). With periodic measurements, a statistically significant (p < .005) increase in ODDLuc activity was detected within 60 minutes of treatment (Figure 2A). The activity of ODCLuc showed a much smaller but statistically significant increase (p < .02) as well. In contrast, the activity of wtLuc did not change over the first 4 hours of treatment (see Figure 2A). To verify that these effects were not unique to bortezomib, we used a saturating dose of the peptide aldehyde MG132, which resulted in a significant increase in ODDLuc activity within 30 minutes, whereas neither LucODC nor wtLuc values changed over the first 4 hours of treatment (Figure 2B). Similar results were observed in U87 glioma cells treated with either bortezomib (Figure 2C) or MG132 (Figure 2D).

ODDLuc is stabilized by proteasome inhibitors. Time course experiments measuring the effects of (A) 64 nM bortezomib or (B) 42 μM MG132 on ODDLuc, LucODC, and wtLuc activity in HCT116 cells. Time course in stably transduced U87 glioma cells showing the effects of (C) 64 nm bortezomib or (D) 42 μM MG132 on ODDLuc, LucODC, and wtLuc activity. Significance was calculated by the Student t-test: **first point at which p < .005 and *p < .02. All data are represented as mean ± SD of triplicates. Graphs are representative of at least three independent experiments.
In Vivo Assessment of Proteasome Inhibition
We used a xenograft tumor model to determine the utility of ODDLuc as an in vivo proteasome sensor. To establish tumors, 5 × 106 HCT116 cells expressing either wtLuc-mCherry or ODDLuc-mCherry were subcutaneously injected into the flanks of nude mice. Six days after injection, mice with palpable tumors (30–50 mm3 in volume) underwent fluorescence imaging to verify equal tumor burden as assessed by mCherry fluorescence (Figure 3A). Cohorts of mice with equal ODDLuc- and wtLuc-expressing xenograft tumors (by mCherry imaging) underwent bioluminescence imaging to assess baseline luciferase reporter activity, followed by injection with vehicle (normal saline). Serial imaging 3, 6, and 24 hours after normal saline injection was used to determine the effects of vehicle on ODDLuc and wtLuc activity (see Figure 3A). The same animals were then injected with bortezomib, followed by serial imaging, to determine the effects of proteasome inhibition on reporter activity (see Figure 3A).

In vivo imaging of proteasome activity. A, Schematic of dosing and imaging regimen for HCT116 xenograft tumors. B, Representative fluorescence images demonstrating equivalent HCT116 tumor size based on mCherry expression from tumors 6 days after implantation. C, Representative bioluminescence images of wtLuc and ODDLuc reporter tumors. Images shown were at baseline and 3 hours after injection of vehicle or bortezomib. Note that the maximum of the color scale for wtLuc animals is 50-fold higher than that for ODDLuc.
Representative images from mice with wtLuc- and ODDLuc-expressing HCT116 xenografts demonstrate that tumor burden was similar on the basis of mCherry fluorescence imaging (Figure 3B). At baseline, bioluminescence imaging revealed that average wtLuc activity was 94-fold greater than ODDLuc activity, as would be expected given the destabilized nature of ODDLuc (Figure 3C). In response to vehicle treatment, there was no significant change in either wtLuc or ODDLuc activity (see Figure 3C). After treatment with bortezomib, bioluminescence intensity from ODDLuc tumors clearly increased, whereas the signal from wtLuc tumors remained stable (see Figure 3C).
When averaged over multiple animals, relative reporter activity from ODDLuc xenograft tumors was indistinguishable from wtLuc controls after treatment with vehicle (Figure 4A). When the same animals were treated with bortezomib, there was a rapid and highly significant increase in ODDLuc reporter activity that peaked within 3 hours after injection of drug (Figure 4B). As would be expected based on the pharmacokinetics of bortezomib, ODDLuc reporter activity returned to baseline by 24 hours after drug injection (see Figure 4B). By comparison, wtLuc activity did not significantly change over the same time course. These results demonstrate that ODDLuc is a sensitive and rapid biomarker of proteasome inhibition in vivo.
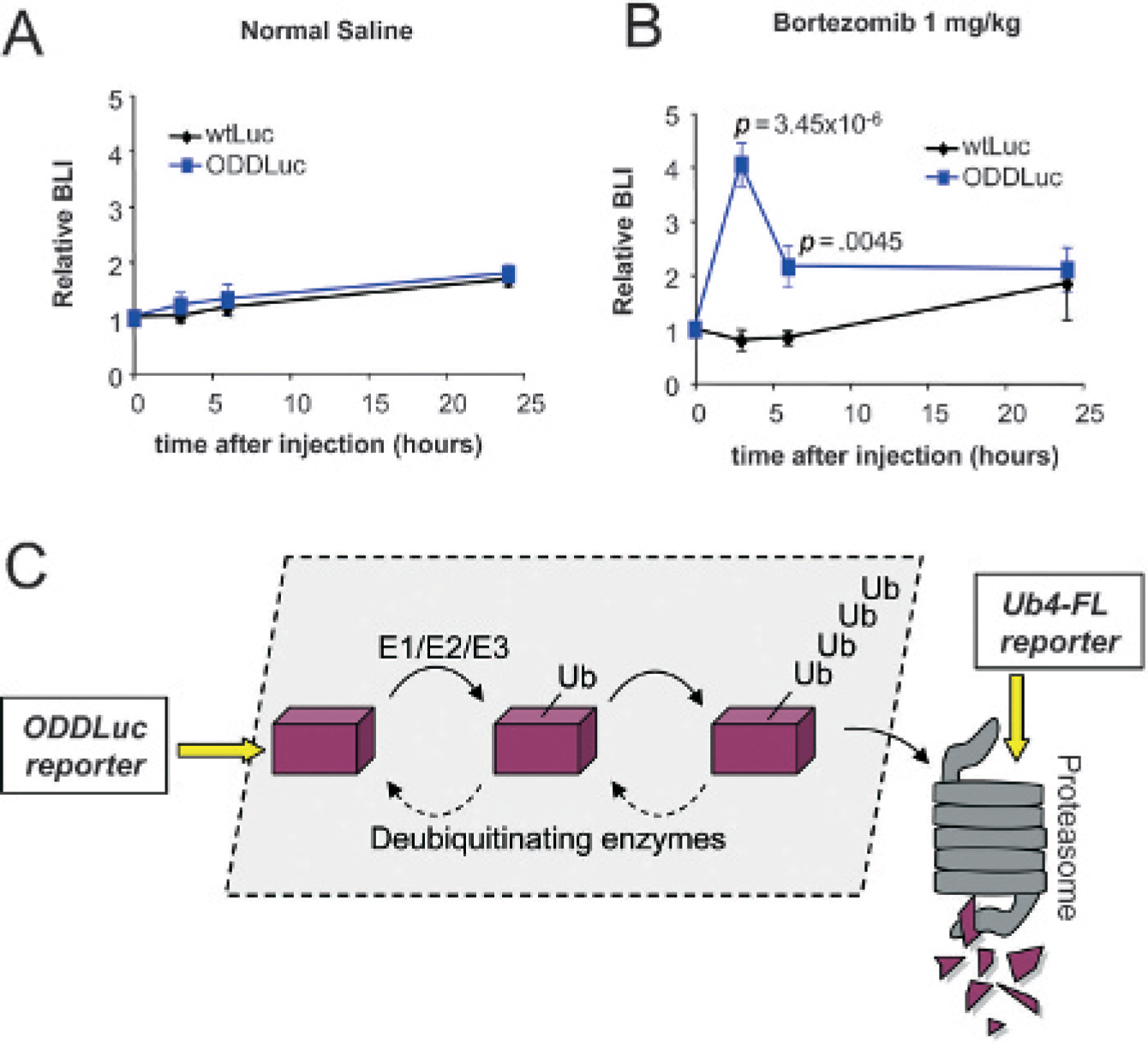
In vivo pharmacodynamics of proteasome inhibition with bortezomib. Dynamic imaging of the effects of a single dose of vehicle (A) or bortezomib (B) on bioluminescence intensity from wtLuc- or ODDLuc-expressing HCT116 tumors. Each point on the graphs represents the average peak bioluminescence intensity for eight mice, and error bars indicate the standard error of the mean. Significance was calculated by the Student t-test. C, Schematic of ubiquitin conjugation and proteasome-mediated degradation pathway. Positions where ODCLuc and Ub4-FL reporters enter this pathway are indicated by yellow arrows.
Conclusions
In this study, we demonstrated a novel application for the use of the ODD from HIF-1α as a surrogate marker of proteasome function. Fusion of ODD to luciferase allows for the rapid and sensitive detection of proteasome inhibition both in vitro and, importantly, in vivo in a xenograft tumor model. Owing to its shorter half-life, ODDLuc displayed faster kinetics and a more dynamic range in vitro compared with native luciferase or the previously characterized luciferase fusion to ODC.12, 13 In vivo, peak ODDLuc activity was apparent within 3 hours after injection of bortezomib, and normalization to wtLuc provides a good control for potential nonspecific effects since both reporters are expressed in the same context (identical promoter, selectable marker, and mCherry internal control).
The ODDLuc reporter was originally developed as a sensor of hypoxia dependent on the rapid proteasomal degradation under conditions of normal oxygenation. 19 22 For in vitro applications, cells are grown in ambient oxygen (21% O2), which is supraphysiologic compared with that found in normal tissue (3–5% O2). Given this, hypoxic stabilization of ODDLuc should not confound reporter activity. Destabilization of the ODDLuc reporter also requires the VHL protein. VHL is ubiquitously expressed in all cell types, except renal cell carcinoma cells, where mutation of VHL is a central determinant of transformation. 25 For in vivo applications, tumors are almost universally hypoxic owing to growth beyond the passive diffusional capacity of oxygen, disordered tumor vasculature, and impaired cardiopulmonary function in sick tumor-bearing mice. The blunted dynamic range of ODDLuc we observed in vivo (approximately 4-fold) compared with in vitro (approximately 15-fold) may in part be due to intratumoral heterogeneity in terms of oxygenation and partial stabilization of ODDLuc. However, using the ODDLuc reporter as a dynamic readout largely normalizes this confounding factor. Whereas the initial baseline reporter reading is influenced by the degree of tumor hypoxia, all subsequent readings are normalized to baseline. Changes that alter tumor oxygenation (eg, angiogenesis) take much longer to become physiologically relevant (days) than the acute time course of a pharmacodynamic study (hours). For this reason, immediate changes largely reflect alterations in proteasome activity rather than tumor oxygenation. Experimentally, our results demonstrate that even within hypoxic tumors, the ODDLuc reporter has sufficient dynamic range to read out proteasome inhibition.
Various types of degradation signals have been fused to reporter genes to assess proteasome function in previous studies. The most dynamic and sensitive of these is the ubiquitin firefly luciferase reporter (Ub4-FL) described by David Piwnica-Worms's group. 26 This reporter contains four ubiquitin molecules directly tethered to firefly luciferase, thus circumventing the need for a functional ubiquitin-conjugation system to direct its proteasome-mediated degradation. Additionally, by virtue of using a mutant form of ubiquitin, the ubiquitin chain on Ub4-FL is resistant to endogenous hydrolases that may cleave it off. As such, the Ub4-FL polypeptide is inherently primed for proteasome degradation and is a nearly direct readout of proteasome activity (Figure 4C). In contrast, the ODDLuc reporter requires an intact ubiquitin conjugating system, including E1/E2/E3 and polyubiquitinating activities prior to proteasome degradation (see Figure 4C). Although ODDLuc is a more indirect measure of proteasome activity, it may have utility in the phenotypic screening for compounds that impinge on the pathway leading to the proteasome. For example, recent studies have shown that targeting E3 ligases such as VHL or deubiquitinating enzymes such as UCH-LI may have clinical utility 27 29 and may act synergistically with proteasome inhibitors. Thus, the ODDLuc reporter may have utility in efforts to identify compounds that alter proteosomal degradation by targeting the ubiquitination machinery,27, 28 in addition to the 26S proteosome per se.
The ability of the ODD domain to independently promote protein degradation was first biochemically described over 10 years ago, 20 yet more recent studies have described its utility for in vivo reporter assays examining oxygen deficiency, HIF activity, and tumor biology.19,21,30 In this study, we showed that ODD also has utility for the in vitro and in vivo study of proteasome function when fused to a luciferase reporter. The outstanding dynamic range and rapidity of response of ODDLuc to proteasome inhibition make it well suited for high-throughput screening for proteasome inhibitors and/or inhibitors of the ubiquitin conjugation pathway. Likewise, its use as a sensor of proteasome activity in vivo provides a near real-time readout for assessing pharmacodynamic efficacy of novel proteasome inhibitors.
Footnotes
Acknowledgment
This work was supported by the Dana-Farber Cancer Institute and Novartis Drug Discovery Program (ALK) and the Dunkin Donuts Rising Stars Program (JEB).
Financial disclosure of authors and reviewers: None reported.