
Abstract
The purpose of this study was to track fluorophore-labeled, tumor-targeted natural killer (NK) cells to human prostate cancer xenografts with optical imaging (OI). NK-92-scFv(MOC31)-zeta cells targeted to the epithelial cell adhesion molecule (EpCAM) antigen on prostate cancer cells and nontargeted NK-92 parental cells were labeled with the near-infrared dye DiD (1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine). The fluorescence, viability, and cytotoxicity of the labeled cells were evaluated. Subsequently, 12 athymic rats with prostate cancer xenografts underwent OI scans before and up to 24 hours postinjection of DiD-labeled parental NK-92 cells or NK-92-scFv(MOC31)-zeta cells. The tumor fluorescence intensity was measured and compared between pre- and postinjection scans and between both groups using t-tests. OI data were confirmed with fluorescence microscopy. In vitro studies demonstrated a significant increase in the fluorescence of labeled cells compared with unlabeled controls, which persisted over a period of 24 hours without any significant change in the viability. In vivo studies demonstrated a significant increase in tumor fluorescence at 24 hours postinjection of tumor-targeted NK-92-scFv(MOC31)-zeta cells but not parental NK cells. Ex vivo OI scans and fluorescence microscopy confirmed a specific accumulation of NK-92-scFv(MOC31)-zeta cells but not parental NK cells in the tumors. Tumor-targeted NK-92-scFv(MOC31)-zeta cells could be tracked to prostate cancer xenografts with OI.
Recently, immunotherapies against prostate cancer have shown promising results.1–5 A new therapeutic approach is based on natural killer (NK) cells, which exert a high, selective cytotoxicity against tumor cells without toxicity against nonmalignant cells.3–5 Cancer therapy with NK cells can be performed by activation of endogenous NK cells through systemic administration of cytokines or by adoptive transfer of ex vivo expanded and activated autologous or donor-derived NK cells.5–10 The latter approach includes the use of cytotoxic NK cell lines that are suitable for clinical use. 11 Based on the high intrinsic cytotoxic activity of NK cells against a wide range of malignancies and the lack of toxicity, phase I/II clinical trials have been initiated.11–14
The efficacy of NK cell therapy in patients is currently evaluated by indirect measures, such as reduced tumor markers and improved survival of patients.11–14 Polymerase chain reaction-based monitoring of NK-92 kinetics in the peripheral blood of patients revealed that a large portion of NK-92-cells apparently leave the circulation within minutes after transfusion. 11 However, a homing of NK-92 cells to the site of the tumor could not be confirmed in clinical studies, and one could argue that an apparent tumor response could have been due to cytotoxic drugs, which had been given in conjunction with the NK cells.
New molecular imaging techniques that provide noninvasive in vivo cell tracking of NK cells could directly monitor the accumulation of NK cells in the tumor tissue15–17 and thus confirm or disprove one major requirement for a tumor response to this new therapy. In addition, a direct in vivo NK cell tracking technique could help further evaluate activating and inhibitory mechanisms on NK cell tumor accumulation and its cytotoxic activity. Various imaging techniques are available for NK cell tracking, such as single-photon emission computed tomography (SPECT), positron emission tomography (PET), optical imaging (OI), and magnetic resonance imaging (MRI).18–20 OI provides several advantages compared with other imaging techniques, such as absence of radiation exposure, high sensitivity, and quick and easy data acquisition, as well as being inexpensive.21,22 The purpose of our study was to develop an OI technique for in vivo tracking of fluorophore-labeled NK cells to human prostate cancer xenografts. This technique could be applied to diagnose an absence or presence of NK cell tumor accumulation as an imperative prerequisite for NK cell–mediated tumor cytotoxicity.
Material and Methods
NK Cells
The interleukin (IL)-2-dependent NK cell line NK-9211,23 (obtained from American Type Culture Collection [ATCC]) and the transduced NK-92-scFv(MOC31)-zeta cells 24 expressing a chimeric antigen receptor specific for the tumor-associated epithelial cell adhesion molecule (EpCAM) antigen were used in this study. NK-92-scFv(MOC31)-zeta cells were generated by retroviral transduction of NK-92 cells with a construct encoding a chimeric antigen receptor. This receptor consists of a humanized single-chain Fv (scFv) antibody fragment of EpCAM-specific antibody MOC31 for tumor cell recognition 25 (kindly provided by Uwe Zangemeister-Wittke, University of Berne, Switzerland), genetically fused via a flexible hinge region to transmembrane and intracellular domains of the CD3 zeta chain as a signaling moiety. An NK-92-scFv(MOC31)-zeta cell population with high surface expression of the chimeric antigen receptor was selected by sorting with magnetic beads as described previously.26,27
The NK-92-scFv(MOC31)-zeta cells were propagated in X-VIVO 10 medium (Lonza Biosciences, Walkersville, MD) supplemented with 5% heat-inactivated human serum (Lonza Biosciences), human IL-2 (100 units/mL, National Cancer Institute [NCI], Rocksville, MD), and G418 (0.6 mg/mL, InvivoGen, San Diego, CA). The NK-92 parental cells were propagated in RPMI 1640 (Sigma, St. Louis, MO) supplemented with fetal calf serum (10%), bovine calf serum (10%), 100× glutamine (1%), 100× nonessential amino acids (1%), 100 × Na pyruvate (1%), 100× penicillin-streptomycin (1%) (Sigma, St. Louis, MO, and Gibco, Carlsbad, CA) (all solutions prepared and distributed by the Cell Culture Facility, University of California-San Francisco [UCSF]), human IL-2 (100 units/mL, NCI, Rocksville, MD), and β-mercaptoethanol (0.005% of 200 mM stock solution, Sigma)
In Vitro EpCAM Expression by Flow Cytometry and NK Cytolytic Activity
A confluent T-75 flask of prostate cancer cell lines DU145 or PC3 cells was trypsinized, harvested cells were resuspended in Stain Buffer (catalog #554656, BD Pharmingen, Franklin Lakes, NJ), and 1.0 × 106 cells/sample was transferred to tubes for antibody staining. Fluorescein isothiocyanate (FITC) anti-EpCAM (catalog #347197, BD Pharmigen) or FITC mouse IgG1 k isotype control (catalog #555748, BD Pharmingen) was added to a final concentration of 0.5 μg/mL. Cells were incubated at 4°C for 30 minutes and centrifuged, and the supernatant with unbound primary was removed. Cells were resuspended in 500 μL of Stain Buffer, and 105 cells were analyzed with a cytometer (FACS Calibur, Beckton Dickinson, Franklin Lakes, NJ). Data analysis was performed with FlowJo version 7.2.4 (Tree Star Inc, Ashland, OR).
This was followed by a qualitative coincubation study to determine the cytolytic activity of NK-92-scFv(MOC31)-zeta cells against DU145 cells. DU145 cells were plated overnight at a density of 4 × 104 cells/well in a six-well plate. Either NK-92-scFv(MOC31)-zeta cells or NK-92 parental cells were added to each well at an effector to target ratio of 3:1. Triton-X 100 (0.1%) and DU145 culture media were added to the wells as positive and negative controls, respectively. The cells were then coincubated at 37°C in 5% CO2 for 16 hours. The study was performed in triplicate. The NK cell activity was determined by reassessment of the number of intact target cells with light microscopy at 1 hour and 16 hours after coincubation.
To confirm the specificity of cell killing, a fluorescence-activated cell sorting (FACS)-based cell killing experiment was performed as described by Muller and colleagues. 27 A single cell suspension of DU145 cells was prepared by trypsinization. Following trypsinization, the cells were incubated for 5 minutes at room temperature with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE) (Sigma, Deisenhofen, Germany), and then the target cells were washed twice with 15 mL of phosphate-buffered saline (PBS). Labeled DU145 cells (5 × 104 in 100 μL growth medium/tube) were seeded in polypropylene tubes before the addition of 100 μL/tube of medium to determine spontaneous lysis, or 100 μL of NK-92 or NK-92-scFv(MOC31)-zeta cells at an effector to target ratio of 10:1 in the presence of 15 μg/mL of murine monoclonal antibody MOC31 as a specific competitor or an isotype-matched control antibody. The tubes were incubated for 2 hours at 37°C. Cells were centrifuged for 5 minutes at 500g, supernatant was removed, and 200 μL/tube of a propidium iodide (PI) solution (1 μg/mL in PBS) was added. After incubation for 5 to 10 minutes at room temperature, fluorescence was determined using a FACScan. Specific cytotoxicity was calculated using CellQuestPro software (BD Biosciences, Bedford, MA). Dead target cells were determined as CFSE and PI positive. The number of spontaneously lysed target cells was subtracted.
Cell Labeling
NK-92 parental and NK-92-scFv(MOC31)-zeta cells were labeled with DiD (C67H103CIN2O3S; 1,1′-dioctadecyl-3,3,39,39 tetramethylindodicarbocyanine, vibrant cell labeling solution, Molecular Probes, OR), a nontargeted, lipophilic, carbocyanine near-infrared (NIR) fluorochrome with a molecular weight of 1052.08 Da and excitation and emission maximum of 644 and 665 nm, respectively. NK cells were incubated for different incubation time intervals with 5 μL of DiD dye/mL of serum-free media. The cells were then washed three times with PBS (pH 7.4) by sedimentation (5 minutes, 400 rcf, 25°C) and used for in vitro or in vivo studies.
In Vitro Study
In a pilot study, varying concentrations of DiD (0, 0.1, 0.2, 0.4, and 0.5 mL of DiD per 100 mL of solution) in different media were investigated with OI to determine the fluorescence of the dye, when it is not integrated with cells. Subsequently, triplicate samples with decreasing quantities of NK-92 parental and NK-92-scFv(MOC31) -zeta cells were incubated with 5 mL of DiD dye in 250 mL of Dulbecco's Modified Eagle's Medium (Sigma) for 5, 10, 15, and 20 minutes. The labeled and nonlabeled NK cells underwent OI scans starting at 2 hours up to 24 hours postlabeling. The cell samples subsequently underwent complementary fluorescence microscopy using a Nikon TE2000 microscope equipped with a Coolsnap HQ2 camera (filter sets, Cy5 and DIC; objective X40) to prove that the DiD dye was, in fact, integrated into the cells. Complementary trypan blue viability tests of the labeled cells and nonlabeled controls were obtained to prove that the labeled cells retained their viability.
Animal Model
This study was carried out in 12 6- to 8-week-old male athymic rats (Harlan Teklad, USA). The animals received subcutaneous implantations of DU145 (ATCC, Manassas, VA) human prostate cancer cells into the right lower flank or right abdominal wall. Tumor cell implantation was performed by injections of 5 × 106 tumor cells suspended in 250 μL of PBS mixed with 250 μL of Matrigel (BD Biosciences) using a 26-gauge needle and a 1 mL disposable syringe. When the tumors reached a diameter of about 1.2 cm (range 0.6–1.8 cm, average 1.2 cm), the rats were divided into two groups: six rats received intravenous injections of 15 × 106 DiD-labeled anti-EpCAM-directed NK-92-scFv(MOC31)-zeta cells and six rats received intravenous injections of 15 × 106 DiD-labeled nontargeted NK cells. All animals underwent OI before and after NK cell injection.
Optical Imaging
All OI studies were performed using the IVIS 50 small-animal imaging system (Xenogen, Alameda, CA) and Cy5.5 (excitation: 615–665 nm; emission: 695–770 nm passbands) filter set. For in vitro studies, cell samples were placed in a nonfluorescing black polystyrene 96-well plate. One week prior to OI, all rats were put on nonfluorescent chow free of alfalfa (Rodent diet 2014, Harlan Teklad), except one rat of the NK-92-scFv(MOC31)-zeta group, which accidentally received fluorescent food 2 days before the start of the imaging experiments. However, since the tumor of this animal was located in the lateral abdominal wall without overlap by bowel loops, this animal was not excluded from the study. For in vivo imaging, the rats were anesthetized with isoflurane and placed in a light-tight heated (37°C) chamber. The rats were imaged in two positions, with the anterior and left lateral abdomen facing the charge-coupled device camera. Identical illumination parameters (exposure time 2 seconds, lamp level high, filters Cy5.5 and Cy5.5bkg, f/stop 2, field of view 12 cm, binning 4) were selected for each acquisition. Grayscale reference photographic images were also obtained under low-level illumination. Rats were imaged before and at 1.5, 8, and 24 hours postinjection of the NK cells. At 24 hours, rats were sacrificed by an overdose of pentobarbital, followed by bilateral thoracotomy. The tumor and organs (liver, spleen, lungs, bone) were dissected and evaluated by ex vivo OI scans using the same parameters as for in vivo studies. The tumors were preserved for fluorescence microscopy.
Data Analysis
Optical images were analyzed using Living Image 2.5 software (Xenogen, Alameda, CA). All images were displayed and analyzed as an overlay of the fluorescent image on the photographic image. All images were measured in units of average efficiency and corrected for background tissue autofluorescence (ie, the fluorescent image was normalized by a stored reference image; thus, images are unitless). For in vitro image analysis, regions of interest (ROI) were defined as the circular area of the cell suspension. For in vivo image analysis, ROI were defined as an area encompassing the whole tumor. For ex vivo images, ROI were defined as an area encompassing the tumor and a representative portion of the organs. The ex vivo fluorescence data were analyzed as a ratio between the fluorescence intensity of the tumor and the fluorescence intensity of the various organs to compensate for potential interindividual variations in delivered cell or contrast agent quantity.
Confocal Microscopy
The tumors were explanted 24 hours after NK cell injection and preserved in tissue freezing medium (Tissue Tek OCT compound, Sakura Finetek, Torrance, CA) at — 80°C. Five-micrometer thick slides were processed for immunostaining. CD94 immunostaining (clone HI30, eBioscience, San Diego, CA), specific for human NK cells, was performed to visualize the NK cells in the tumor. The tumor nuclei were mounted with a mounting medium containing DAPI (Vectashield Mounting Medium with DAPI, Vector Laboratories, Burlingame, CA). Fluorescence microscopy was performed to visualize the DiD dye (red/Cy5 channel), FITC (green channel), and DAPI (blue channel) using a Zeiss LSM510 confocal microscopy system (Carl Zeiss GmbH, Germany) equipped with krypton-argon and ultraviolet lasers, and the images were acquired using LSM version 5 software (Carl Zeiss).
Statistical Analysis
A statistician at our institution performed the statistical analyses. Data were displayed as means ± standard deviations of each independent measurement. Statistical significance was assigned for p values #x003C; .05. All in vitro experiments were performed in triplicate. The results were tested for significant differences with the Student t-test. The power calculations were done as follows: the minimal number of animals needed in each experimental group, as estimated by our statistician by power calculations, is six, assuming a two-way analysis of variance for comparisons of experimental groups with a .05 significance level. A two-way analysis of variance for comparisons of experimental groups that received different NK cells (EpCAM targeted or nontargeted) will have 91% power to detect a variance among the two investigated NK cell populations by means of 25% of a standard deviation and will have 97% power to detect a variance among the presence or absence of leukocytes in the tumor tissue by means of 25% of a standard deviation, assuming that the common standard deviation is 1.000, when the sample size in each group is six. Six rats per group will have 80% power to detect differences of more than 1.5 SD. All of these assumed differences are far less than the expected differences.
Results
Efficient Cytolysis of EpCAM-Expressing DU145 Cells by NK-92-scFv(MOC31)-zeta Cells
NK-92-scFv(MOC31)-zeta cells express a chimeric antigen receptor that fuses an EpCAM-specific single-chain antibody to the transmembrane and intracellular domains of a receptor that activates a killing response. 24 To verify the presence of the EpCAM antigen in the prostate cancer cell lines used in this study, flow cytometry was used to measure cell surface EpCAM expression. The staining of both DU145 and PC3 cells with an EpCAM-specific antibody was significantly above an isotype control (Figure 1A). Furthermore, relative quantification showed that DU145 cells stained more intensely than PC3 cells (see Figure 1A). This finding is in agreement with the literature 28 and motivated the selection of DU145 cells for subsequent in vitro and in vivo studies.
Semiquantitative evaluations of NK cell cytotoxicity with light microscopy showed that NK-92-scFv(MOC31)-zeta cells effectively lysed the DU145 cells after 16 hours of coincubation with only cell fragments detectable by light microscopy and no intact target cells remaining (Figure 1B). In contrast, the parental NK-92 cells did not affect the viability of DU145 cells, and the number of intact target cells remained the same directly after and at 16 hours after coincubation (see Figure 1B). Both NK cell lines showed no cytolytic activity after 1 hour of coincubation (see Figure 1B). The positive and negative controls showed complete DU145 cell lysis and no cell lysis, respectively.
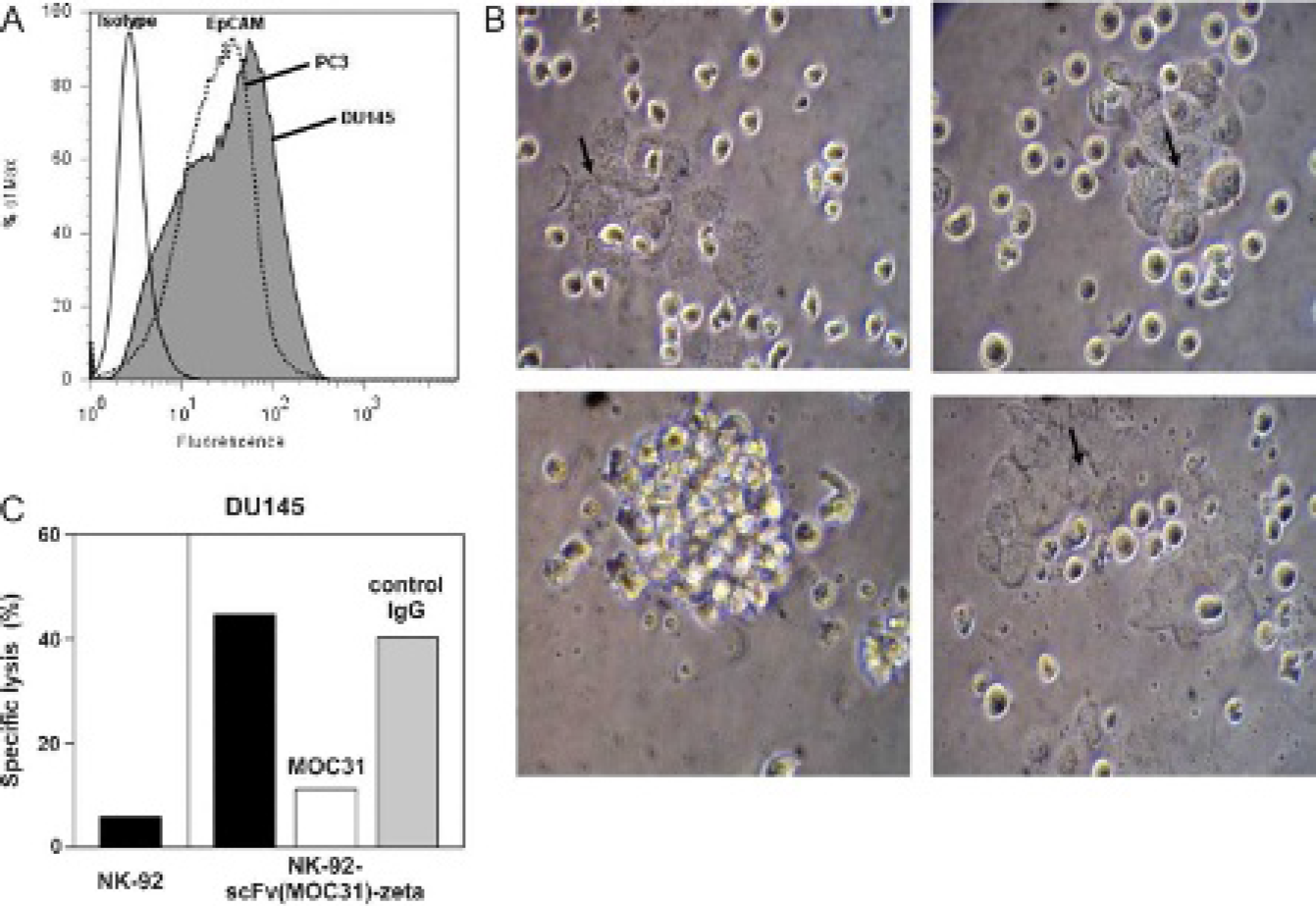
A, Flow cytometry analysis of epithelial cell adhesion molecule (EpCAM) expression in DU145 and PC3 cells. The solid, unfilled profile represents staining with an isotype control, whereas both the dashed, unfilled (PC3 cells) and solid, filled (DU145 cells) profiles represent staining with an EpCAM-specific antibody. Isotype controls for both DU145 and PC3 cells were performed and overlaid with one another (data not shown). The DU145 isotype control is shown for presentation. B, In vitro cytotoxicity study. Light microscopic evaluation shows early cytolysis of EpCAM-positive DU145 cells (arrows) by EpCAM-targeted NK-92-scFv(MOC31)-zeta cells at 1 hour after coincubation (top left) but effective cytolysis at 16 hours (bottom left). Conversely, nontargeted natural killer (NK) cells show minimal cytolysis at 1 hour (top right) and 16 hours (bottom right) after coincubation. Initial effector to target ratio 3:1; × 20 original magnification. C, Specificity of target cell recognition and cell killing. DU145 cells stained with CFSE were incubated with NK-92 or NK-92-scFv(MOC31)-zeta cells for 2 hours at an effector to target ratio of 10:1 in the absence of either competitor (filled bars)or 15 μg/mL MOC31 (open bar) or an irrelevant control antibody (shaded bar). Cells were stained with propidium iodide, and dead target cells were quantified as double positive cells by flow cytometry. EpCAM-positive DU145 cells are specifically lysed by NK-92-scFv(MOC31)-zeta cells.
To further confirm the specificity of cell killing by NK-92-scFv(MOC31)-zeta, EpCAM-expressing DU145 prostate carcinoma cells were stained with CFSE and incubated for 2 hours with NK cells at an effector to target ratio of 10:1 in the presence of parental MOC31 monoclonal antibody. Samples were analyzed for dead target cells by flow cytometry after staining with PI. As shown in Figure 1C, MOC31 antibody efficiently prevented EpCAM binding of the chimeric receptor and reduced specific lysis by NK-92-scFv(MOC31)-zeta from 45% to 11%, close to the level of cell killing by parental NK-92 (6% specific lysis). The addition of an irrelevant control antibody had no effect on NK-92-scFv(MOC31)-zeta activity (41% specific lysis).
In Vitro Studies
The pilot study showed no fluorescence of free DiD in media. However, DiD-labeled NK cells showed a marked fluorescence. Quantitative fluorescence signal intensities of labeled NK cells were significantly higher compared with unlabeled controls (p #x003C; .05; Figure 2A). There was a slightly higher fluorescence for NK cells incubated with DiD for 15 and 20 minutes compared with those incubated for 5 and 10 minutes, but this difference was not significant (p > .05). The fluorescence of DiD-labeled cells decreased with decreasing cell numbers (see Figure 2A). There was no evidence of quenching effects within the range of evaluated cell quantities. Follow-up OI scans showed that the fluorescent signal of DiD-labeled cells remained stable for 24 hours after labeling (see Figure 2A).

In vitro cell labeling study. A, Fluorescence signal intensity of decreasing quantities of natural killer (NK) cells that had been incubated with DiD for 15 minutes. Data are displayed as means of three samples in each group with standard deviation of the mean. *Significant difference between the fluorescence signal of labeled and unlabeled cells (p #x003C; .05). The fluorescence signal of labeled NK cells remaineds Table 24 hours after cell labeling. B, Comparison between NK92 parental and NK-92-scFv(MOC31)-zeta cells. Fluorescence signal intensity of decreasing quantities of NK-92 parental and NK-92-scFv(MOC31)-zeta cells that had been incubated with DiD for 15 minutes is compared at 2 hours postlabeling. The NK-92 cells were more efficiently labeled with the dye for the given period of incubation.
The fluorescence signal of the NK-92 parental and NK-92-scFv(MOC31)-zeta cells was analyzed and compared at 2 and 24 hours postlabeling. The NK 92 parental cells were noted to have a higher efficiency of labeling, as shown in Figure 2B. This is in accordance with previous studies by Daldrup-Link and colleagues, in which there was improved labeling of the parental cells compared with the genetically modified cell line. 17
Fluorescence microscopy confirmed that the dye was integrated into the cell membrane (image not shown). Viability tests with trypan blue showed that cell viability was preserved and that there were no significant differences between labeled and nonlabeled groups directly after labeling and at 24 hours after labeling (p #x003C; .05).
In Vivo and Ex Vivo Studies
Rats with implanted EpCAM-positive prostate cancers showed a marked tumor fluorescence on OI scans after intravenous injections of DiD-labeled anti-EpCAM-directed NK-92-scFv(MOC31)-zeta cells (Figure 3A). Corresponding quantitative fluorescence signal intensity data of the tumors were significantly higher on postinjection scans compared with preinjection scans at all investigated time points (p #x003C; .05) (Figure 3B). The tumor fluorescence increased on OI scans at 1.5 and 8 hours and remained subsequently stable on 24-hour scans.
On the other hand, all rats that received DiD-labeled nontargeted NK parental cells remained nonfluorescent (see Figure 3A). There was no significant increase in fluorescence signal of the tumors on postinjection scans compared with preinjection scans at any time (p > .05) (Figure 3B).
When comparing tumor fluorescence data after injection of tumor-targeted and nontargeted NK cells, the tumor fluorescence after injection of NK-92-scFv(MOC31)-zeta cells was significantly higher compared with the tumor fluorescence after injection of nontargeted NK parental cells (p #x003C; .05).
Ex vivo OI confirmed that the fluorescence intensity of the tumors, normalized to the fluorescence of various organs (liver, spleen, lungs, and sternum), was significantly higher in the NK-92-scFv(MOC31)-zeta group compared with the NK parental group (p #x003C; .05, n = 6 in each group) (Figure 4).
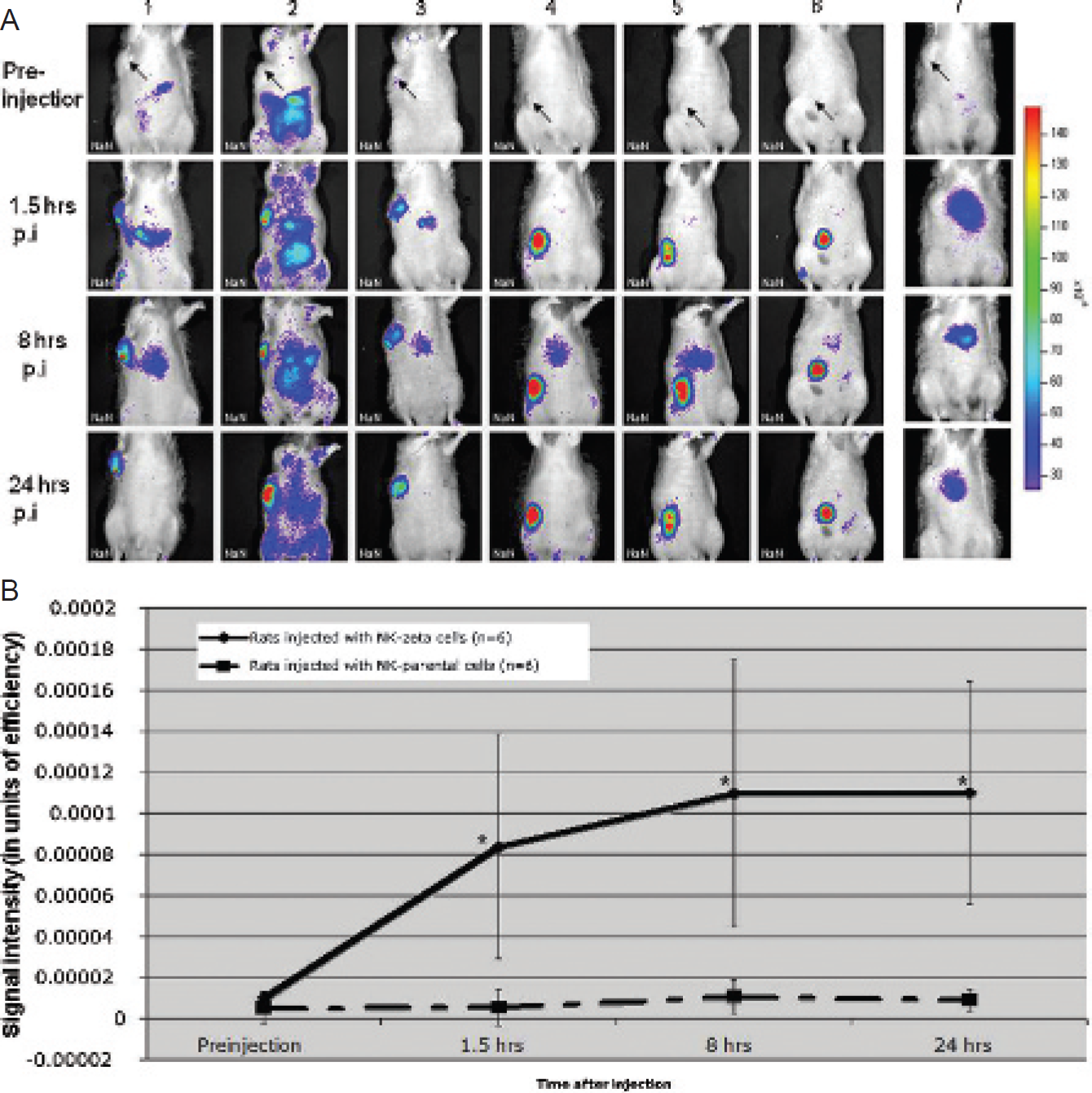
A, In vivo natural killer (NK) cell tracking. Optical imaging scans of rats with EpCAM-positive DU145 prostate cancer xenografts (arrow) before and at various time points after intravenous injection of EpCAM-targetedNK-92-scFv(MOC31)-zetacells (rats 1–6) show a progressive increase in fluorescence signal of the tumors at 1.5 and 8 hours postinjection, followed by a stable signal at 24 hours postinjection. Conversely, control animals injected with NK parental cells did not show any significant change in tumor fluorescence (rat 7, representative image). B, Quantitative in vivo analysis confirms a significant increase in tumor fluorescence signal after injection of NK-92-scFv (MOC31)-zeta cells compared with preinjection scans. The fluorescence signal of control tumors remains unchanged after injection of parental NK cells. Data are displayed as means of six animals in each group with standard deviations. *Significant difference between the fluorescence signal of animals that received NK-92-scFv(MOC31)-zeta cells or NK-92 parental cells (p #x003C; .05).
Fluorescence Microscopy
Fluorescence microscopy confirmed that the NK-92-scFv(MOC31)-zeta cells accumulated in the tumors, whereas the NK parental cells did not (Figure 5). DiD-labeled NK-92-scFv(MOC31)-zeta cells could be visualized in the Cy5 (red) channel by fluorescence microscopy. This was also confirmed by FITC-conjugated anti-CD94 staining specific for human NK cells (green channel). Superimposing the Cy5, FITC, and DAPI (for the nuclei; blue) channels confirms the stability of the DiD dye in the NK cell membrane and accumulation of the NK-92-scFv(MOC31)-zeta cells in the tumor at 24 hours postinjection. The control group did not show any fluorescence in the Cy5 and FITC channels, confirming the absence of NK parental cells (see Figure 5).
Discussion
Our data demonstrate that genetically engineered NK cells can be labeled with the lipophilic NIR fluorochrome DiD without affecting cell viability and that the accumulation of fluorochrome-labeled NK cells in prostate cancers can be tracked in vivo with OI. Fluorochrome-labeled NK-92-scFv(MOC31)-zeta cells that express a chimeric receptor against the EpCAM antigen could be tracked to EpCAM-expressing prostate cancers after their intravenous injection into athymic nude rats. Conversely, fluorochrome-labeled nontargeted NK-92 cells could not be tracked to EpCAM-positive prostate cancers.
Tumor therapies with NK cells are extensively investigated in preclinical and initial clinical trials because NK cells provide a well-defined target specificity, a lack of inhibitory receptors, highly effective cytotoxicity against malignant cells, and low toxicity against nonmalignant cells of normal organs.11,23 However, the level of tumor cytotoxicity has been reported to vary with different NK cell populations and different tumor types. 11 The described noninvasive NK cell tracking technique could be applied for preclinical assessments of these new immunotherapies and help improve the design of related clinical trials.
To the best of our knowledge, our experiments describe for the first time an in vivo trafficking of fluorochrome-labeled NK cells to tumors by fluorescence reflectance imaging. Fluorescence imaging has the following advantages over other modalities: it is very sensitive, inexpensive, fast, and not associated with radiation exposure and allows longitudinal studies.29,30 Other investigators used bioluminescent imaging for NK cell tracking.31,32 Luc-expressing cytokine-induced killer cells have been tracked to subcutaneous tumors by bioluminescent imaging. 33 This technique provides a higher sensitivity compared with fluorescence imaging but required more expensive transduction methods. In addition, the applied fusion proteins may also be associated with a risk of immunogenicity. MRI was recently used to track genetically engineered NK-92-scFv(FRP5)-zeta cells to HER2/neu-positive breast cancers. 17 MRI has the advantage of a high anatomic resolution and superior soft tissue contrast but the disadvantages of a limited sensitivity and being an expensive modality. Techniques to track indium 111-oxine-labeled leukocytes are established in clinical practice, mainly for the purpose of detecting inflammation. 34 Although in principle readily clinically available, these techniques have not been applied for NK cell tracking. PET and SPECT/computed tomography (CT) have been applied to track other immune cells, such as T lymphocytes, dendritic cells, and monoctyes, to fibrosarcoma, glioma, lymphoma, and colon carcinoma.19,20,35 These techniques have the advantage of being readily clinically applicable; however, they have the disadvantages of being expensive and associated with radiation exposure and decay of the label.

A, Ex vivo optical imaging (OI) study. OI of explanted tumors and organs of two representative animals at 24 hours after NK-92-scFv(MOC31)-zeta cell injection (upper panel) or NK-92 cell injection (lower panel). Following NK-92-scFv(MOC31)-zeta cell injection, a marked fluorescence of the tumor was noted, which was higher in intensity compared with the explanted organs. Following NK-92 cell injection, the tumor did not show an increased fluorescence. Li = liver; Lu = lungs; Sp = spleen; St = sternum; T = tumor. B, Quantitative ratio of tumor to organ fluorescence at 24 hours postinjection of NK-92-scFv(MOC31)-zeta cells (black bars)or NK-92 cells (grey bars). Data are expressed as means and standard deviations of six animals in each group. *Significant difference between the fluorescence signal of animals that received NK-92-scFv(MOC31)-zeta cells or control cells (p #x003C; .05).
Various techniques for labeling of immune cells have been studied in the past. Our described protocol provides effective NK cell labeling by simple incubation with a commercially available NIR lipophilic dye for a few minutes. Our technique is less complicated and less expensive than previously applied labeling techniques with radionuclides,36–38 magnetic resonance agents,17,39,40 bioluminescent labels,41,42 or reporter genes.31,43,44 Bioluminescent labels and reporter genes have the advantage that the label is not diluted or excreted over time, whereas exogenous labels have the advantage of being closer to potential clinical applications.
Adoptive immunotherapies provide a new treatment option for patients with hormone-refractory prostate cancer, which is very difficult to control and impossible to cure with standard chemotherapy regimens. To direct NK cells to prostate cancers, these immune cells need to be targeted to unique antigens on prostate cancer cells. Various approaches of adoptive immunotherapies and EpCAM-targeted antibody therapies have been previously investigated in prostate cancers.23,45–48 However, to the best of our knowledge, ours is the first study that combines these two approaches.
Prostate cancer immunotherapies comprise cellular immunotherapy with CD8+ T lymphocytes modified to target prostate-associated antigens 45 and dendritic cell-mediated vaccination therapy.46,47 Recently, natural cytotoxic receptors fused with human IgG1 known as NKp30-Ig have been used as “targeted missiles” for the treatment of prostate cancer cell lines DU145 and PC3 in vivo. 48 The toxicity of NK cells against prostate cancer has not been studied extensively before. The NK-92 cell line investigated in our study has been previously used for adoptive immunotherapy of breast carcinoma, ovarian carcinoma, myeloma, and leukemia but not prostate cancer.23,26,27 NK cell therapy has the advantage over other immunotherapy approaches with cytotoxic T cells and targeted antibodies of providing a stronger cytotoxic effect and is not immunogenic. 11
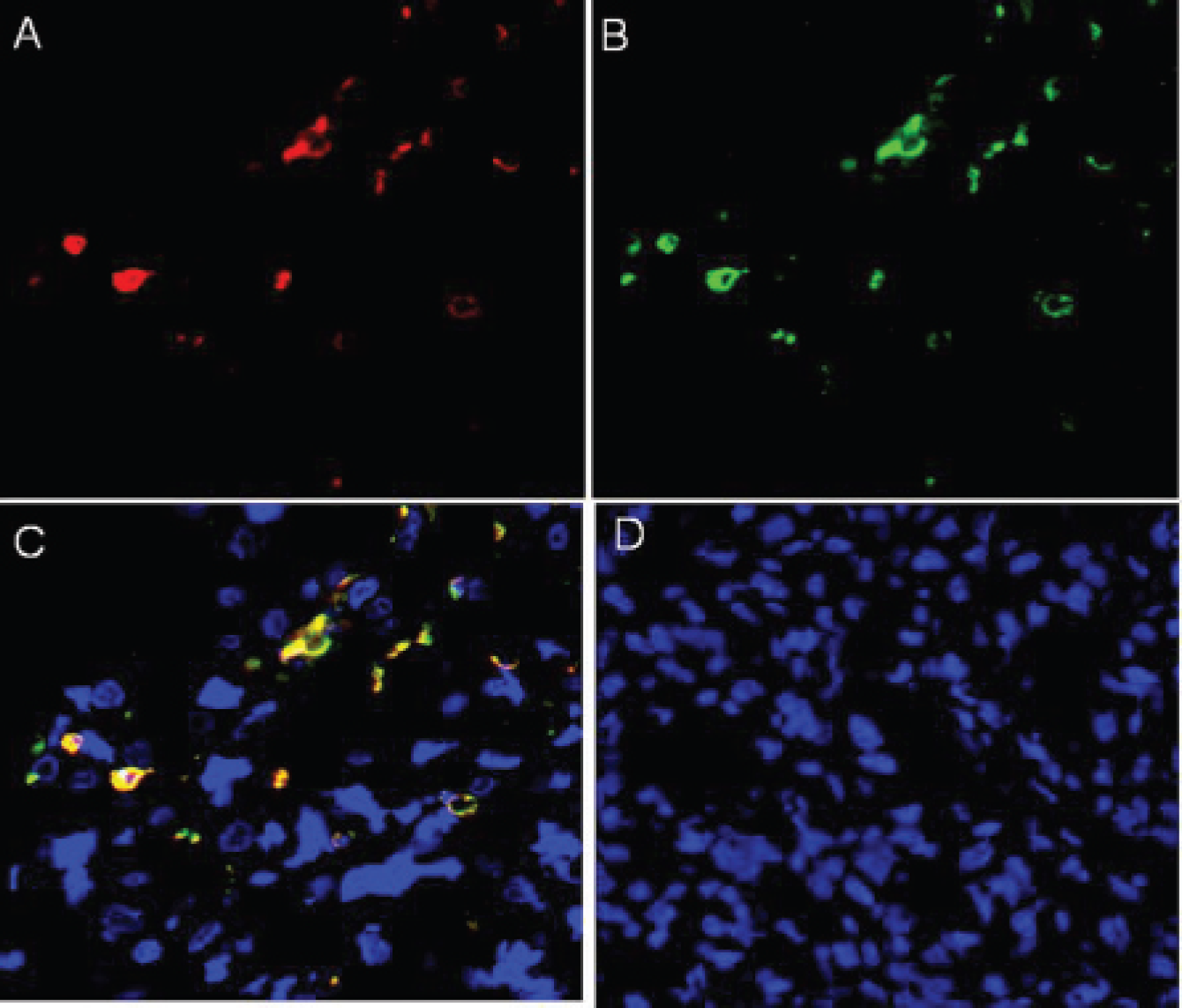
Confocal microscopy of a representative tumor at 24 hours after injection of NK-92-scFv(MOC31)-zeta cells (A–C) or control NK-92 cells (D). DiD-labeled NK-92-scFv(MOC31)-zeta cells are visualized in the tumor tissue with the Cy5 (red) channel (A). Anti-CD94 immunostaining conjugated with FITC confirmed the presence of NK-92-scFv(MOC31)-zeta cells in the FITC (green) channel (B). Colocalized images of DiD- and FITC-labeled NK-92-scFv(MOC31)-zeta confirm that the DiD-containing cells are indeed the applied NK-92-scFv(MOC31)-zeta cells (C, red and green channels). The tumor from the control group did not show any NK cell accumulation (D, red and green channels). (Blue = DAPI staining nuclei in all figures; X40 original magnification.)
The EpCAM antigen has become a major target for prostate cancer immunotherapy because about 87% of human prostate cancers show high levels of EpCAM expression, individual tumor cells in a given tumor show a high quantity of EpCAM antigens, EpCAM-negative prostate cancers are rare, and EpCAM expression does not vary significantly with different tumor grades or histopathologies. 49 EpCAM is expressed in different epithelial tissues such as the pancreas (ductal and acinar epithelial cells), jejunum (epithelial cells of villi and crypts), colon (epithelial cells of crypts), kidney (epithelial cells of collecting ducts, Henle loops, and distal tubulus), and salivary glands. Therefore, it cannot be excluded that normal tissues can be affected by retargeted NK cells. We would also like to emphasize here that EpCAM-specific antibodies have been tried in humans with acceptable toxicities. Anti-EpCAM Ag immunotherapy has reached phase II clinical trials in patients with prostate cancers. Recent therapy protocols include ING-1, 50 adecatumumab51,52 and edrecolomab, 53 as well as an anti-EpCAM-directed immunotoxin.25,54 An EpCAM transgenic mouse tumor model has also been described to study the effects of anti-EpCAM-directed immunotherapy. 55
In our study, we tracked combined NK cell immunotherapy and EpCAM-directed therapy by investigating genetically engineered NK-92-scFv(MOC31)-zeta cells that express a chimeric antigen receptor specific for the EpCAM antigen. 24 This immunotherapy approach with genetically engineered, tumor-targeted NK cells provided a highly efficient tumor cell lysis in other types of cancers, such as breast cancers, ovarian cancers, leukemia, and myeloma. 23 Our cytotoxicity studies showed an effective cytolysis of EpCAM-expressing DU145 prostate cancer cells by EpCAM-targeted NK-92-scFv(MOC31)-zeta cells. Additional quantitative europium release cytotoxicity assays by Uherek and colleagues confirmed that NK-92-scFv(MOC31)-zeta cells specifically and efficiently lysed EpCAM-expressing tumor cells, even at low effector to target ratios. 24 We performed flow cytometry–based cytotoxicitiy tests and confirmed the specific and efficient killing of DU145 cells by NK-92-scFv(MOC31)-zeta cells (see Figure 1C). Control experiments showed that the same EpCAM-expressing tumor cells (DU145) were resistant to parental non-EpCAM-targeted NK-92 cells and that both NK-92-scFv(MOC31)-zeta and NK-92 were not cytotoxic against EpCAM-negative targets. 24
The benefit of retargeted NK cells is that they can be expected to kill their targets independently from endogenous immune effector mechanisms; that is, they are NK cells with built-in antibody-dependent cell-mediated cytotoxicity (ADCC) activity. No components of the host immune system are required for effective activity. Hence, in this regard, at least initially, the cells should act in immunocompetent hosts (and humans) in a manner similar to that in immunodeficient animals. Nevertheless, we would stress that if NK-92 are to be used as allogenic cells in humans (or xenogenic cells in immunocompetent animals), antibody and T-cell responses of the host immune system may develop against the NK cells and limit the time period in which they can be used. This needs to be addressed in future studies in immunocompetent animals and in clinical trials in humans
We recognize several limitations of our study:
We evaluated a small group of 12 animals because power calculations determined a required minimal number of 6 animals per experimental group to confirm a superior tumor recognition of NK-92-scFv(MOC31)-zeta cells compared with NK-92 cells with our OI technique. Further studies in larger cohorts may evaluate the extent of interindividual and intraindividual variations of the acquired data.
We used the human DU145 prostate cancer cell line for our tumor model because this cell line is characterized by high EpCAM expression and can be grown as xenografts in athymic nude rats. The efficacy of our immunotherapy and imaging technique has to be confirmed in other human prostate cancers with higher (eg, LnCAP cell line) or lower (eg, PC3) levels of EpCAM expression. 28
3. We evaluated human prostate cancers and human NK cells in a xenograft animal model. Although in principle clinically applicable, our imaging technique faces several challenges for translational applications:
The fluorochrome DiD is not approved by the Food and Drug Administration (FDA). We chose this NIR dye because it provided easy and quick cell labeling without interference with NK cell function. In addition, the DiD dye could be visualized with the Cy5 filter of our fluorescence microscope and thereby allowed histopathologic confirmations of our imaging findings. An alternative FDA-approved fluorescent dye, which could be used for NK cell labeling in a clinical setting, is indocyanine (ICG). However, the high excitation and emission wavelength of ICG in the range of 710 to 750 nm and 810 to 875 nm, respectively, restricts comparative investigations with standard fluorescence microscopy, which usually operates at lower wavelengths.
Another constraint for immediate clinical applications of our technique is the limited penetration depth of fluorescence reflectance imaging. Recent developments of tomographic fluorescence systems (fluorescence molecular tomography [FMT], FMT-CT, FMT-MRI, fluorescence protein tomography) that are capable of three-dimensional reconstructions may overcome these technical limitations.
Noninvasive imaging techniques for in vivo depiction and tracking of NK cells are crucial for the development of new immunotherapies for cancer treatment. Prostate cancers are frequent, EpCAM-positive tumors with a poor response to conventional chemotherapy and a critical need for new treatment options, such as the presented NK cell therapy. Our data show that OI can diagnose the presence and time point of NK cell accumulation in prostate cancers. Potential applications of this OI-based NK cell tracking technique comprise preclinical treatment protocol optimizations, such as (a) comparative investigations of the in vivo distribution and tumor accumulation of different NK cell subtypes; (b) comparisons between NK cells and other immune cells, such as dendritic cells or T cells; and (c) investigations of the efficacy of these immune cells against various different tumor types, such as androgen receptor–positive and -negative tumors, as well as assessment of efficacy of different combination therapies. Further technical developments, such as handheld OI devices, fluorescence tomographic systems, and intraoperative/endoscopic OI devices, may facilitate translational clinical applications.
Footnotes
Acknowledgments
Financial disclosure of authors: This study was supported by a Prostate Cancer Foundation grant.
We thank Ms. Annemarie Schimpf for help with NK-92-scFv(MOC31)-zeta cell culture and analysis; Nikon Imaging Center, UCSF, for fluorescence microscopy equipment for the in vitro study; and the NCI for providing us with IL-2.
Financial disclosure of reviewers: None reported.