
Abstract
The use of phage-displayed peptide libraries is a powerful method for selecting peptides with desired binding properties. However, the validation and prioritization of “hits” obtained from this screening approach remains challenging. Here, we describe the development and testing of a new analysis method to identify and display hits from phage-display experiments and high-throughput enzyme-linked immunosorbent assay screens. We test the method using a phage screen against activated macrophages to develop imaging agents with higher specificity for active disease processes. The new methodology should be useful in identifying phage hits and is extendable to other library screening methods such as small-molecule and nanoparticle libraries.
Introduction
Macrophages and monocytes play key roles in development, maintenance, and progression of most disease processes. For example, in cancer, macrophages are involved in host response [1,2], are responsible for clearing apoptotic tumoral cells [3], and play a key role in subversion of adaptive immunity, thereby promoting tumor growth and progression [4]. In atherosclerosis, monocytes are recruited into plaques by specific molecular stimuli, become activated, engulf oxidized lipids, and then secrete proinflammatory cytokines, amplifying the inflammatory response [5]. Indeed, activated macrophages have a critical role in plaque rupture by producing matrix metalloproteinases that degrade the extracellular matrix that stabilizes the plaque. In rheumatoid arthritis, macrophages engulf apoptotic cells and produce proinflammatory cytokines and chemokines that contribute to bone and cartilage destruction [6]. Given their fundamental role in host response and importance for disease progression, macrophages have been exploited as an imaging target in the aforementioned diseases, primarily using nanoparticles [7,8], proteins (annexin V) [9], and other polymeric constructs [10]. Some of these approaches have been useful for detecting tumor infiltration in glioma [11], initiation of insulitis [12], nodal staging in cancer [13], and assessment of therapeutic efficacy involving macrophages, among others.
Despite these recent advances, an underexplored concept has been the development of imaging ligands that are able to differentiate between resting macrophages and those activated by stimuli such as granulocyte-macrophage colony-stimulating factor (GMCSF), oxidized low-density lipoprotein (oxLDL), or lipopolysaccharide (LPS). Such imaging agents would be highly useful in phenotyping disease more accurately and in providing clinically relevant information (e.g., discrimination of vulnerable vs. stable atherosclerotic plaques). Given the general unavailability of such imaging agents, we set out to develop ligands that could serve as discriminators between resting and activated macrophages.
Phage display technology has been used extensively to develop novel imaging agents [14–16] and was therefore considered as a primary development tool for the current research. However, the rapid identification and validation of “hits” from a selection process often remains a technical challenge because most screens yield 10–50 of such hits. The primary goal of this research was to develop and validate a computational approach to display data from high-throughput enzyme-linked immunosorbent assay (ELISA) screens to visualize display affinity and specificity for identified peptides. Applying this approach, we show here the discovery and validation of two novel peptide sequences specific for activated macrophages.
Materials and Methods
Cell Culture Methods and Macrophage Stimulation
U937 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and passaged according to the distributor's instructions. To promote macrophage formation, phorbol 12-myristate 13-acetate (PMA) was added to cell culture media prior to plating cells in gelatin-coated microtiter plates. PMA-treated U937 cells were stimulated (“activated”) by direct addition to culture media of 1 µL/mL of an aqueous solution of 50.0 mg/mL of LPS (Sigma-Aldrich, St. Louis, MO) 24 hr before use.
Phage Selection and Counterselection
Phage selection and counterselection were performed using macrophages derived from PMA treatment of the human monocytic cell line U937, either activated with LPS or treated with vehicle. A total of 1010 plaqueforming units (PFU) of phage, displaying random disulfide-constrained seven-amino-acid peptides (C7CPhD, New England Biolabs, Beverly, MA), were incubated with LPS-activated U937 cells for 1 hr at 37°C to allow time for internalization. Extracellular-restricted phage particles were removed with 0.2 M glycine (pH 2.2) in three 8-min washes. Internalized phage particles were recovered by lysing cells with 0.1% triethanolamine (Sigma Chemical Company, St. Louis, MO) in phosphate-buffered saline (PBS, pH 7.4) for 4 min at room temperature. Cell extracts were neutralized with 500 µL of 0.5 M Tris-HCl (pH 9.0). Subtractive counterselection was performed by exposing initially internalized isolates to three successive rounds of incubation for 30 min at 37°C with monolayers of nonactivated U937 cells, followed by separation of internalized and extracellular phage particles as described above. Phage particles that were internalized by LPS-activated U937 cells but not by nonactivated U937 cells were amplified in Escherichia coli, retitered, and subjected to three additional rounds of positive selection using LPS-activated U937 cells, after which individual phage clones were selected for sequence determination and ELISAs.
Enzyme-linked Immunosorbent Assay of Phage Clones
Amplified and purified stocks of individual phage clones (109 PFU) in 1× Dulbecco's phosphate-buffered saline (DPBS) with Ca2+ and Mg2+ (Cambrex BioSciences; Walkersville, MD), supplemented with 0.1% (w/v) bovine serum albumin (BSA, Sigma-Aldrich), were incubated with U937 cells at 37°C for 1–2 hr. After incubation, cells were washed five times with 1× DPBS containing 0.1% BSA and 0.05% (v/v) Tween 20 (Aldrich, Milwaukee, WI) and then once with DPBS containing 0.1% BSA to remove noninternalized phage. Cells and internalized phage were fixed in 2.0% paraformaldehyde (Fisher Scientific, Fairlawn, NJ) for 5 min and then permeabilized with 0.1% (v/v) Nonidet-P40 (NP-40) in DPBS containing 0.1% BSA. Permeabilized cells were blocked for 30 min at room temperature with 1.0% (v/v) normal goat serum (NGS, Vector Laboratories, Burlingame, CA) and 1.0% (v/v) normal horse serum (NHS, Vector Laboratories) in DPBS containing 0.1% BSA. After blocking, cells were incubated for 1 hr at room temperature with 0.15 µg/mL anti-M13-biotin (ExAlpha, Maynard, MA). Cells were then washed three times with DPBS containing 0.1% BSA and incubated for 30 min with streptavidin-horseradish peroxidase conjugate (Amersham Life Sciences, Piscataway, NJ), followed by three additional washes with DPBS containing 0.1% BSA. One hundred microliters of tetramethylbenzidine (Sigma-Aldrich) was added to each well of microtiter plates, which were incubated 30 min at room temperature before reading absorbance at 650 nm on a Molecular Devices (Sunnyvale, CA) Emax ELISA microtiter plate reader.
Multidimensional Analysis of Screening Data
Raw plate reader outputs corresponding to LPS-treated or -untreated U937 cells and consisting of ordered 8× 12 arrays of values corresponding to each 96-well plate were unpivoted to afford a denormalized table of values. Each well position was then associated with similar arrays of metadata labels encoding the experimental design, namely, the phage clone sample number (or a “mock” label to indicate the absence of a phage clone) and phage clone sequence, if known. Values from each well were background-subtracted using the median value of mock-treatment wells from each assay plate. Background-subtracted (Bsub) values for mock-treatment wells were accumulated across multiple assay plates to afford two mock-treatment distributions reflecting assay noise, one corresponding to LPS-treated cells and one corresponding to LPS-untreated cells, and trimmed according to Chauvenet's criterion as previously described [17]. These mock-treatment distributions were used to normalize independently each value corresponding to a phage-treated well, affording Z-normalized (Znorm) values for each well. All data formatting, manipulation, and normalization was implemented using Pipeline Pilot (Scitegic, Inc., San Diego, CA), sequence similarity analysis of phage clones was performed using the Jotun-Hein similarity method implemented in MegAlign (DNAStar, Inc., Madison, WI), and data visualizations were prepared using DecisionSite (Spotfire, Inc., Somerville, MA).
Immunocytochemistry of Phage-display Peptides
U937 cells treated with either PMA alone or PMA plus LPS were plated on glass slides (Nalge Nunc International, Rochester, NY) and incubated with PTAPTST-, PAPRSGS-, or PAIAHRT-displaying phage for 1 hr at 37°C. After incubation, cells were washed, fixed with 2% paraformaldehyde, and permeabilized with 0.1% NP40. Fixed, permeabilized cells were blocked with 3% NGS, 1% BSA, and 0.1% Triton X-100 in PBS, then incubated with 0.15 µg/mL anti-M13-biotin (ExAlpha) for 1 hr at room temperature. Next, cells were washed and incubated with streptavidin-fluorescein isothiocyanate (FITC) (1:1000 dilution) for 30 min at room temperature followed by a final wash. Slides were mounted using VectaShield (Vector Laboratories) containing 4′,6-diamidino-2-phenylindole (DAPI) or without DAPI and visualized by fluorescence microscopy or bright-field/fluorescence for inset using a Nikon Eclipse TE2000-S Insight QE (MVI, Avon, MA) equipped with a 40× objective.
Quantitation of Phage Clone Uptake by Fluorescence Plate Reader
Phage were FITC-labeled as previously described [18], with the following modifications: a solution of 1 × 1012 PFU of phage was resuspended in 100 µL of a 0.3 M NaHCO3 solution containing 3 mg/mL of FITC. The phage/FITC reaction was allowed to continue for 1 hr at 4°C in the dark. Subsequent to incubation, the volume of the labeled phage was brought up to 1 mL with DPBS and then the phage were purified by polyethylene glycol precipitation. The FITC-labeled phage were then resuspended in 200 µL of DPBS. FITC-labeled phage were incubated with U937 + LPS-treated cells or U937 cells for 1 hr at 37°C. Following incubation, cells were washed six times with DPBS+ then analyzed for FITC-phage cellular mediated uptake via fluorescence plate reader (Spectramax GeminiXS, Molecular Devices).
Results
Selection of Phage-displayed Peptides Specific for Activated Macrophages
U937 cells are monocyte-like hematopoietic precursor cells that can be induced to differentiate into phagocytic macrophages by stimulation with phorbol esters [19]. Further treatment of these cells with the bacterial endotoxin LPS, oxLDL, or GMCSF results in a population of activated macrophages [20]. The ability to chemically manipulate the activation status of these cells makes them ideal candidates for a phage-display selection approach to identify peptides specific for activated versus nonactivated macrophages. In the current study, we used LPS as a stimulant because it mimics tissue infections and because other preliminary data had shown similar macrophage response to varied stimuli (data not shown). We thus incubated LPS-activated U937 cells with 1011 PFU of phage, corresponding to an average of 100 copies of each unique peptide sequence in the linear random 7-mer phage-display library, C7CPhD (New England Biolabs). To select for peptide sequences that mediate phage internalization, cells pre-incubated with phage were washed with glycine buffer at low pH to elute noninternalizing phage. Peptides capable of cell-mediated internalization are desirable because they chaperone and trap imaging agents inside the cells, resulting in increased target-to-background ratios. To deplete phage that bound to markers present on both LPS-activated and nonactivated U937 cells, the phage pool isolated after one round of selection was subjected to three rounds of subtraction on macrophages (U937 cells + PMA) to remove from the phage library peptides that bind to or are nonspecifically internalized by these cells regardless of the macrophage activation status. Phage remaining after subtraction were amplified and applied to activated macrophages three more times, for a total of four rounds of selection. Phage selection and subtraction procedures resulted in a 100-fold increase in the ratio of cell-internalized phage particles to extracellular phage particles (Figure 1).
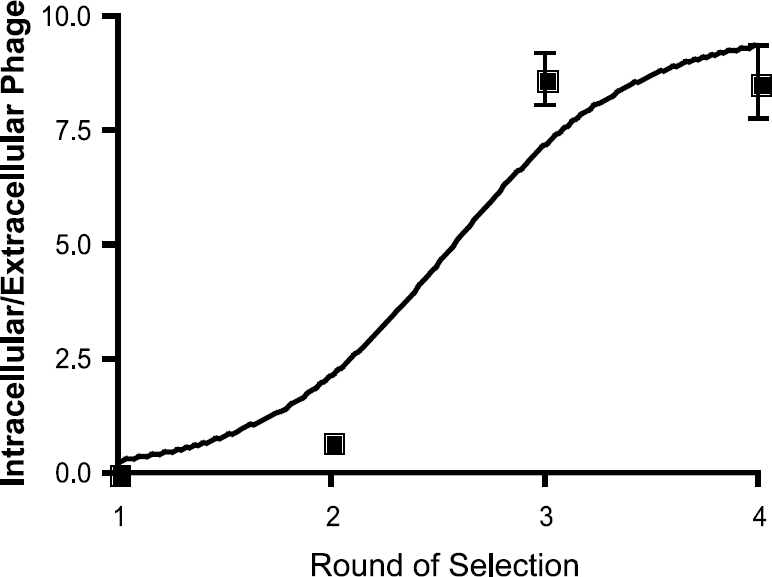
Selection of phage-display library targeting LPS-activated U937 cells. The ratio of intracellular phage to extracellular phage increased >100-fold after four rounds of iterative selection on LPS-activated U937 cells.
Prioritization of Screening Results by Multidimensional Data Analysis
One of the difficulties associated with phage-display screening is to validate which clones are true hits. Analyzing all selected phage clones would be costly in terms of time and resources. Conversely, if few phage clones are analyzed, the chances of obtaining a clone with the correct binding properties are diminished. To balance these competing limitations, we developed a cell-based, high-throughput screen based on ELISA coupled with multidimensional data analysis to enable the facile analysis of large numbers of phage clones. Plaques (N = 50) corresponding to individual peptide sequences were picked and amplified for use in cell-based ELISAs. Individual phage clones were exposed either to PMA-treated U937 cells (resting macrophages) or PMA/LPS-treated U937 cell (activated macrophages), which were then fixed and permeabilized to allow immunodetection of internalized phage particles.
We assessed the affinities and specificities of internalization using background-subtracted and Z-normalized scores (see also Materials and methods) for each phage clone following incubation of assay microtiter plates with a colorimetric substrate. Using mock-treated wells from each plate as an independent normalization standard for each phage clone, we generated background-subtracted values (Bsub) and Z-normalized (Znorm) values for four replicate experiments involving both resting and activated macrophages. As a preliminary view of the data, we assembled the Znorm values from each of these eight experiments to produce an outcome “signature” for each phage clone. Unbiased hierarchical clustering of these data revealed a collection of outcomes with a range of affinities and specificities, which were visualized as a heat map (Figure 2A).

Identification of phage clones specific for LPS-activated U937 cells. Performance of 50 selected phage clones in quadruplicate high-throughput binding experiments with both LPS-untreated and -treated U937 cells. (A) Unbiased hierarchical clustering of Z-normalized (Znorm) scores for phage-treated wells, colored relative to the mean score (+20; black blocks) for all 400 measurements to depict relative distance below (red blocks) or above (green blocks) the mean score. Maximal intensities for red or green blocks correspond to scores of −35 and +75, respectively. (B) Composite scores for affinity (i.e., for LPS-treated cells) and specificity (i.e., comparing LPS-treated and -untreated cells). Data are displayed in terms of higher rankings (green blocks) or lower rankings (red blocks) of affinity or specificity relative to all clones tested. Also included are overall rankings (italicized numerals at right), derived as described in the text for the top five clones. The sequences of three phage clones isolated for further analysis are depicted.
To effect more quantitative scoring of phage clones, we constructed composite scores across identical replicate experiments for both affinity and specificity. To assess binding affinity as judged by the colorimetric readout of the ELISAs, we first averaged the four Bsub values associated with activated macrophages, then scaled this average according to the (common) variance of the signal from activated macrophages in the absence of phage particles. Across all 50 clones, these signal-to-noise ratios were quite robust (median value = 11.3) and ranged from 1.51 to 90.3. To assess peptide specificity from the ELISA data, we performed an analogous signal-to-noise calculation for resting macrophages (i.e., using the variance associated with LPS-untreated cells) and then computed specificity as the ratio of these signal-to-noise measurements. As expected, most phage clones were not specific for activated macrophages (median specificity value = 1.04), although several phage clones were quite specific for LPS-treated cells (maximum specificity value = 14.2). Each of these replicate-composite affinity and specificity metrics were used independently to rank the 50 phage clones in descending order of either affinity or specificity, and these ranks were used to construct a second heat map containing the results of composite scoring (Figure 2B). Finally, an overall score was assigned to each phage clone by combining the independent rankings for that clone in both affinity and specificity, with equal emphasis on each property of the phage clone. The results of this overall score agreed well with the hierarchical clustering results, and were highly consistent with visual inspection of the heat map depicting the affinities and specificities ranked independently. Using this method, two phage clones (PTAPTST and PAPRSGS) were selected for further experimentation on the basis of having excellent affinities and good specificities; an additional phage clone (PAIAHRT) was selected on the basis of moderate sequence similarity to the first two clones.
Confirmation of Screening Results Using Fluorescence Microscopy
Phage clones identified using the above screening procedure were specific for LPS-treated macrophages (Figure 3). After incubation with phage displaying the peptides PTAPTST or PAPRSGS and subsequent incubation with fluorescent antibodies to phage, LPS-treated U937 cells exhibit bright fluorescence attributable to phage internalization (Figure 3A and inset). In contrast, little fluorescence due to phage internalization was observed with similarly incubated, nonactivated U937 cells, demonstrating the specificity of these clones for LPS-treated U937 cells (Figure 3B). In keeping with the data obtained from the phage ELISA, a clone of similar sequence to the others, PAIAHRT, was specific for LPS-treated U937 cells, but fluorescence was less pronounced under identical treatment conditions, suggesting a lower affinity of this clone for U937 cells. To effect a more quantitative method of determining cellular-mediated phage uptake and validation of the multidimensional analysis, phage clones displaying the indicated peptides or an aliquot from the unselected library were labeled with fluorescein then incubated with U937 or U937 + LPS-treated cells. Following incubation, phage uptake was quantified via fluorescence plate reader (Figure 3C). Results from this experiment demonstrate that the peptides identified by multidimensional analysis are indeed specific for activated macrophages, with the sequence PAIAHRT having the lowest specificity for the activated macrophages. In contrast, FITC-labeled phage from the unselected library demonstrated no appreciable uptake above background levels (Figure 3C), suggesting that phage uptake is specific to the selected peptide sequences and not mediated through nonspecific phagocytosis of phage.

Validation of phage clones by fluorescence microscopy. U937 cells treated with either LPS (A) or vehicle (B) were incubated with the indicated phage clone. Microscope images indicate selective labeling of LPS-activated cells with green fluorescence corresponding to the labeling of incorporated phage particles. Nuclei are stained blue with DAPI in both experiments to identify cell nuclei (scale bar = 5 µM). Inset depicts a higher magnification bright-field/fluorescence image of U937 cells treated with LPS then incubated with the indicated phage clone. Image was taken at 40 ×. (C) FITC-labeled phage clones or unselected phage uptake into U937 cells treated as above in A and B was quantitated by fluorescence plate reader.
Discussion
The use of phage-displayed peptide libraries is a powerful method for selecting peptides with desired binding properties [15]. However, the validation and prioritization of hits obtained from this screening approach remains challenging. We have used a cell-based ELISA coupled with multidimensional data analysis to facilitate rapid selection and prioritization of peptide sequences that have desired binding properties. The mathematical manipulation of the data and subsequent graphical visualization is beneficial in a number of ways: First, normalizing raw absorbance data relative to appropriate mock-treated controls facilitates the direct formal comparison of scores obtained from multiple plates (or experiments run at different times), increasing the total number of clones that can efficiently be analyzed. Second, representing scores from activated macrophages in terms of the variance of scores from resting macrophages permits direct determination of the specificity of results, as judged by absorbance, in terms of the likelihood that a positive result from activated macrophages can be explained by the control experiments using resting macrophages. Finally, graphical visualization of data using heat maps allows convenient prioritization of clones meeting desired parameters; such selection is more difficult when surveying data tables that include large numbers of clones with multiple replicates. The approach utilized in this study is readily extensible to other forms of high-throughput screening, such as small-molecule libraries [17] or conjugated nanoparticle-based libraries [21].
The peptide sequences (PTAPTST and PAPRSGS) discovered using this approach were shown to be specific for LPS-activated macrophages with a remarkable difference in staining patterns between activated and resting macrophages. Using fluorescence uptake data, we show that the difference between cellular states is at least 7:1, quite surprising given that the parental cell line is the same. Another remarkable observation was that the peptide sequences triggered internalization of phage particles into activated macrophages. This cellular trapping is highly desirable for subsequent conversion of the peptide sequences into imaging agents. The developed molecular framework and subsequent data analysis and visualization could ultimately be applied to other targets and other libraries, making it a useful scaffold for the development of novel targeted therapeutics or diagnostic tools.
Footnotes
Acknowledgments
We acknowledge the following funding sources: P50 CA86355 (R. W. and K. A. K.) and the National Cancer Institute's Initiative for Chemical Genetics (ICG) (P. A. C.).