
Abstract
Rapid and efficient delivery of imaging probes to the cell interior using permeation peptides has enabled novel applications in molecular imaging. Membrane permeant peptides based on the HIV-1 Tat basic domain sequence, GRKKRRQRRR, labeled with fluorophores and fluorescent proteins for optical imaging or with appropriate peptide-based motifs or macrocycles to chelate metals, such as technetium for nuclear scintigraphy and gadolinium for magnetic resonance imaging, have been synthesized. In addition, iron oxide complexes have been functionalized with the Tat basic domain peptides for magnetic resonance imaging applications. Herein we review current applications of permeation peptides in molecular imaging and factors influencing permeation peptide internalization. These diagnostic agents show concentrative cell accumulation and rapid kinetics and display cytosolic and focal nuclear accumulation in human cells. Combining methods, dual-labeled permeation peptides incorporating fluorescein maleimide and chelated technetium have allowed for both qualitative and quantitative analysis of cellular uptake. Imaging studies in mice following intravenous administration of prototypic diagnostic permeation peptides show rapid whole-body distribution allowing for various molecular imaging applications. Strategies to develop permeation peptides into molecular imaging probes have included incorporation of targeting motifs such as molecular beacons or protease cleavable domains that enable selective retention, activatable fluorescence, or targeted transduction. These novel permeation peptide conjugates maintain rapid translocation across cell membranes into intracellular compartments and have the potential for targeted in vivo applications in molecular imaging and combination therapy.
Keywords
Introduction
Efficient delivery of diagnostic compounds to the cell interior using permeation peptides has enabled novel applications in molecular imaging. Peptide-based imaging agents in particular are desired for molecular imaging applications because of their potential for specific detection of a target, great flexibility in design, facile synthesis, and the availability of well-characterized chelation cores for incorporation of imageable metals, such as technetium (99mTc) and gadolinium (Gd) [1–4]. Peptide-based imaging agents targeting cell-surface receptors are well known [1,2,5–8]. Fragments of antibodies (single-chain fragments, diabodies, or minibodies) [9], small proteins [10], and peptides [2,5] have been used successfully to image cell surface targets such as carcinoembryonic antigen, phosphatidylserine, and somatostatin type 2 receptors, respectively. Targeting of peptide-based imaging agents largely has been limited to extracellular or externally oriented membrane receptors because size, charge, and pharmacokinetic considerations of conventional peptide sequences as well as the dielectric constant of the plasma membrane do not enable translocation across cellular membranes [2,11]. In addition, detection of diagnostic agents that are confined to the cell surface is potentially limited because signal amplification through transport, sequestration, or enzymatic activity does not occur. Delivering imaging agents to intracellular compartments would allow for selective retention and signal amplification, thereby opening up a broad range of novel molecular imaging applications.
Permeation peptides have been extensively utilized to achieve intracellular delivery. Early observations revealed that these permeation peptides rapidly translocate into various cell types. Included among such peptides are the third helix of the homeodomain of Antennapedia [12], polyarginines [13], guanidinium peptides [13,14], and viral proteins such as herpes simplex virus VP22 [15], HIV-1 Rev protein [16], and the basic domain of HIV-1 Tat protein [17–19]. Studies of the HIV Tat protein in the late 1980s [17,18] showed the feasibility of utilizing Tat-derived polypeptides for rapid transport of various cargoes into cells. Subsequent work with Tat basic domain and related sequences has shown transport and intracellular delivery of diagnostic molecular imaging and therapeutic agents, 99mTc chelates, fluorescent probes, macrocyclic Gd chelates, and derivatized nanomaterial particles into various cell types and tissues [8,20–24]. Although abundant research exists utilizing permeation peptides for therapeutic applications [25–28], this review will focus on the use of permeation peptides for molecular imaging applications.
Intracellular Delivery Strategies: Metal Complexes
Radioisotopes for Scintigraphy
Technetium-99m (99mTc) labeling of permeation peptides potentially would provide a clinically viable molecular imaging agent through convenient and well-established techniques. 99mTc is currently a widely used radioisotope in medical imaging because it is readily available from 99Mo/99mTc generator systems, has an ideal photon energy for imaging applications (140 keV), and possesses a short half-life (6.02 hr) [1,3,29]. 99mTc can be stably coordinated by a variety of chelators including various tetradentate N or S ligands (N2S2, N3S, etc. donor cores) and diethylenetriaminepentaacetic acid (DTPA) [2]. These and related chelators also coordinate other isotopes (such as indium-111, gallium-67, gallium-68, copper-64, and yttrium-90 [4]) that broaden the spectrum of radioisotopes that can be exploited for diagnostic and radiotherapeutic applications. Chelation chemistry for 99mTc also has received much attention because 99mTc donor cores enable radiation therapy using chemically analogous rhenium (186Re/188Re) species [30], radioisotopes with emission spectra that deposit significant local energy within the cell, thereby producing DNA damage. Other radiotherapeutic isotopes also are under investigation [30]. More importantly, in contrast to utilizing fluorescence intensity analysis to compare differences in permeation peptide transduction between cell types or conditions, radiotracer analysis is not modulated by the many factors that affect fluorescence detection (i.e., nonlinear signals, aggregation, and quenching) [31] and can enable direct quantitative analysis of peptide content associated with target cells. Thus, to quantitatively study permeation peptides, we designed prototypic peptides that constituted in their simplest form two functional domains: a membrane permeant peptide sequence derived from HIV-1 Tat basic domain residues 48–57 (GRKKRRQRRR) and a peptide-based chelator for coordination of radioisotopes useful in medical imaging and therapy (Figure 1A).
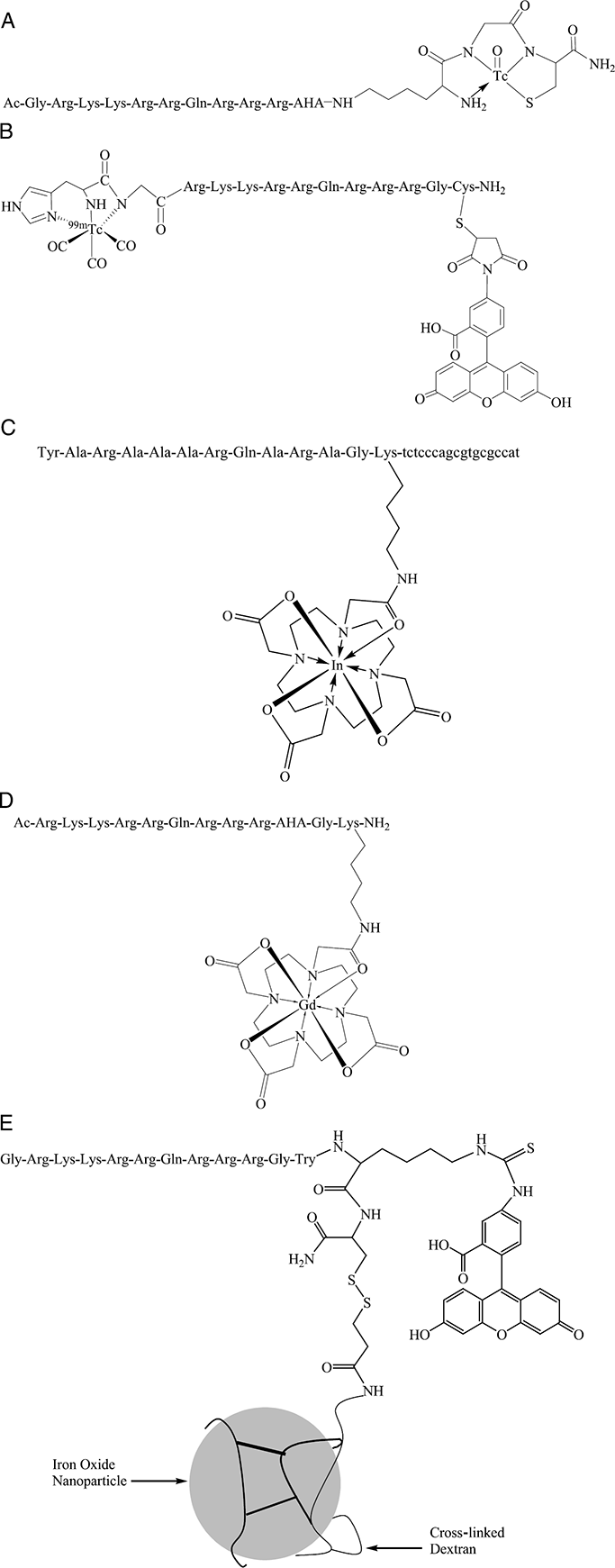
Structures for 99mTc-N3S-Tat peptide (A), dual labeled 99mTc(CO)3-Tat peptide-fluorescein complex (B), 111In-PTD-anti-bcl-2-PNA (C), Gd-DOTA-Tat peptide (D), and a cross-linked iron oxide particle derivatized with Tat peptide (E).
Peptide conjugates were prepared by standard solid-phase peptide synthetic methods. A typical sequence is GRKKRRQRRR-AHA-***ε-KGC, wherein GRKKRRQRRR represents the Tat48–57 permeation domain, ε-KGC provides an amino diamido thiol (N3S) chelating core for incoming TcO3+ or ReO3+ species [6,32], and AHA represents 6-aminohexanoic acid as an aliphatic linker between the permeation domain and the chelator. Among several choices available for the chelator moiety [2], ε-KGC was a convenient starting point for these prototypic peptides. Advantages of this sequence include(1) an appropriate protected residue (ε-K) for orthogonal coupling during solid-phase peptide synthesis, (2) amino acid chain extension off the ε-amine of K confers rotational freedom between the membrane permeation and chelating domain of the peptide conjugate, (3) the chelation moiety is readily radiolabeled at room temperature using a facile transchelation reaction, and significantly, (d) this chelation moiety has proven favorable pharmacokinetic and metabolic properties in the clinical development of other targeted peptide agents [6,33]. The Tat peptide conjugates are labeled with 99mTc by ligand exchange using 99mTc-glucoheptonate as the ligand exchange reagent [7,22]. Incorporation of a metal into the N3S donor core has been shown to result in the formation of three five-membered rings and a chiral center [32]. Such structures are known to be stable and typically resemble a square pyramid with the peptide atoms at the base and the oxygen atom in the apical position [32,34]. Corresponding cold ReO3+-Tat peptide complexes are obtained by ligand exchange methods using Re-glucoheptonate as the ligand exchange reagent [7,22]. Of note, these Tat peptide conjugates also can be labeled on the C-terminal thiol with fluorescein maleimide for comparative studies with the metal chelates [20,22].
We have shown that peptides based on the basic domain of Tat and a conjugated chelator for metals can be used to deliver 99mTc into cells in culture [22]. These peptides quickly (#x003C;2 min) accumulate and concentrate within cultured cells while maintaining stable chelation of the radioisotope (Figure 2). Washout experiments of Tat peptides from cells also demonstrate similar rapid kinetics that is ~80% complete within 20 min. However, a residual compartment of slowly exchanging or retained activity is observable for long times and is a function of extracellular peptide concentration. This residual compartment is used to nonspecifically label cells for in vivo cell-trafficking studies [8]. Given the nuclear localization and RNA-binding properties of Tat [35,36], an appealing potential use of these novel <sp>99mTcO3+-Tat peptide complexes would entail chelation of 186Re or 188Re for delivery of therapeutic radiopharmaceuticals directly to the cell nucleus. Combined with appropriate targeting constructs, this might enable efficient delivery of gamma irradiation doses directly to RNA/DNA pools within tumors.
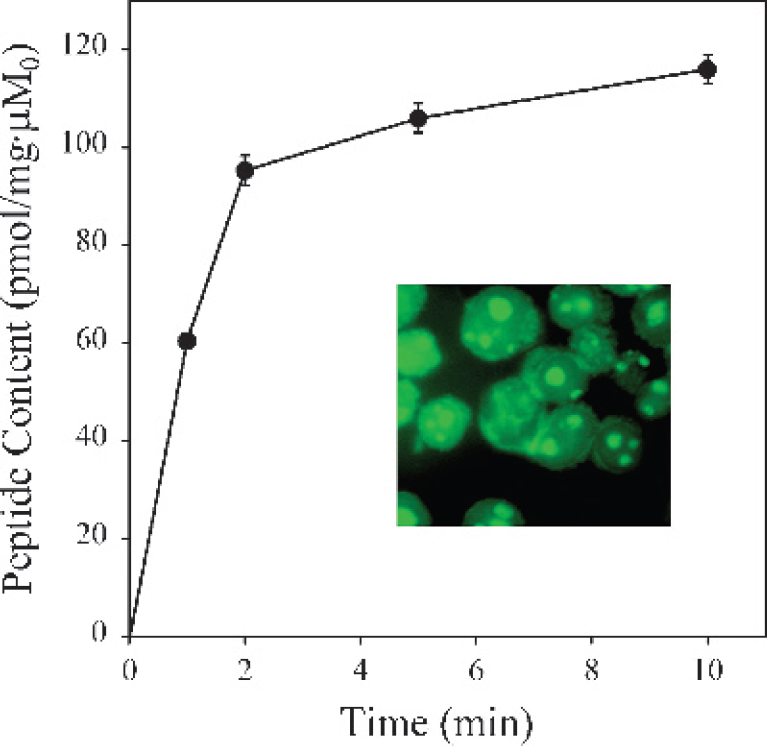
Kinetics of 99mTcO3+-Tat peptide complex accumulation into human Jurkat cells. Cells were incubated in physiologic loading buffer for the indicated times, washed, and cell-associated activity determined. Each point represents the mean of three to four determinations; bars represent ± SEM when larger than the symbol. Inset: Cellular accumulation of Tat peptide-fluorescein conjugate in human Jurkat tumor cells. Cells were incubated with Tat peptide for 10–20 min at 37°C followed by fixation. Note that green fluorescence from labeled peptide resides in both cytosolic and nuclear (nucleolar) components. The cytosolic compartment containing fluorescent peptide appears as a rim surrounding the Jurkat cell nuclei, typical of lymphocytic cell types. (Modified from Polyakov et al. [22]).
Recently, metal-triaquo-tricarbonylcomplexes[M(CO)3 (OH2)3]+, where M = 99mTc or Re, have been incorporated into peptides using chelators such as phosphines [37] and alpha-amino moieties of N-terminal histidine, an amino acid that is readily added to any peptide during solid-phase synthesis or tagged to polypeptides in common cloning vectors [38]. These 99mTc-triaquotricarbonyl species have been shown to be highly stable [39–41], thereby providing another promising alternative chelation strategy for coordination of 99mTc to Tat peptides. Coordination is carried out at atmospheric pressures and reasonable temperatures, a necessity for practical and widespread use of radiolabeling [39,42]. Additionally, 99mTc(CO)3(OH2)3+ has a stable and small donor core [40] that is less likely to disrupt the secondary structure of the peptide compared to conventional macrocyclic chelators. Furthermore, by utilizing N-terminal histidine for labeling, a less restrictive metal coordination site is established compared to macrocyclic chelators. Thus, Re, a slightly larger metal than 99mTc, can easily be accommodated by the peptide tag and enable a more direct comparison between technetium and rhenium complexes. Using this labeling strategy, an N-terminal histidine 99mTc(CO)3-Tat peptide was reported (Figure 1B) [43].
When injected into mice, radiolabeled 99mTcO3+-Tat peptides distribute throughout the body, reaching peak accumulation in organs and tissues within minutes [22]. Significant radioactivity is detected in all organs, except the brain. The peptide subsequently clears primarily through the kidneys during the first hour after injection. Previous studies have indicated that after glomerular filtration, radiolabeled, low molecular weight standard peptides and antibody fragments are likely resorbed by proximal tubules via luminal endocytosis [44,45]. The long residence times of nontransported radiometabolites generated after lysosomal proteolysis of radiolabeled fragments in renal cells also are reported to contribute to persistent renal radioactivity levels [45,46]. In contrast, the rapid renal clearance observed for 99mTcO3+-Tat peptide complexes suggests that lysosomal proteolysis is not relevant to the overall excretion pathway of these peptides in vivo and may favorably impact whole-body imaging applications with 99mTcO3+-Tat peptide complexes.
However, other published pharmacokinetic data suggest that not all Tat peptide applications in vivo will be readily executed. For example, using a radioiodinated biotinylated Tat48–58 conjugated to streptavidin, Lee and Pardridge [47] found that Tat conjugation of large molecular weight proteins resulted in a marked increase in the rate of removal of the protein from the circulation compared to nonconjugated proteins, resulting in a reduced plasma area under the curve (AUC). Thus, following intravenous injection of the fusion protein, they concluded that Tat conjugation produced opposing effects on membrane permeation and plasma AUC, resulting in no net increase in organ uptake and delivery in vivo.
The mode of delivery also may impact pharmacokinetics, organ accumulation, and, in particular, the ability to penetrate the blood-brain barrier. For example, following intraperitoneal injection of a similar Tat basic domain fused to β-galactosidase, brain uptake and blood-brain barrier penetration were observed [48]. Note that in contrast to the radiolabeled peptides in the above studies, the β-galactosidase permeation peptide fusion protein is an enzymatic reporter, which, given enough time, is capable of generating detectable signal-to-noise ratios ex vivo with small amounts of enzyme. In particular, the biodistribution study in vivo using β-galactosidase as a reporter resulted in brain tissue slices that were faintly but distinctly stained for β-galactosidase following a 16-hr incubation ex vivo with the β-galactosidase substrate, X-Gal. In contrast, other organ tissue slices were darkly stained for β-galactosidase following a 4-hr incubation ex vivo with X-Gal. A comparison of these results indicates that uptake of β-galactosidase into the brain was greatly diminished as compared to other tissues. Consequently, the blood-brain barrier still remains a formidable obstacle for stoichiometric reporters, such as radiolabeled or fluorescently labeled peptides, that are not capable of enzymatic amplification (see Targeted Imaging Strategies).
Permeation peptides are also being utilized for targeted imaging of mRNA, providing a noninvasive and efficient strategy for intracellular delivery compared with traditional techniques of microinjection and transfection [49–51]. In one study, 99mTc-antisense DNA targeted to the alpha regulatory subunit of protein kinase A was delivered into the cytoplasm of ACHN cells through electrostatic interactions with cationic Tat peptide [52]. Intracellular DNA hybridization was demonstrated by an in situ hybridization assay indicating that once internalized, the antisense DNA escaped from the electrostatic interaction with Tat peptide. In another study, Tat peptide was coupled to a molecular beacon and specific cellular internalization and targeting to the mRNA of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was reported [51]. Improved specificity toward the mRNA of GAPDH was demonstrated with the permeation peptide construct as compared to results utilizing standard transfection protocols. 111In has also been utilized for diagnostic imaging of mRNA. A combination diagnostic imaging and therapeutic construct was synthesized through conjugation of a bcl-2-peptide nucleic acid (PNA) to a peptide transduction domain (PTD) followed by chelation of 111In with a goal to image and knock down expression of bcl-2 in non-Hodgkin's lymphoma (Figure 1C) [53]. Cellular efficacy remains to be reported. Although the above studies demonstrate successful imaging of mRNA in live cells, challenges remain for applications in vivo due to a variety of issues such as serum protein binding, delivery, low signal-to-noise ratios, and stability.
Gadolinium and Iron Oxide Nanoparticles for Magnetic Resonance Imaging
Contrast agents using paramagnetic metals such as gadolinium are routinely used in magnetic resonance imaging (MRI) to shorten longitudinal relaxation times (T1) of water protons and enhance contrast of MR images [54]. Contrast agents that shorten T1 generate signal enhancement and decrease the overall imaging time by allowing use of shorter recycle delays. To reduce the toxicity of free gadolinium, the metal must be stably complexed with polydentate ligands in a manner that preserves water relaxivity properties. Because gadolinium allows up to nine coordinate sites, chelating moieties are typically octadentate with either acyclic or macrocyclic polyaminopolycarboxylate ligands that form kinetically and thermodynamically stable complexes with gadolinium. The ninth coordination site of Gd (not occupied by the polydentate ligand) is used by fast-exchanging water molecules to transmit the paramagnetic relaxation effect to bulk solvent. Clinical MR contrast agents are confined to extracellular spaces and distribute nonspecifically throughout vascular and interstitial tissues to noninvasively image perfusion and vascular abnormalities [54]. Other agents under development are responsive to environmental changes, such as the decreased pH seen in extracellular spaces around tumors [55,56]. To increase sensitivity and specificity, targeted MR contrast agents have been explored that produce an increase in relaxivity that may arise from (1) enzymatic activation [57,58], (2) target or ligand binding [59–61], or (3) cationic concentration [62,63]. However, these approaches are limited by poor cell-permeation properties of the compounds. Thus, an efficient and generalizable method for intracellular delivery of targeted contrast agents would expand the realm of potential imaging applications in MRI.
Peptide transduction domains have been explored to carry MR contrast agents into cells [64,65]. As an example of MR contrast agent delivery, reports using (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)gadolinium(III) (Gd-DOTA) conjugated to a Tat basic domain peptide show the feasibility of rapid delivery of a T1 contrast agent into intracellular compartments (Figure 1D) [8,64]. For these applications, DOTA is conjugated to a Lys appended to Tat using orthogonal synthetic strategies and stably complexed with Gd under standard conditions [64]. T1-weighted spin-echo MR images of cells embedded in agar indicated the presence of enhanced intracellular relaxivity, consistent with transduction of Gd-DOTA-Tat peptide into the cell interior. The intracellular localization of the Gd complex was confirmed with a fluorescein-conjugated Tat peptide that showed cytosolic and punctate nucleolar localization characteristic of other small Tat peptide conjugates. Furthermore, intracellular delivery of the Gd(III)-DOTA complex using other short permeation peptides such as poly-Arg peptides has been reported. The permeation, relaxivity, and enhanced retention properties of the Gd-DOTA poly-Arg peptide were well characterized in several cell types using inductively coupled plasma mass spectrometry (ICP-MS) and T1 relaxivity [65].
Tat basic domain and related permeation sequences also have been used to mediate delivery of biocompatible cross-linked iron oxide particles (CLIO-Tat) and superparamagnetic nanoparticles into the cell interior for applications in MRI (Figure 1E) [21,66–69]. The derivatized particles have been shown to internalize into mouse spleen lymphocytes greater than 100-fold more efficiently than nonmodified particles [21]. By labeling CLIO particles with fluorescein isothiocyanate (FITC), cytoplasmic and nuclear localization was demonstrated. CLIOs affect the transverse relaxation time (T2) by introducing large susceptibility effects, which alter the local magnetic field homogeneity and result in signal loss where CLIOs are located. The strong decrease in signal by MR imaging enables the magnetically labeled cells to be tracked in vivo.
Using high-resolution MR microscopy and conventional MRI, CLIO-Tat-labeled immune cells infiltrating diabetic mouse pancreas were visualized [70]. In addition, CLIO-Tat loaded T cells have been visualized by MRI homing into the spleen. Here, the CLIO-Tat loaded T cells produce a tissue-specific decrease in signal intensity [71]. CLIO-Tat particles also have been shown to efficiently internalize into hematopoietic and neural progenitor cells [72]. Following intravenous injection into immunodeficient mice, magnetically labeled CD34+ cells home to bone marrow and can be detected by MR imaging of tissue samples. Interestingly, the magnetically labeled cells that homed to marrow can be recovered by magnetic separation columns. Iron particle incorporation was at first reported to not impact cell viability, differentiation, or proliferation of CD34+ cells [72]. However, careful analysis in a recent report, using similar labeling protocols, has suggested that non-derivatized iron oxide particles do not efficiently load into CD34+ stem cells derived from umbilical cord blood and may impact engraftment [73].
Intracellular Delivery Strategies: Optical Techniques
Although radiotracer techniques can quantitatively determine cell-associated radioactivity, the exact subcellular location of permeation peptides cannot be evaluated by these population-based gamma-counting techniques. Permeation peptides dual-labeled with fluorescein and 99mTc would enable both qualitative subcellular localization by fluorescence microscopy and quantitative scintigraphic imaging. Thus, several novel dual-labeled Tat peptides derivatized with N-terminal 99mTc/Re(CO)3 chelation and C-terminal fluorescein-5-maleimide conjugation have been synthesized to study cell tracer kinetics, intracellular localization, and murine biodistribution (Figure 1B) [43].
Overall, dual-labeled Tat peptide complexes are comparable to corresponding single-labeled Tat peptides in uptake kinetics and net accumulation levels. Both complexes also retain about the same percentage of residual radioactivity following a 30-min washout from cells in culture [43]. Subcellular localization is unaffected by labeling both termini of the peptide with dual-labeled Tat peptides resulting in the same fluorescence localization to the nucleus and cytoplasm as seen previously with single-labeled Tat peptides (Figure 2, inset). Transport assays and fluorescent cell uptake studies collectively suggest that the mechanism of Tat peptide translocation is not wholly dependent on free C- or N-termini.
To determine the utility in vivo of dual-labeled Tat peptides, fluorescence biodistribution studies have been performed in mice utilizing dual-labeled Re(CO)3-Tat peptide-fluorescein conjugates. In contrast to the 99mTcO3+-labeled peptide biodistribution, but in accord with radiometric 99mTc(CO)3-peptide biodistribution and imaging data, the highest fluorescence intensity following intravenous bolus administration is observed in liver and kidney tissue with no significant fluorescence intensity in brain tissue (Figure 3) [43]. These results and those of other studies [22,47,74] are consistent with the observation that these Tat peptides are characterized by high blood clearance rates, further indicating the need for strategies, such as introduction of binding moieties or poly(ethylene glycol) (PEG) motifs to slow blood clearance rates, or cleavable domains to increase target tissue signal accumulation.

Fluorescent distribution in vivo of a dual-labeled Re(CO)3-Tat peptide-fluorescein complex. Following intravenous bolus injection of the complex, organs were harvested after 30 min and analyzed by epifluorescence microscopy. Panels compare fluorescent signal in liver (A) and brain (B). (C) Phase contrast microscopy of the identical brain tissue section as in B. Liver and brain fluorescent pictures were obtained at identical magnification and exposure times. Note relative lack of brain penetration in vivo following intravenous administration of the Tat peptide. (Modified from Bullok et al. [43]).
Targeted Imaging Strategies
Targeting strategies would enable the monitoring of novel tailored therapies, including gene therapy and new molecularly targeted chemotherapeutics, by detecting specific enzyme activities, gene expression, and tumor responses, such as apoptosis. Two major targeting strategies exist to improve molecular imaging in vivo with permeation peptides: activation-induced intracellular retention and activation-induced membrane transduction.
Activation-induced Retention
For targeting via activation-induced retention by a protease, for example, a proteolytic sequence specifically recognized by an intracellular enzyme would be inserted between the permeation peptide and the radioisotope chelation core or fluorophore. Following membrane permeation, interaction with and cleavage by the target enzyme would result in intracellular trapping and accumulation of the imaging moiety. This strategy for selective targeting with therapeutic permeation peptides has been exploited in vitro by incorporating the recognition sequence for HIV protease between the Tat peptide and a precursor form of caspase-3 [75]. Therein, only cultured cells infected with HIV cleave the Tat peptide and release functional caspase-3, resulting in specific killing of infected cells.
Pilot experiments using a targeted strategy for imaging the final commitment of cells to death in vivo involved incorporating a caspase-3 recognition sequence between the C-terminus of the Tat peptide-based permeation peptide and a 99mTcO3+ chelation core [76]. Upon permeation into apoptotic cells, the probe should be cleaved by active caspase-3, leaving the C-terminally chelated 99mTcO3+ trapped inside the cell. To test this caspase-3-cleavable 99mTcO3+-Tat peptide probe, a fulminate liver apoptosis mouse model was generated using a published procedure [10]. Because the Fas receptor is highly expressed on the liver, as well as the kidney, thymus, gonads, and subsets of leukocytes [77], stimulation of the receptor through intravenous injection of an anti-Fas antibody leads to massive hepatocyte apoptosis. FVB mice were induced to undergo liver apoptosis upon treatment with purified hamster anti-Fas monoclonal antibody (mAb) by intravenous injection 45 min prior to imaging. Following intravenous injection of the cleavable 99mTcO3+-Tat peptide, mice were immediately positioned for imaging on a gamma scintillation camera (Figure 4). In untreated mice, the 99mTcO3+-Tat peptide initially showed a whole-body microvascular distribution, followed by rapid and abundant renal localization and excretion. By 30 min postinjection in untreated mice, the only site of imageable radioactivity was the urinary bladder (Figure 4). Note the absence of liver activity or other significant background activity. In contrast, mice pretreated with anti-Fas Ab showed abundant hepatic and renal retention of radioactivity 30 min postinjection, consistent with caspase-3-mediated peptide cleavage and enzymatically induced accumulation of the imaging fragment [76]. These images represent an early example of molecular imaging of caspase-3 activity in the whole animal and provide “proof of principle” for this approach to targeted imaging of apoptosis in vivo.

Scintigraphic images of organ distribution of caspase-3-cleavable 99mTcO3+-Tat peptide in FVB mice 30 min post injection. Using a published procedure [10], FVB mice were administered purified hamster anti-Fas mAb (Jo2, PharMingen, San Diego, CA; 8 µg/animal) by intravenous injection and allowed to recover for 45 min prior to imaging. Following metofane anesthesia, 99mTcO3+-Tat peptide (200 µCi) was administered by tail vein injection and mice were positioned immediately for imaging on a gamma scintillation camera (Siemens Basicam, New York, NY; 5-mm pinhole collimator; 20% energy window centered over 140 keV). Posterior images of mice were collected with a 256 × 256 matrix. Images were corrected for radioactive decay, but no corrections were made for scatter or attenuation. Left, untreated control mouse; right, mouse pretreated with anti-Fas mAb. Note focal radioactivity only in the urinary bladder of the control mouse, but abundant retention of radioactivity in the liver and kidneys, two organs that express the Fas receptor, in the pretreated animal. The nose of the animal is toward the top of the image, the tail toward the bottom.
Protease-mediated Fluorescence Activation
A second targeted retention strategy involves protease-mediated fluorescence activation. Using this strategy, the incorporated proteolytic peptide sequence, positioned adjacent to the permeation peptide, is flanked by a fluorescence donor and a fluorescence quenching acceptor to allow for resonance energy transfer (RET) between the chromophores. Upon enzymatic cleavage, fluorescence quenching would be relieved and fluorescence signal would be detected.
More importantly, incorporation of quenched fluorescence into a permeation peptide imaging probe should allow for greater imaging sensitivity as the intact quenched probe contributes essentially no background signal. This advantage allows use of activatable optical probes over a broader range of administration routes and infusion protocols. For example, with small molecular weight radiopharmaceuticals, agents are generally administered as an intravenous bolus [2,78]. Tracer pharmacokinetic principles indicate that all tissues are rapidly exposed to a high concentration of the radiotracer followed by washout from nontarget sites [78]. High affinity and specific interactions (binding) are then revealed over time as the nontarget radioactivity clears from the system. However, it was recently reported that the membrane permeation properties of Tat basic domain and related permeation peptides in cells are offset by their very rapid blood clearance in vivo [47]. Thus, area-under-the-curve analysis shows no significant net gain in tissue delivery and retention with model polypeptides following intravenous bolus injections of radiolabeled Tat peptides. Strategies to prolong and enhance tissue delivery could entail use of deposition methods (subcutaneous or intraperitoneal instillation) or use of intravenous infusion protocols. However, with radiolabeled compounds, this would greatly increase the blood pool compartment and background, thereby reducing image quality [78]. In contrast, unlike radiotracers that are continually emitting detectable signal, activatable optical agents that are silent or quenched under baseline conditions enable deposition or infusion protocols. Only when activated by the target enzyme will the agents begin emitting a detectable signal while both the circulating and the retained nonactivated probe will ideally remain optically silent and produce little background signal. This strategy can be used as a delivery advantage for enzyme-targeted permeation peptide conjugates.
For example, to achieve intracellular protease fluorescence activation, a permeation peptide probe, TcapQ647, was synthesized [79]. The quenched-fluorescent probe consisted of a C-terminal executioner caspase recognition sequence, DEVD (Figure 5A), flanked by the far-red quenching chromophore, QSY 21, and the fluorescence donor, Alexa Fluor 647. High quenching efficiencies (92–99%) were achieved in both low-serum buffer and in 100% serum. Consequently, very little fluorescence was detected in solution or in cells in the absence of active caspase-3. Upon induction of apoptosis using doxorubicin or vinblastine, significant increases in intracellular fluorescence were detected by confocal microscopy. An increase in cellular fluorescence was not observed in apoptotic cells incubated with the all

A schematic comparison of molecular imaging strategies involving intracellular protease-mediated fluorescence activation and retention (A) and extracellular protease-induced transduction (B). For strategies involving intracellular protease-mediated retention (A), the permeation peptide remains in a quenched state and is free to transduce into and out of cells in the absence of the target enzyme. Upon transduction and intracellular proteolysis, the charged fluorescence tag is cleaved from the permeation peptide, leading to fluorescence dequenching and signal accumulation. For strategies involving extracellular protease-induced transduction (B), penetration of the fluorescent permeation peptide is inhibited by charge neutralization through interactions between the cationic arginine and the anionic glutamine side groups. Upon cleavage by the target extracellular enzyme, the two fragments are separated, permitting the permeation peptide to transduce into (and out of) the cell. Note that with this latter strategy, no specific mechanism maintains retention of the peptide within target cells. In (B), “U” indicates a succinoyl group.
Activation-induced Membrane Transduction
Another recently reported approach to targeted delivery involves proteolytic cleavage of an “activatable cell-penetrating peptide” (ACPP) in the extracellular spaces surrounding cells [81] (Figure 5B). The ACPP was synthesized by coupling a Cy5-labeled polyarginine (R9) permeation peptide to a polyanionic polyglutamate peptide via a matrix metalloprotease 2 (MMP-2) proteolytic sequence. Coupling the polyanion to the permeation peptide inhibited cell membrane translocation and intracellular accumulation, evidenced by an 18-fold reduction of uptake into cells compared to a control peptide lacking the polyanion. Following preincubation of the ACPP with MMP-2, inhibition was relieved and a subsequent 17-fold increase in cell membrane translocation over uncleaved peptide was observed. Initial NMR studies suggested that the permeation inhibition is due to charge neutralization through ion pairing of the Arg and Glu side groups. Following incubation of the ACPP with MMP-2 expressing HT-1080 cells, the cleaved peptide, R9-Cy5, could be detected within the cytosol and nucleus of the examined cells by fluorescence microscopy. The utility in vivo of the ACPP was also examined using a tumor xenograph mouse model. HT-1080 cells were implanted subcutaneously into mice prior to peptide administration. Images taken 1 hr post ACPP injection demonstrated an approximately twofold signal increase over background at the tumor, which was statistically different from the fluorescence detected with control peptides (1.3- to 1.5-fold increase over background). To determine if the probe internalized into tumor cells, tumors were excised and analyzed by microscopy. Evidence of tumor-associated (extracellular and perhaps intracellular) fluorescence was observed.
Comparison of Activatable Retention and Activatable Transduction Strategies
The advantages of intracellular protease-mediated fluorescence activation or retention strategies include the theoretical signal gains resulting from trapping the imaging agent inside of the cell following permeation peptide removal. Although permeation peptides on their own do maintain a trapped component [20], a significant fraction eventually washes out [22]. Thus, by removing the permeation sequence in the intracellular space, washout of the retained imaging fragment would be significantly reduced. In addition, when applied to quenched optical imaging strategies, background fluorescence from the uncleaved probe in the blood and extracellular spaces does not contribute significant background fluorescence to the detected signal. This allows interrogation of a broad range of tissues, especially excretory tissues, where the background fluorescence from a nonquenched probe would be unacceptably high.
By comparison, the advantage of extracellular protease-induced transduction is the ability to interrogate extracellular enzymes that are often linked to metastasic potential or cellular damage [82,83]. However, one disadvantage of activatable-transduction approaches is that nonspecific extracellular binding or accumulation of the uncleaved probe may contribute significant background signal to the overall signal. Another potential disadvantage relates to the transient extracellular localization of the cleaved permeation peptide. In theory, after cleavage, there is kinetic competition between rates of transmembrane transduction into cells and rates of tissue washout of the peptide fragment in vivo, potentially reducing cellular signal-to-noise ratios. In addition, because permeation peptides possess a significant bidirectional membrane flux, the lack of an intracellular trapping or retention motif will allow further washout of any intracellular permeation peptide over time, again leading to loss of signal. Thus, activated delivery necessarily requires the probe to accumulate nonspecifically and be retained sufficiently in the target location in vivo to be detected over non-target-tissue accumulation.
Both activation-mediated intracellular retention and activation-induced transduction strategies are in the initial stages of development and can be refined for improved in vivo imaging. As these strategies advance, it will likely become clear which approaches are best suited for various applications in vivo.
Clues to Internalization Mechanisms
Little can be concluded regarding the mechanisms of membrane transduction and subcellular localization for all classes of permeation domains. Historically, it had been generally thought that the membrane translocation mechanism(s) involves a passive membrane diffusive or destabilization process that does not require binding to cell surface receptors [84,85]. Though many investigators now dismiss a passive membrane-diffusion mechanism, a number of inconsistencies exist. For example, some have observed that transduction occurs in many cells at 4°C, indicating that conventional endocytosis is not involved [12,15,20]. However, others have not observed uptake at 4°C. In addition, although endosomotropic agents such as chloroquine and monensin generally are without effect on Tat43–60 peptide-mediated transduction, inhibitors of caveolae formation (nystatin and fillipin) inhibit 50% of a luciferase reporter signal arising from Tat43–60 peptide-mediated phage penetration in COS-1 cells [86]. Secondly, studies using full-length VP22 indicate that cytoskeletal function is necessary for penetration [15], but studies using Tat49–57 and Jurkat cells in the presence of microtubule- and filament-disrupting agents (colchicine, taxol, nocodozole, and cytochalasin D) [22] demonstrate persistent membrane permeation. Another recent study in reporter T cells correlates uptake of large Tat-fusion proteins with macropinocytosis activity and concludes that the mechanism of membrane transduction involves lipid raft-dependent macropinocytosis [87]. Although much remains to be determined regarding the general mechanism(s) of penetration, several factors that influence permeation peptide penetration can be gleaned from the various permeation peptide studies: cargo size, the permeation peptide itself, and cell type. Extensive reviews of mechanistic studies exist [28,88], but here we will highlight only a few studies that discuss factors influencing penetration of small imaging permeation peptides.
Many discussions regarding mechanism of penetration within the literature are confounded by lack of directly comparable experiments. In particular, experiments suggest that peptides containing small cargoes such as a radiolabel or fluorophore may enter the cell by a different mechanism than large cargoes, such as proteins or antibodies. For example, one study using extracellular full-length Tat protein coupled to GFP concluded that cell-associated heparan sulfate (HS) proteoglycans were required for cell penetration [89]. However, studies using Tat49–57 coupled to fluorescein demonstrated with HeLa and CHO cell mutants deficient in proteoglycan biosynthesis that HS proteoglycans are not involved in cell penetration [90]. It is possible that there is a threshold at which transduction mechanisms differ, but this threshold remains unknown.
Studies that directly compare uptake of permeation peptides with comparably sized cargo, usually a fluorescence molecule such as fluorescein, have indicated that penetration is affected by the permeation peptide sequence per se. Differential uptake is observed among the various permeation peptides, with flock house virus (FHV) peptide having the lowest penetration and polyarginine and Tat-based peptides having the highest [91,92]. Additionally, transduction of permeation peptides can be affected by different conditions. Comparative studies with penetratin, polyarginine, and Tat peptide revealed that transduction of polyarginine into unfixed PC-12 cells is minimally affected at 4°C or with depletion of intracellular ATP [93]. In contrast, penetratin transduction was largely inhibited.
The observation that cell type can impact peptide penetration is a relatively new concept. Early studies putatively indicated that permeation peptides could indiscriminately penetrate into all cells. However, recent reports indicate intercellular transduction of full-length VP22-GFP fusion proteins into cultured human carcinoma A549 and H1299 cells as well as monkey COS-1 cells is limited [94] and that transduction of a Tat49–57-EGFP fusion protein into muscle cells in vivo is poor [95]. Fusion proteins comprising Tat49–57 peptide or full-length VP22 linked to the N-terminus of diphtheria toxin A fragment were also inefficiently transduced, although found to bind to the surface of Vero cells [96]. In addition, Madin-Darby canine kidney (MDCK) and CaCo-2 cells, a colonic carcinoma cell line, grown in a semipermeable membrane (transwell) configuration are impermeant to small 99mTcO3+-Tat peptides under physiologic conditions [97]. MDCK epithelial cells represent a model vectorial cell system to study transport mechanisms in distal renal tubule epithelia [98]. CaCo-2 cells, when grown into a confluent monolayer, form tight junctions and express other characteristics of enterocytic differentiation with important morphological and biochemical similarities to small intestinal columnar epithelium [99]. To confirm that 99mTcO3+-Tat peptides were not able to enter MDCK or CaCo-2 cells, uptake studies using fluorescein-conjugated L- and D-Tat49–57 were analyzed by fluorescence microscopy. Both cell lines failed to show any intracellular fluorescence under physiological conditions. In contrast, KB 3-1 and HeLa tumor cells, under identical conditions, show high uptake with a cytosolic and focal nuclear pattern of localization shown immunohistochemically to coincide with nucleoli [97]. Interestingly, internalization of fluorescein-labeled peptides is clearly evident in MDCK and CaCo-2 cells after treatment with plasma membrane permeabilizing reagents, such as digitonin and acetone-methanol. This indicates that acetone-methanol fixation leads to artifactual uptake of permeation peptides into otherwise impermeant cells. Of note, β-methyl-cyclodextrin, a cyclic oligosaccharide reagent that depletes cholesterol and disrupts membrane rafts [100], did not stimulate internalization of these small Tat peptides, thus indicating that cholesterol is not responsible for 99mTc-Tat peptide exclusion from these cells. In addition, HS proteoglycans are reported to be expressed in MDCK [101,102], CaCo-2 [103,104], and HeLa cells [105]. Our studies confirm expression of HS antigen in all these cells as well as in KB 3-1 cells [97]. As Tat peptide internalization is identified only in HeLa and KB 3-1 cells, we concluded that HS proteoglycans are neither sufficient for permeation nor for exclusion of small imaging Tat peptides from cells, a result that differs from larger fusion proteins. These results also coincide with the physiologic observation of bladder retention of 99mTcO3+-Tat peptides during imaging experiments in vivo.
Finally, as indicated above, fixation protocols have been shown to influence the analysis of either the intracellular uptake or subcellular localization of permeation peptides [53,106]. Independent of the permeation peptide literature, critical experiments regarding fixation-induced artifacts were reported previously in the general microscopy literature [107]. In a direct comparison of fixation techniques utilizing paraformaldehyde, glutaraldehyde, acetone, methanol, and paraformaldehyde/methanol, methanol and acetone were shown to disrupt intracellular compartments and destroy plasma membrane integrity. This disruption of the plasma membrane was shown to allow proteins as large as Bcl-2 and Bcl-xL to equilibrate with the extracellular space. These data were corroborated when permeation peptide conjugates were shown to artificially enter previously impermeant cells following a methanol/acetone fixation [97]. Based on these data, future studies of permeation peptide cellular internalization should not be performed with methanol or acetone fixation as false-positive results for internalization or cellular redistribution can arise. Consequently, fluorescence microscopy studies determining the localization or extent of internalization of permeation peptide conjugates in methanol- or methanol/acetone-fixed cells [89,108–112] may need to be reevaluated.
To address the issue of permeation peptide internalization and subsequent subcellular localization, several groups have compared peptide internalization in both live and paraformaldehyde-fixed cells [20,93,97,113–115]. Vives et al. [20] demonstrated that in HeLa cells, nucleolar and nuclear localization were found in both paraformaldehyde-fixed cells and live HeLa cells. Potocky et al. [115] also reported that within a population of live cells, discreet subcellular localization patterns exist, some having punctate staining only and others having a combination of punctate, diffuse, and nucleolar staining (almost 50%). Interestingly, a study comparing fluorescein-labeled Tat peptide and penta-Arg uptake into live PC-12 cells demonstrated that only penta-Arg localized to the nucleolus. Thus, permeation peptide uptake and eventual distribution to varying degrees into the cytosol or nucleus is not an artifact. In summary, confocal microscopy of live cells incubated with permeation peptides definitively confirms a heterogeneous intracellular localization, including cytosolic staining, that appears dependent upon permeation peptide sequence and cell type.
To conclude, the exact mechanism(s) for membrane translocation of small permeation peptides remain under investigation, and selected factors that affect translocation have been documented. Nonetheless, despite uncertainty regarding translocation mechanisms, successful utilization of permeation peptides not only for molecular imaging, but also for therapeutic and combination applications, is progressing in numerous fields of research.
Footnotes
Acknowledgments
We thank colleagues of the Molecular Imaging Center for technical assistance and helpful discussions. These studies were supported by grants from the National Institutes of Health (RO1 CA82841 and P50 CA94056) and the Department of Energy (DE FG02 94ER61885).