
Abstract
The enzyme heme oxygenase (HO) is an important regulatory molecule present in most nucleated mammalian cells which functions to break down the pro-oxidant molecule heme into three products, carbon monoxide (CO), biliverdin and free iron. The HO system has been associated with many physiologic functions, including vascular tone, regulation of inflammation and apoptosis, angiogenesis and antioxidant capabilities. Deficiencies in HO are associated with several pregnancy disorders, including preeclampsia. With no present cure, this disorder continues to affect 5–7% of all pregnancies worldwide, leading to maternal and fetal morbidity and mortality. Researchers continue to strive for therapeutic potentials and this review will outline the possible use of the HO/CO system as a target treatment/prevention of preeclampsia in the future.
Pregnancy is a state of a series of intricate and complex events that lead to the proper development of the placenta and fetus, in addition to maternal cardiovascular alterations that accompany these happenings. It is well established that the appropriate development of the placental vascular system is necessary for adequate blood flow to the fetus, allowing for proper nutrition and fetal growth. Unsurprisingly, abnormalities in placental development can lead to inadequate delivery of blood and production of factors that spill into the maternal circulation, contributing to complications of pregnancy, such as preeclampsia (PE). PE is a life-threatening disorder that affects roughly 5–7% of all pregnancies worldwide [1]. It is well established that this syndrome arises from inadequate placental development [2], but to date, the exact etiology as well as possible treatment or prevention options remain elusive. A molecule of particular recent interest in the proper development and function of the placenta is heme oxygenase (HO).
HO exists in two isoforms: HO-1, the inducible form; and HO-2, the constitutive isozyme. While HO-2 generally controls HO activity during steady-state conditions [3], HO-1, a heat shock protein, is induced by diverse stress-related conditions, and is capable of cytoprotective and anti-apoptotic actions, protection against inflammation and oxidative stress [4]. Using the substrate heme, HO creates three products in equimolar ratios: carbon monoxide (CO), biliverdin, which is converted to bilirubin, and iron (Figure 1), each possessing biological activity. Both biliverdin/bilirubin have potent antioxidant effects [5], while elemental iron leads to the production of ferritin, an iron sequestering protein, reducing free iron levels and thereby oxidative stress [6]. CO has been implicated in maintaining vascular tone, increasing angiogenesis, and reducing inflammation and apoptosis [7]. This review will aim to highlight the current literature regarding the contribution of HO to uncomplicated pregnancies and its possible role in the development and treatment of PE.
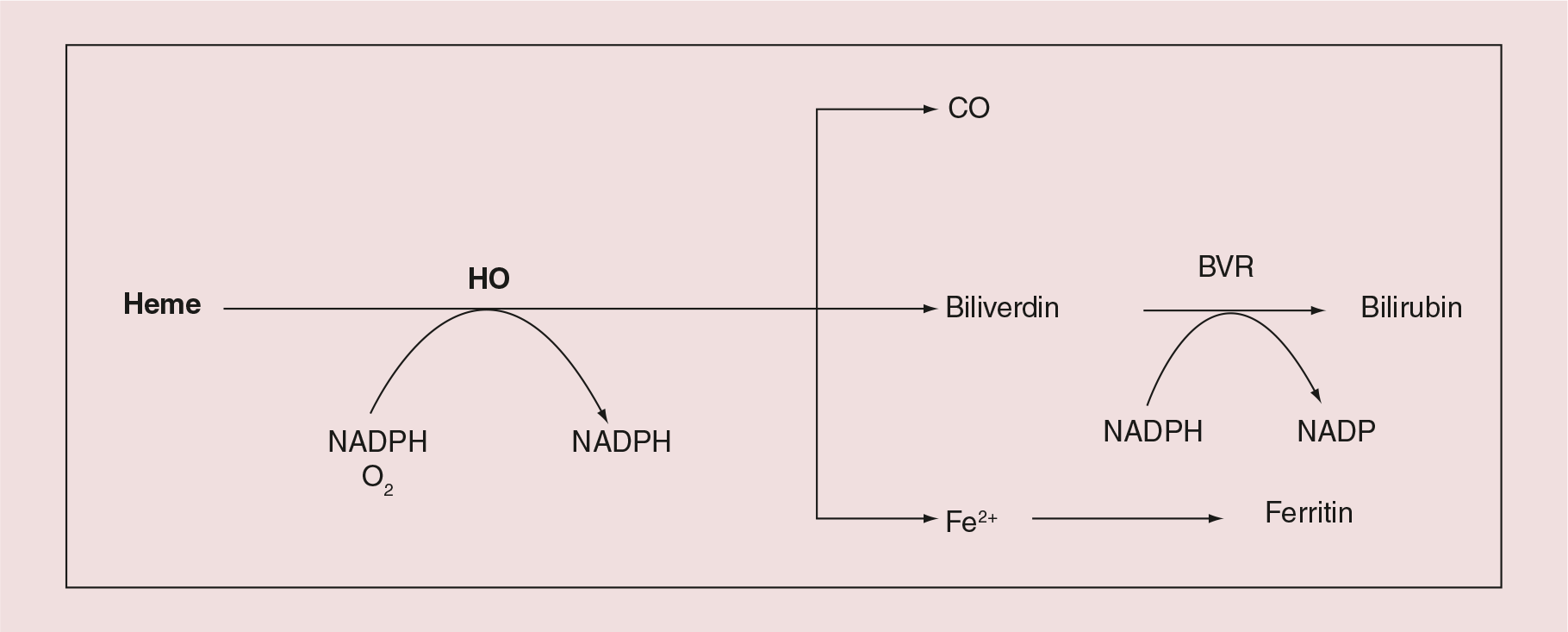
Localization of HO in the placenta & its necessity for uncomplicated placentation & maintenance of pregnancy
In pregnancy, a woman's total HO activity level is increased compared with that of a nonpregnant woman [8]. In the myometrium, both HO-1 and HO-2 have been identified and at a 15-fold increase in pregnant women versus nonpregnant [9]. Several groups have reported finding each isoform in the placenta of humans [10–12], rats [13,14] and mice [15]; however, study results regarding location, temporal fluctuation and relative amounts are inconsistent between groups. There is no doubt that both mRNA [10–12] and protein levels [11,12,16] of HO-1 and HO-2 have been identified in the placenta. The literature is inconsistent when discussing relative amounts, location and temporal changes in each isoenzyme, and this has been reported in recent reviews [17–19]. Using the rat and mouse as placental models, researchers have shown that HO-1 levels are largely elevated at the time of placental vascular development [15] and localized to the feto–maternal interface [13,15] decreasing at term [15]. Additionally, at this time-point transcript levels of HO-1 are still highest in the placenta compared with various organs, functional and actively producing CO [20]; performing at 90% of the levels measured in the spleen [15], the organ known to have the highest HO activity [21]. Therefore, it is evident that HO is not only present in the placenta, but also actively operative. Though discrepancies exist in the literature, HO-1 and HO-2 are spatially and temporally localized throughout the placenta and gestation, indicating a potential role in placenta functionality, proper placenta development and maintenance of healthy pregnancy.
If the HO/CO system is involved in the progression of a healthy pregnancy, then it would be hypothesized that impairment of this system could result in pregnancy complications. Indeed this finding is evident in cases of early pregnancy loss [22] and PE [12,23,24]. Studies have shown lower protein levels of both HO-1 at the feto–maternal interface and HO-2 in invasive trophoblasts of placentas with spontaneous abortion, hydatiform mole, and PE [22]. A significant reduction in HO-1 protein using western blot analysis was shown in the placentas of women with PE at term [12], and a decrease in both isoforms of HO in damaged infarcts of placenta compared with healthy areas, using immunohistochemistry [24]. In addition, HO-1 mRNA is decreased in the blood of women with PE [25]. This data suggest a relation between complications of pregnancy and an alteration in the HO system, but whether it is a cause or consequence of the disorder is not clear by these results. While it is evident that changes in the HO system occur in abnormalities of pregnancy, the localization of both isoenzymes to the chorionic villi, the underlying cytotrophoblast (CTB) and the vascular endothelium [24], in addition to strong expression of HO-1 at the distal ends of cells columns [26] suggest a role in trophoblast invasion and regulation of placental function.
Furthermore, placental HO protein expression is decreased in PE patients [12,26–27], suggesting a role for reduced HO expression in the pathogenesis of PE. Importantly, Farina and colleagues [23] measured HO-1 levels in chorionic villi of women at 11 weeks of gestation and noted that those women who went on to develop PE had decreased levels of HO-1 in these localized areas of the placenta, a finding supporting the role of the HO system in PE.
Rodent models of pregnancy have been used to elucidate the role of HO in maintaining a healthy pregnancy. Researchers have been able to partially (HO-1+/-) [28] and fully (HO-1−/-) [9,29–30] knockout HO-1 in mice, and, in both cases, have reported adverse effects in pregnancy. Both HO-1−/- and HO-1+/- mice exhibit a prolonged time for blastocyst attachment versus regular mice [30]. Maintenance of pregnancy is poor in HO-1−/-mice, as extremely low birth rates [29–31] and poor survival are reported [29]. Furthermore, HO-1+/- mice demonstrate insufficient spiral artery remodeling, evident through vascular corrosion casting of the utero–placental unit [28]. Studies, in vitro, have shown that human trophoblast invasion is inhibited by the HO inhibitor zinc protoporphyrin-9 (ZnPP), or specifically by HO-2 antibodies [32], further supporting the role of HO in facilitating trophoblast invasion of the spiral arteries. HO-1−/- mice also exhibit macromolecular oxidative damage, tissue injury and chronic inflammation [33], in addition to anemia, and accumulation of hepatic and renal iron [33]. This is not unlike a clinical case reported in 1999, a rare double deletion of the maternal and paternal allele for HO-1, leading to no HO-1 production [34]. In this case, a young boy was identified with severe growth restriction and developmental delay, erythrocyte fragmentation and persistent intravascular hemolysis, and an abnormal coagulation/fibrinolysis system [34]. The close comparison of HO-1 knockout mice and the clinical case of HO deletion supports the use of these mouse models for comparison of the effects of HO in human pregnancy.
It is clear that the HO/CO system is important in maintenance of an uncomplicated pregnancy, and its deficiency is associated with gestational complications. The HO−/- and HO+/- mouse models provide evidence that the HO system is necessary for an uncomplicated pregnancy. Studies have shown fetal abortions and low birth weight in HO-1−/- are rescued by maternal low dose (50 ppm) exposure to CO early in pregnancy, gestation day (GD)3–8 [30], demonstrating that this molecule is at least partly responsible for HO/CO's role in pregnancy. It is conceivable then that the increased levels of CO in women who smoke during pregnancy may be providing some beneficial property, perhaps through vasodilatory, anti-inflammatory, or angiogenic properties of CO [7], in reducing the incidence of PE.
The development of PE
As per the new American College of Obstetrics and Gynecology 2013 guidelines, the diagnosis of PE has been altered from new-onset hypertension (HTN) (>140/90 mmHg) and proteinuria (>300 mg/24 h) [35–38]. A diagnosis of PE is not dependent on the presence of proteinuria, but rather hypertension and one of the following: thrombocytopenia, impaired liver function, renal insufficiency (serum creatinine greater than 1.1 mg/dl), pulmonary edema or new-onset cerebral or visual disturbances [35]. PE continues to be one of the leading causes of preterm birth and significant maternal morbidity [39]. It is widely accepted to be a disorder of placental origin which ultimately has an adverse impact on the maternal vascular endothelium, leading to the clinical signs of hypertension and proteinuria. We now know that several pre-existing maternal factors place a woman at increased risk of developing PE, including underlying hypertension, pregestational diabetes, young/advanced maternal age, nulliparity, high BMI, a history of PE and multifetal gestation, as explained in a review by Young et al. [40].
While the exact etiology of PE is not fully understood, it is widely considered to be a disorder of placental origin. In the traditional two-stage theory of PE pathophysiology, first described by CW Redman et al. [2] in 1991, an inefficient utero–placental circulation is thought to initiate hypoxic stress and damage within the developing placenta (stage 1). Damaged placental debris and placentally derived factors are released into the maternal circulation where they result in inflammation and widespread endothelial dysfunction leading to the clinical condition of PE (stage 2). In addition, it is thought that predisposing maternal factors, rendering the mother's endothelium more sensitive to the circulating placental constituents [1], are also necessary to develop PE (Figure 2). The remainder of this review will delineate the two stages of PE, the effects of smoking cigarettes on these stages and potential roles of the HO/CO system in the decreased incidence of PE.

Cigarette smoking in pregnancy, but not smokeless tobacco, reduces the incidence of PE, possibly through increased combustible by-products, such as CO
Ironically, the only external factor shown to decrease the incidence of developing PE is cigarette smoke, reducing the risk by 33% [41,42], in a dose-dependent manner [43,44]. While this seems paradoxical, the finding has been reported in multiple countries and over many years of published reports [41,45–47]. It is well known that smoking cigarettes during pregnancy is associated with a multitude of adverse fetal outcomes, perinatal death [48], ectopic pregnancy [49] and stillbirth [50] to name a few; an average of 10.5% of all pregnant women in Canada (2005–2006) [51] and the USA (2010) [52] smoke annually. Furthermore, babies born to mothers who smoke cigarettes are often preterm [53] and of low birth-weight, on average 200 g lighter per pack per day smoked than a baby born to a mother who does not smoke cigarettes in pregnancy [54]. The question is which of the 4000 toxins found in cigarette smoke [55] may be affecting the maternal/placental development of PE?
Studies have demonstrated the importance of combustible by-products of cigarette smoke. Women who use smokeless tobacco (ST), or snus, do not demonstrate a similar reduced risk of PE compared with women who smoke cigarettes in pregnancy, and in fact are associated with a slight increased risk of developing PE [43,56]. While ST products have comparable concentrations of several of the addictive (i.e., nicotine) [57,58] and toxic compounds [55] found in traditional cigarettes, they do not create combustible by-products of cigarettes smoke, such as CO (Tab le 1) [55]. It is well known that smoking leads to higher levels of CO in one's body [55], suggesting that this increased CO contributes to the lower incidence of PE in women who smoke during pregnancy. In 2004, Kreiser and colleagues [59] published findings indicating that women with gestational hypertension or PE have end-tidal breath CO levels (1.36 ± 0.30 ppm) significantly lower than control, normotensive women (1.72 ± 0.42 ppm, p = 0.001) and nonpregnant women (1.78 ± 0.54 ppm, p = 0.002) [59]. This study was completed in two different medical centers and was controlled for maternal factors, such as age, ethnicity, parity and importantly household smokers. These results led the authors to conclude that the lower CO levels may be a deficient compensatory response to the hypertensive state of PE, or that it may be a primary alteration involved in the pathogenesis of the disorder [59]. These findings also support the idea that the lower incidence of PE among smokers may in fact be due to the production of CO, potentially replacing a deficient level of the endogenous gas. Our research group has also shown that there is an inverse relationship between exposure to environmental ambient CO and the incidence of PE [60]. Maternal data were evaluated retrospectively for all births in Ontario, Canada from 2004 to 2009, and ambient CO levels were obtained from the Ontario Ministry of Environment, Air Quality Ontario website [60]. After adjustment for several confounding factors, the lowest incidence of PE was associated with the highest quartile of CO exposure [60]. The idea for use of end-tidal CO measurements as a predictive marker of pathological conditions in pregnancy has been patented as a possible method to determine the onset of a pregnancy complication, such as PE [61].
CO is produced endogenously in the body through the actions of HO and this review will outline the potential effects of the HO/CO system on the developing placenta and maternal vascular adaptation to pregnancy, highlighting evidence of how these effects may contribute to a reduced risk of PE in pregnant woman who smoke cigarettes.
Stage 1: the impaired development of the placenta contributes to the development of PE
The effect of cigarette smoking on placental development and how it may inhibit the development of PE (summarized in Box 1).
The first stage of PE is thought to be initiated by the compromised establishment of the utero–placental circulation at the maternal–fetal interface. In a healthy pregnancy, the extravillous invasive cytotrophoblast placental cells invade the decidualized maternal endometrium, infiltrating the uterine spiral arteries and replacing the endothelial layers of these vessels. In doing so, they transform these vessels into high-flow, low-resistance conduits, insensitive to vasoactive factors and capable of transporting the expanded blood volume necessary for fetal growth late in gestation [69]. In a pregnancy complicated by PE, there is often shallow extravillous CTB invasion and minimal remodeling of the uterine spiral arteries; an observation noted in many placental bed biopsies of PE patients [70]. These poorly modified arteries continue to respond to vasoactive stimuli in the maternal circulation, creating a pulsatile blood flow to the placenta [70], and maintaining a low-flow, high-resistance structure, significantly reducing utero–placental perfusion [1,71]. The consequences of disrupted blood flow to the placenta are localized areas of placental oxidative stress and hypoxic injury. The pulsatile blood flow causes a reintroduction of oxygen on an inconsistent basis, leading to a production of reactive oxygen species (ROS), resulting in ischemic-reperfusion injury within the placenta. The PE placenta demonstrates evidence of increased apoptosis and necrosis [72], increased shedding of placental debris into the maternal circulation, and leading to an inflammatory response [2]. It is well known that trophoblast invasion and establishment of the utero–placental circulation are established by 20–22 weeks of gestation [73], therefore, although PE is usually diagnosed later in pregnancy, it appears that development of PE begins in the first half of pregnancy.
HO/CO attenuates placental apoptosis
The villous syncytiotrophoblast cells lining the inter-villous space are especially sensitive to the hypoxia-reoxygenation type of injury, resulting in increased shedding of apo-necrotic syncytiotrophoblast debris into the maternal circulation [72,74]. Indeed, an increased load of trophoblast tissue has been measured in the circulation of women with PE [74,75]. Furthermore, the placentas have also been shown to have more overall placental apoptosis compared with controls [76], and more specifically to have an increase in syncytiotrophoblast proliferation and apoptosis [77]. Important to note, in an uncomplicated pregnancy, apoptosis is necessary for habitual growth and development, even increasing near term [78]. However, it is the significant increase observed in PE pregnancies that may lead to negative effects.
The first stage of preeclampsia, the impaired placental development could allow for involvement of the heme oxygenase/carbon monoxide system in Figure 2.
1.1 placental apoptosis
1.2 villous development
1.3 placental hemodynamics
Women who smoke cigarettes (≥10 cigarettes per day) in pregnancy have a decreased level of apoptosis in the syncytiotrophoblast of their term placenta [79,80]. A decrease in placental trophoblast apoptosis (using Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling assay and DNA fragmentation quantification) was observed in term placentas of women who smoke during pregnancy, with an overall decrease in apoptosis from first to third trimester [80]. These two studies used placental tissue exposed to maternal cigarette smoking throughout pregnancy. However, when placental villous explants were exposed to cigarette smoke extract (CSE; the bubbling of lit cigarettes into media) at increasing concentrations, no difference in cell death was apparent between control and CSE exposure groups [81]. This disparity in results may be due to the methodology in creating CSE, as compared with in vivo maternal cigarette-smoke exposure. CSE was created by passing air through lit cigarettes and bubbling this air into media, which was subsequently frozen in aliquots. Though the authors measure nicotine levels in the CSE media, they do not measure CO. It is known that the solubility of CO in water is very low [82], and we have measured the rapid reduction of CO in media using gas–solid chromatography (Venditti CC & Smith GN, Unpublished Data.). If CO is the agent in cigarette smoke leading to a decrease in placental apoptosis, its absence in CSE media would result in differing results. Though the findings differ between the studies, neither case led to an increase in apoptosis in the placenta, indicating that cigarette smoking does not lead to apoptosis of the placental trophoblast cells.
Using an in vitro model of placental hypoxia-reoxygenation injury, we were able to inhibit syncytiotrophoblast apoptosis and secondary necrosis with CO treatment, using concentrations of CO similar to that measured in the blood of women who smoke cigarettes [83]. Furthermore, in a mouse model of abortion, both the introduction of cobalt protoporphyrin (CoPP) [84], a potent HO inducer, and an adenovirus with an HO-1 insert [85] led to decreased apoptosis through the induction of Bag1 (a marker of anti-apoptosis) mRNA measured at the feto–maternal interface. CoPP also decreased markers of cell death (caspase-3 and phosphorylation of JNK and STAT1) in a reduced utero–placental perfusion (RUPP) model in rats [86]. Furthermore, induction of HO by CoPP also led to increased phosphorylation and activation of ERK and STAT3, mediators of prosurvival, in the RUPP rat model [86], resulting in increased fetal survival rates and reduction in the incidence of miscarriage in murine pregnancies [84,85].
Outside of pregnancy, CO has been shown to have potent anti-apoptotic and cytoprotective effects in several organ systems in both physiological and pathological settings [87–91]. It is impossible to determine exactly which of the many chemicals in cigarette smoke affect the apoptosis noted in early or late trimester placentas of women who smoke cigarettes [80]. A decrease of term placental apoptosis has been measured in placentas of women who smoke cigarettes, and this corresponds to a time of increased apoptosis in placentas of women with PE [92]. In addition, when the combustible product CO was analyzed for its effect on placental apoptosis, a significant decrease was reported [83]. It is possible that the elevated maternal CO levels in women who smoke offer cytoprotection for the syncytiotrophoblast from hypoxia-reoxygenation injury and subsequently limit apoptosis and shedding of placental debris into the maternal circulation of women with PE.
HO/CO contributes to standard & adaptive placental villous development
Burton et al. [93] reviewed terminal villi histologically within term placental tissue of women who smoke cigarettes during pregnancy and those who do not, noting that all tissue (in both groups) appeared healthy, and capillaries were well perfused, each with smooth regular endothelium. In the placental villi of women who smoked cigarettes, a decrease in capillary lumen volume fraction, as a percentage of the entire villi volume, was observed [93]. This decrease was dose dependent, as women who smoked more heavily had a more pronounced decrease in capillary lumen volume fraction [93]. In an uncomplicated pregnancy, growth of the placenta increases after 20 weeks of gestation, mainly due to the expanding villous tree [94], the major component of the placental size [95]. Howe and colleagues [96] reported that the placentas of women who smoke greater than 15 cigarettes per day are larger in volume than those of nonsmokers. Pfarrer et al. [95] prepared plastic casts of fetal capillaries within placental cotyledons of four women who smoked cigarettes during pregnancy (>20 cigarettes per day). Using scanning electron microscopy, they observed that the density of the terminal capillary convolutes was increased, as was the branching of the convolutes (adaptive angiogenesis), and they hypothesized that possible compensatory mechanisms were aimed at increasing the oxygen and nutrient exchange surface area of the placenta [95].
Using a pregnant mouse model, our research group demonstrated that exposure to chronic CO (250 ppm) in ambient air (throughout pregnancy) led to an increase in vessel branching and diameters in several of the arteries in the utero–placental unit [97]. This increase was associated with an augmentation of maternal uterine artery blood flow to the placenta [97]. An increase in the number of capillaries per villous compartment was noted in pregnant anemic women, and the authors of this study suggest a possible adaptation to a maternal hypoxic situation [98]. Perhaps the same situation is occurring in the placentas of women who smoke cigarettes, but requires further work to elucidate the effects of smoking on the placental villous development.
HO/CO alteration of the placental hemodynamics
Smoking in pregnancy has been shown to positively affect placental blood flow in a dose-dependent manner [99]. Muller et al. [100] reported a decrease in uterine systolic/diastolic ratio, and they suggested that it was most likely an increase in blood flow, not velocity; as no change in umbilical arterial systolic/diastolic ratio was noted. Nicotine is one of many chemicals in cigarettes and its effect on the vasculature has been consistently shown to be vasoconstrictive [101], as shown specifically in the isolated perfused placenta by our group [102] and others [103,104]. Using the placental perfusion model, our research group [105] has also shown that CO (at levels both below and well above those measured in the blood of women who smoke cigarettes) was able to elicit a concentration-dependent decrease in placental perfusion pressure. We demonstrated that the vasodilatory properties of CO on the placental vasculature were likely mediated through the soluble guanylyl cyclase (sGC) pathway [105], as has been reported in other systems of the body [106]. Using 1H-(1,2,4) oxadiazole(4,3-a)quinoxalin-1-one (ODQ), an sGC specific inhibitor, the CO-induced vasodilation was abolished, while the addition of 3-(5-hydroxymethyl-2-furyl)-1-benzylindazole (YC-1) augmented the CO-induced decrease in perfusion pressure [106]. In mice, the injection of a CO-releasing molecule (CORMA1) lead to an increase in renal blood flow [107], a finding that was abolished when blocked with an sGC-specific inhibitor (ODQ) [107]. Therefore, HO in the placenta may act to increase blood flow through vasodilation of utero–placental arteries. Our mouse model of CO exposure showed an increase in uterine artery blood flow [97], indicating a possible role for HO-mediated vasodilation through CO. In isolated human placentas, a hemin-induced HO-1 upregulation reduced pre-constricted placental artery vascular tension by more than 50% [12], further supporting the idea that HO may be involved in the regulation of placental vascular resistance and subsequent decrease in maternal uterine vascular tone.
HO activity is increased in many maternal tissues in pregnancy, and it is believed to be an adaptive mechanism for the increased circulating blood mass and erythropoiesis in pregnancy, as this would lead to increased red blood cell turnover and an elevation in free heme [8]. When tin mesoporphyin (SnMP; an inhibitor of HO) is administered to pregnant mice, a significant increase in systolic, diastolic and mean arterial pressure (MAP) is observed [8]. This finding was also observed in rats whereby inhibition of HO-1 by SnMP in late pregnancy also leads to a hypertensive phenotype [108]. In a rat model of renal hypertension, administration of SnMP further exacerbated the elevated blood pressure [109]. The administration of CoPP, an inducer of HO, reduces the MAP in a rat model of placental ischemia [110] and renal hypertension [109]. Mice with an HO-1+/- genotype are characterized with increased diastolic blood pressure [29], further implicating the HO system in the maintenance of normal vascular tone. This was observed in mouse fetal umbilical arteries as well, as the addition of SnMP also increases umbilical artery blood velocity acutely, returning to normal within 24 hours [29]. Not only does the effect on HO specifically act on maternal blood pressure, but the exposure of mice [111] and rats [112] to CO significantly attenuates hypertension, leading to a possible mechanism by which the HO/CO system may reduce the development of PE.
George et al. [113] infused pregnant rats with recombinant sFLT1 from GD14 to GD19 using implanted miniosmotic pumps. This procedure increased maternal circulating sFLT1 levels almost twofold, decreased unbound VEGF by 17% and increased the MAP by 17 mmHg on GD18 [113]. In a subset of animals with sFLT1 administration, the researchers then injected CoPP (HO-1 inducer) via the intraperitoneal cavity on GD14 of pregnancy. This induction of HO-1, as measured by western-blot analysis, led to an increase in VEGF levels by 50% and a normalization of MAP [113]. Additionally, the sFLT1 administration led to augmentation of aortic pre-proendothelin mRNA, and this was reduced with HO-1 induction [113]. Activation of the endothelin system leads to potent vasoconstriction [114] and endothelin 1, a marker of endothelial activation, is increased in maternal plasma of women with PE [115]. Statins have been shown to induce HO-1 expression and activity in tissues (as measured by CO) in vivo [116] and in vitro [117], and the exposure of mice to pravastatin reduced the pre-activated contractile responses of carotid arteries to phenylephrine [118]. Furthermore, pravastatin increased the vasorelaxant response to acetylcholine [118]. These studies provide a possible mechanism by which the HO system is implicated in blood pressure regulation and normalization of vascular abnormalities in rodent models of hypertension.
Taken together, the in vitro and in vivo data demonstrate an HO-induced vasodilatation in maternal and placental vessels. These results suggest that the increased CO and induction of the HO system through maternal cigarette smoking may decrease hypertension, improve intraplacental perfusion and enhance oxygenation of the placental tissue. Perhaps, through activation of the HO system and the anti-apoptotic, angiogenic and vasodilatory actions of CO, as we have just described, the incidence and the progression of PE is able to be attenuated.
Stage 2: the maternal endothelial system and PE
The effects of cigarette smoking on maternal adaptation to pregnancy: how it may inhibit the progression of PE (summarized in Box 2).
While the impaired placental development is of central importance in the establishment of PE, we now know that maternal pre-existing or contributing factors are of equal importance in the progression of this disorder [1,104,119,120]. Vascular impairment in PE patients is evident through increased reactivity to vasopressors [121] and by impairment of endothelium-derived vasorelaxation [122]. As reported by several researchers, the hypertension, proteinuria, coagulopathy and liver dysfunction of PE can all be explained by systemic endothelial dysfunction [69,123]. In fact, one of the best characterized abnormalities of PE is glomerular endotheliosis, found in up to 80% of women with the disease [124], and is characterized by glomerular endothelial damage [124]. This renal abnormality is not seen in any other form of hypertension, and disappears following delivery, suggesting that it is not a secondary occurrence to hypertension or hypoperfusion [120]. Following delivery, the clinical signs of PE largely dissipate; however, maternal impaired vascular function can remain for years [125]. We have shown that women with PE demonstrate an alteration in number and function of endothelial progenitor cells (markers of endothelial reparative capacity) at 2 months postpartum and endothelial colony forming units at 2 and 6 months postpartum [126]. Research has shown that several risk factors for the development of PE are akin to those of cardiovascular disorders: obesity [127], hypertension [127,128], diabetes mellitus [129], familial cardiovascular disease [130]; and that a diagnosis of PE identifies women with underlying cardiovascular risk factors [131]. Taken together, these results support the hypothesis that underlying maternal vascular dysfunction may have a role in the pathology of PE specifically sensitizing the mother's vasculature to respond to the increased placental factors abnormally.
The second stage of preeclampsia, the maternal influence on the development of preeclampsia, could involve the heme oxygenase/carbon monoxide system (Figure 2).
2.1 vascular adhesion molecules
2.2 inflammatory dysfunction
2.3 vascular angiogenic molecules
HO/CO system on the maintenance of vascular adhesion molecules in endothelial injury
In PE, there is evidence of increased markers of endothelial injury as early as 20 weeks of gestation, suggesting that endothelial dysfunction is a major contributor to the clinical presentation of PE [38]. Among many markers of endothelial dysfunction, both vascular cell adhesion molecule (VCAM)-1 [132] and the intercellular adhesion molecule (ICAM)-1 [133] are increased in the serum of women with PE. These two molecules mediate adhesion between endothelial cells and leukocytes. Upon exposure of endothelial cells to inflammatory mediators, cellular adhesion molecules recruit and bind lymphocytes, monocytes and neutrophils in the circulation [134], which can lead to vascular damage, endothelial cell destruction, membrane lipid peroxidation and increased vascular permeability, through the release of reactive oxygen species and leukotrienes [135].
It has been proposed that cigarette smoking acts through the actions of the HO/CO system to blunt the immune system [17]. Song et al. [136] stimulated human umbilical vein endothelial cells (HUVECs) with TNF-α, inducing an inflammatory response. They subjected the cells to high concentrations of CORM-3 and reported a downregulation of VCAM-1 expression, as shown with western blotting and flow cytometry [136]. The reduction was mediated through CO, as the use of degassed CORM-3 (no CO within the metal) did not result in VCAM-1 decrease [136]. While CORM-3 addition was also able to induce HO-1 expression levels, blocking HO-1 with sodium nitroprusside continued to reduce VCAM-1 to the same extent, indicating that CO was working independently of HO-1 induction [136]. Zhang et al. [137] exposed HUVECs to oxidized low-density lipoprotein, leading to induction of oxidative stress in the cells and upregulation of both VCAM-1 and ICAM-1 protein levels and macrophage/monocyte chemo-attractant protein, also an adhesion molecule of the endothelium. The oxidative stress also led to increased levels of HO-1 activity, as measured by bilirubin levels [137]. Administration of genistein, a soy-based product which further increased HO-1 mRNA, protein and activity levels, reduced the protein levels of all three adhesion molecules. This decrease was augmented by addition of CoPP (an HO-1 inducer) and abrogated by addition of ZnPP, an HO-1 inhibitor [137]. Using the same cells (HUVECs), Bergstraesser et al. [138] induced inflammatory stress by addition of TNF-α, leading to increased mRNA levels of several adhesion molecules, including VCAM-1 and ICAM-1. The addition of CORM3, which releases CO into the media, led to the decreased mRNA measurement of VCAM-1 and ICAM-1 in HUVECs first stimulated with TNF-α.
Exogenous CO has immunosuppressive effects on macrophages, monocytes and endothelial cells [139,140] and the in vitro studies discussed [136–138], indicate that vascular endothelial injury, introduced following inflammatory responses and recruitment of adhesion molecules, may be reduced and improved, through the actions of the HO system. They also offer a possible mechanism by which CO decreases the immune response, through the reduction of these adhesion molecules, and possibly independently of HO-1.
HO/CO & inflammation in pregnancy
Successful human pregnancy is thought to be a state of immunological tolerance [141] and associated with an increase in maternal Tregs, a unique subpopulation of T cells [142], that help the fetus, expressing paternal antigens, evade a maternal immune attack [143]. Furthermore, the absence of these cells led to failed gestation, as shown in a mouse model depleted of Tregs [143], and abortion-prone mice also exhibit a diminished number of maternal murine Tregs [144]. Data suggest similar mechanism exists in humans [145]. Zenclussen et al. demonstrated decreased HO-1 and HO-2 protein expression by western blotting and immunohistochemistry at the feto–maternal interface of abortion-prone mice [92]. They then isolated Tregs from spleen and thymus of nonpregnant and GD14 uncomplicated pregnant mice and injected them intravenously into these abortion-prone mice [146]. The Treg therapy prevented abortion and, interestingly, increased HO-1 mRNA levels at the fetal-maternal interface [146]. The inhibition of HO-1 using ZnPP abrogated the protective effects of Tregs [147], suggesting that Treg induction, and acceptance of a fetus, may play a role through the HO system. A study by Lee et al. [148] showed that mice injected with simvastatin had increased HO-1 levels compared with control. Furthermore, this group exposed rat aortic vascular smooth muscle cells to lipopolysaccharide (LPS), inducing an inflammatory response, which was ameliorated with simvastatin treatment [148]. Co-administration of LPS with ZnPP, an HO-1 inhibitor, or hemoglobin, a strong binder of CO, abolished these effects, showing that statin treatment works through the actions of the HO-1 system, and more specifically through its product CO, to reduce inflammatory mediators.
Further evidence for HO-1's role in decreasing inflammation has been documented in acceptance of rodent allograft [149] and xenograft [150,151] experiments, whereby HO-1-deficient mice reject transplanted tissue, allograft [149] or xenograft [150], and induction of HO-1 by administration of CoPP (and HO-1 inducer) prior to transplant leads to a decreased rejection of allograft tissue [149]. This is not surprising, as HO-1−/- mice subjected to hypoxia (10% O2), similar to a hypoxic event from organ transplantation, cannot recuperate from injuries properly and exhibit increased platelet aggregation, apoptosis and reactive oxygen species in their system [31]. Medawar first proposed pregnancy to be similar to an allograft, a fetal allograft [141], and pregnancy has since been described as a state of immune tolerance [152]. Therefore, the beneficial actions of HO in transplants may act in pregnancy in a similar fashion. Several groups have shown the HO/CO system contributes to organ transplant success/reduced rejection; and these are reviewed extensively by Katori et al. [153].
We have shown that women who smoke in pregnancy induce HO protein levels within their placentas [154], which could lead to an increased production of endogenous CO, in addition to the increased exposure to exogenous CO produced by cigarette smoke [55]. It is plausible that women who smoke cigarettes in pregnancy, through the actions of heightened HO/CO levels in their system, reduce the endothelial dysfunction and immune responses, thereby decreasing the incidence of PE in their population.
Angiogenic markers of pregnancy & their alterations in PE
The effect of angiogenic molecules has been proposed as possible contributors to the development of PE [40,41,123]. Two angiogenic factors involved in placental vasculogenesis/angiogenesis and maternal adaptation to pregnancy are VEGF and placental growth factor (PGF) [40,123,155]. Invasive CTBs express VEGF and PGF [156] and these angiogenic factors are thought to play a pivotal role for the success of an uncomplicated pregnancy [40]. The levels of both VEGF and PGF are significantly decreased in women diagnosed with PE, as reviewed by Levine and Karumanchi [123], in both maternal serum [157,158] and by placental immunohistochemistry [156]. The soluble splice variant of the VEGF receptor 1, also referred to as soluble fms-like tyrosine kinase-1 (sFLT1) is an anti-angiogenic factor which binds both VEGF and PGF, reducing their interaction with endogenous receptors on the endothelium, and impairing vascular function [123, 157,159]. In pregnancies complicated by PE, an increase in both placental [156,157] and maternal serum [157–158,160] levels of sFLT1 has been reported. Importantly, measured levels of sFLT1 in maternal serum decrease following delivery [157,160–161], as would be expected if the main source of increased sFLT1 were of placental origin [123]. In pregnant rats, the injection of an adenovirus with the sFLT1 construct leads to an increase of sFLT1 in maternal serum and recreates the maternal clinical signs present in PE patients (hypertension and proteinurea) [157,162]. This model has been recreated by several groups in mice [163–165]. Furthermore, increasing sFLT1 levels in rats, using mini-osmotic pumps [113], also led to PE-like signs, as did the RUPP model in rats, whereby maternal sFLT1 levels were also increased [110]. It has therefore been suggested that therapeutics directed at decreasing the circulating levels of sFLT1 could be a possible treatment for PE patients [166]. In fact, a pilot study directed at extracorporeal removal of sFLT1 in the serum of women with very preterm (<32 weeks) PE revealed an acute decrease in the protein, leading to prolonged pregnancy and improved fetal and maternal outcomes [167]. Furthermore, reductions in sFLT1 by increasing maternal VEGF levels, by either adenovirus [164] injections [162] or mini-osmotic pump release [165], reduce the signs of PE. Induction of HO-1 by CoPP in rats normalized sFLT1-induced HTN, increased VEGF levels and decreased pre-pro-endothelin-1, a molecule of vascular activation [110]. Using the RUPP model in rats, induction of HO-1 by CoPP altered the sFLT1/VEGF ratio to one of angiogenesis [110]. Costantine <i>et al. [118] showed that mice injected with Ad sFLT1, when exposed to water with pravastatin, led to lowered maternal plasma sFLT1 levels. Statins have been shown to increase the HO-1 system in vitro [117,148,168] and in vivo [116], and have also been shown to lower sFLT1 levels in in vitro in HUVECs [168].
Cigarette smoking in pregnancy has been reported to correlate positively with an increase in maternal plasma levels of PGF [169,170] and with a marked decrease in maternal serum sFLT1 levels [161,169–171]. Mehendale and colleagues [81] showed that exposing placental villous explants from pregnant women to CSE led to a pro-angiogenic state with reduced sFLT1 production. Furthermore, they reported a trend toward higher PGF levels in the media of these villous explants, at both 48-and 72-h exposure to the CSE [81]. We have previously demonstrated that protein levels of HO are increased in the placentas (basal plate) of women who smoke cigarettes and in trophoblast cells following an in vitro exposure to CSE [154]. To evaluate the in vitro effect of CSE, we bubbled the smoke from cigarettes into media and exposed trophoblast cells (HTR8–svNeo) to differing dilutions of the CSE. In a dose-dependent manner, 0.5, 1.0 and 2.0% CSE solutions increased the protein levels of HO-1 in the trophoblast cells [154]. A study by Cudmore and colleagues [168] found a decrease in sFLT1 production when HO was overexpressed in endothelial cells using an adenovirus with an inserted HO-1 construct. Furthermore, exposure of placental villous explants to VEGF led to an increase in sFLT1 protein and was further increased following inhibition of HO-1 by SnPP or by electroporation with HO-1 siRNA. Cigarette smoking in pregnancy upregulates the HO/CO system in the placenta, potentially leading to the increase in PGF and the reduction in sFLT1 observed in these women, as in vitro inhibition of HO-1 leads to the augmentation of sFLT1.
Studies suggest that CO may be capable of modulating the angiogenic profile in pregnancy. The exposure of tissues to CO, through the addition of CO-infused media or CORM-2 in media, also led to decreased levels of sFLT1 [168]. George et al. have shown that exposure of rat placental villous explants to a hypoxic environment (1% O2) leads to a significant increase in sFLT1 release and superoxide (a marker of oxidative stress) [172]. Both of these markers of injury, sFLT1 and superoxide, were significantly reduced, and to the same degree, when the tissue media was supplemented with CoPP (an inducer of HO-1) or CORM3 (a CO-releasing molecule) [172]. These data suggest that the reduction in sFLT1 in placental villous explants can be mediated through the HO system and further through the actions of CO.
As the induction of HO decreases the release of sFLT1, it would be expected that VEGF bioavailability would increase. In rat vascular smooth muscle cells, a 30% increase in VEGF protein was observed, following the induction of HO-1 by hemin or in the presence of CORMs [173]. The inhibition of HO (by SnPP) led to a decrease in VEGF levels and further decreased the proliferation (26%), migration (46%), tube formation (48%) and capillary outgrowth (30%) [173]. George et al. [113] used mini-osmotic pumps to infuse pregnant rats with exogenous sFLT1 from GD14 to term, inducing PE-like signs (hypertension and proteinuria). The induction of HO-1 by injection of CoPP did not alter the maternal sFLT1 levels, but a significant increase (˜50%) in free VEGF levels was reported [113]. They also reported similar results in a RUPP model of placental ischemia in rats, whereby induction of HO-1 (by CoPP) led to a decreased placental protein ratio of sFLT1:VEGF, a positive shift in the angiogenic profile [110]. Taken together, these data suggest a positive correlation between the HO/CO system and angiogenesis, whereby sFLT1 is decreased and free VEGF is increased.
The evidence generated to date indicates a pro-angiogenic shift in pregnant women who smoke. The combined effects of increased production and/or increased bioavailability of pro-angiogenic factors by cigarette smoking may contribute to the reduced incidence of PE observed in this population of women. Perhaps it is through pro-angiogenic effects on the placenta itself, as evidenced through increased branching of the utero–placental vasculature in mice exposed to low-dose CO [55] and a shift to placental pro-angiogenic molecules [97,110,113] and/or through the maintenance of maternal vascular endothelial health and function, through the reduction of endothelial adhesion molecules [137–138,174], inflammatory cytokines [150,175] and general apoptosis [150,175]. In the presence of cigarette smoke, HO seems to be upregulated, leading to an increase in angiogenic factors and a decrease in anti-angiogenic molecules. These results suggest an important role for HO and its by-product CO, in modulating the angiogenic profile in pregnant women, and these observations may in part explain the decreased incidence of PE observed in women who smoke during pregnancy.
Use of therapeutics in PE
Use of CO inhalation as a possible treatment in PE
Over the last decade, research on the HO/CO system in disease and therapeutics has garnered widespread interest. Well known for its toxic nature at high levels; CO is lethal when inhaled in air at levels above 30,000 ppm, or leading to percent carboxyhemoglobin (%COHb) levels above 50 [176]. Exposure leading to levels of 30–50% COHb results in hypoxia, leading to headaches and shortness of breath [176]. While extremely high CO exposure is toxic, low-level CO exposure is now thought to have potential for therapeutic use [177]; low-level CO is produced endogenously in almost every nucleated cell of the body [178], leading to endogenous levels of 0.5–1.5% COHb [179]. Smoking cigarettes leads to exogenous production of CO through combustion of cigarette constituents, leading to levels of 400 ppm at the alveolar membrane [180] and resulting in levels up to 17% COHb [181]. Using these %COHb levels as a guide, research in the use of CO as a therapeutic must ensure blood levels of CO are at or below levels of those who smoke, and well below those of toxicity. Pregnancy imparts an added factor into the equation; the fetus. CO passively crosses the placenta and it is well known that fetal hemoglobin has a much higher affinity for CO than that of the mother [182]. Longo et al. [183] measured CO levels in pregnant ewes and their fetuses following maternal exposure to CO (30–300 ppm) and reported that fetal %COHb levels rise much more slowly than those of their mother, but over time reach a level 15–20% higher, with a much longer half-life in the fetus (7 h) compared with their mother (2 h) [183]. This information is important to keep in mind when testing therapeutic potentials of the HO/CO system in pregnancy.
The HO/CO system is thought to be important in the development of an uncomplicated pregnancy and alterations in the system may lead to the development of placenta-related complications such as PE. Women with PE have lower end-tidal breath CO levels compared with healthy pregnant women [184]. Women who smoke in pregnancy have a reduction in the incidence of PE [41,42] and we have reported in the literature possible mechanisms by which the HO/CO system may be implicated in this in decrease. Our group has also shown that exposure to environmental ambient CO is independently associated with a decreased risk of PE [60], further supporting the HO/CO system in the development of PE.
It is possible that manipulation of the HO system and/or administration of exogenous CO may be used in the future as a possible therapeutic, and many researchers have published their findings in support of using this system as a possible therapeutic for pregnancy complications [17,19,39,166,185]. Clinical studies of CO in a pregnant population would only be possible following extensive studies demonstrating its safety first. As a start, we have tested the effect of maternal chronic CO exposure in ambient air in pregnant mice, showing that exposures below 300-ppm CO do not result in any demonstrable fetal or maternal adverse effects [186]. Although the induction of the HO/CO system has been shown to decrease inflammation, induce an angiogenic profile, increase vasodilation and reduce apoptosis in many in vitro and in vivo animal models, studies with human CO inhalation for experimental procedures are few. To our knowledge, there have been two published studies using inhaled CO in a clinical setting, providing a guideline for a safe concentration of CO for administration.
Mayr et al. [187] used healthy human subjects to examine systemic inflammation during experimental endotoxemia, using LPS as an inflammatory inducer. Volunteers were injected with 2 ng/kg of LPS in a randomized, double-blind, placebo-controlled, two-way cross-over design. Following the injection, subjects were randomly assigned to inhale 500-ppm CO, 250-ppm CO or synthetic air for an hour, after which they received another bolus of LPS [187]. Subject %COHb ranged from 1.2 to 7%, using a spectral photometric measuring system; levels below 10% COHb were assumed to be innocuous, as this value was the maximum recorded for a smoker [187]. No effect of CO on the modulation of inflammatory cytokines was observed in this experiment; however, CO inhalation did not affect volunteer vital signs measured by the researchers and this study provides a foundation for future clinical experiments using CO as a therapeutic [187]. In contrast, a study conducted in patients with chronic obstructive pulmonary disease (COPD) demonstrated that exogenous inhaled CO led to decreased leukocytes (eosinophils) in the sputum of these patients [188]. The volunteers were ex-smokers with stable COPD [188]. They were given CO by inhalation (100–125 ppm) for 2 h/day for 4 days. %COHb levels increased to 4.5%, with trends to decreased eosinophils and an improved airway hyper responsiveness [188].
As part of a larger study, our group has examined the effect of 250-ppm CO inhaled over the period of an hour on %COHb level (Figure 3A), breath CO (Figure 3B) and blood pressure (data not shown). During the 4-h wash-out phase, samples were collected to determine %COHb and breath CO (ppm). A further 1 h of CO inhalation then occurred (250 ppm), and again CO in blood and breath was measured an hour after inhalation and the following morning (˜22 h from the first CO inhalation). The half-life of CO in the human subjects was 4.68 ± 1.83 h. The highest level of blood and breath CO levels respectively were 12.0% COHb and 53.5 ppm. Volunteers reported feeling flushed, but no demonstrable health effect was observed in any of the volunteers, as determined by the absence of visual blurriness, headaches or abnormal spO2 levels. This study provides the basis for further studies on the use of exogenous CO as a therapeutic in human disease states. As recent work has demonstrated benefits in treating mice with CO in ambient air at different time-points in pregnancy, to reduce fetal growth restriction and miscarriages [30,189] and to increase blood flow to the utero–placental unit [97], the potential for use of CO in pregnancy is not one without merit.

Conclusion
While the incidence of PE is significantly reduced in women who smoke while pregnant, the toxic effects of cigarettes are significant. The observation that use of snus in pregnancy did not realize the same reduced incidence in PE led to the hypothesis of the combustible product of cigarettes, CO, affording the beneficial effect observed. The HO/CO system is involved in the progression in uncomplicated pregnancy, with a major role observed within the placenta and its proper development and function. Alterations in this system may then lead to the progression of PE through impaired placental development in addition to maternal underlying endothelial activation (Figure 2). The effects of the HO/CO system on the endothelium, inflammatory markers and angiogenic factors offer a secondary approach by which the HO/CO system may benefit women from developing PE. This review has outlined several different approaches that are currently being studied to alter the HO/CO system and could be used as potential therapeutics in the future (Table 2). The proposal for use of the HO/CO system as a treatment for PE is one of evidential merit and an intriguing possibility for a therapeutic in the future.
Identification of important constituent differences between cigarette smoke and oral smokeless tobacco; their effect on the development of preeclampsia.

Modulators of the heme oxygenase/carbon monoxide system.

CO: Carbon monoxide; CoPP: Cobalt protoporphyrin; CORM: CO-releasing molecule; HO: Heme oxygenase; ZnPP: Zinc protoporphyrin. For a more in depth explanation of these and other inducers, please refer to the review written by Li et al. [190].
Future perspective
Clinical trials as pilot runs for possible PE therapeutics Statins & PE
Statins are plasma lipid-lowering drugs [191], which have been shown to offer protection toward cardiovascular disease [192]. The actions of statins are thought to be pleiotropic, with effects including control of endothelial dysfunction and inflammation, mediating anti-proliferation, modulation of the immune system, antioxidant actions [193,194] and angiogenic mechanisms [195]. In vitro studies with cultured cells [148,196] and in vivo animal work [116,148] have shown that statins are able to induce transcription and translation of HO-1. In addition, a study in mice by Muchova et al. [116] identified that the daily injection of both atorvastatin and rosuvastatin over a period of 2–3 weeks led to an increase in tissue-specific HO-1 activity, as measured by CO and bilirubin levels. Furthermore, the mice exhibited antioxidant protection, as measured by lipid peroxidation, which was completely abolished by administration of SnMP, an HO-1 inhibitor [116]. These findings indicate a possible mechanism by which statins may confer their protective effects, through HO-1 and its production of CO and bilirubin, and offer a possible role for statins in the treatment of PE.
The use of statins in pregnancy is contraindicated and designated as pregnancy category X by the US FDA, as the risks in pregnancy are thought to outweigh the potential benefits. Furthermore, studies conducted in early trials for lovastatin revealed fetal anomalies at maternally toxic doses of the drug [197]. More recent studies indicate that use of statin therapy in first trimester is safe and does not lead to increased fetal abnormalities [198–200], in addition to retrospective data reviewed by manufacturers of different statins [201], and the use statins in pregnancy is reviewed by Morton and Thangaratinam [202]. A study conducted by Kumasawa et al. [203] performed in mice showed that placentally derived sFLT1 (by lentiviral injection into the trophectoderm of the blastocyst) led to increased maternal plasma sFLT1 levels and signs of PE. The intraperitoneal injection of pravastatin ameliorated the HTN and proteinuria, reduced sFLT1 levels and induced PGF [203]. Fox et al. [204] found that exposure to water with pravastatin prevented vascular dysfunction and upregulated normal vasodilatory mediators in mice injected with AdsFLT1, exhibiting PE-like signs. Lastly, a study in mice deficient in complement component C1q produces PE-like signs in mice, HTN, albuminuria, endotheliosis, decreased placental VEGF, increased maternal sFLT1 and increased fetal death [205]. Pravastatin injection from GD4 to GD12 completely ameliorated all of the signs of PE [205]. These results further strengthen the idea that pravastatin could be used as a therapeutic for PE, and this idea has been further reviewed by Costantine and Clearly [206]. At this time, two studies involving statins are being conducted in pregnant women. At the Eunice Kennedy Shriver National Institute of Child Health and Human Development, researchers started a double-blinded, placebo-controlled pilot trial to collect pharmacokinetic data and preliminary maternal–fetal safety data for pravastatin in pregnancy [207]. A pilot study for the StAmP trial (statins to ameliorate early-onset preeclampsia) is currently being conducted in the UK, in a multicenter, double-blind, randomized, placebo-controlled trial [208]. To be eligible for the study, women must be between 24+0 and 31+6 weeks gestation, with a singleton pregnancy, diagnosed with early-onset PE, capable of maintaining pregnancy for 48 h and carrying a viable fetus. When recruited, women are randomly allocated to receive either oral pravastatin (40 mg) or placebo, once daily, until delivery. The primary outcome of the study is to evaluate the effect of statins on sFLT1 at 48 h post-randomization, with several secondary measures also being evaluated: BP, proteinuria, total hospital stay and neonatal outcomes, to name a few. This study will provide insight in the possible use of statins for the treatment of PE and allow for development of future therapeutics to modulate the HO/CO system further.
Executive summary
Localization of heme oxygenase (HO) in the placenta and its necessity for normal placentation and maintenance of pregnancy
– HO is an endogenous enzyme which functions to break down the pro-oxidant heme into carbon monoxide (CO), biliverdin and free iron.
– It has many physiologic functions throughout the body, including control of vascular tone, regulation of inflammatory and apoptotic processes, angiogenic modulation and anti-oxidant capabilities.
HO alterations in normal and complicated pregnancies
– HO is increased in the maternal system and the placenta in pregnancy.
– Alterations or deficiencies in the level of HO/CO are associated with pregnancy complications, such as miscarriages and preeclampsia (PE).
The development of PE
– Pregnancy disorder affects 5–7% of all pregnancies worldwide and is a leading cause of maternal and fetal morbidity and mortality.
– It is characterized by de novo hypertension coupled with proteinuria (usually presenting late in gestation).
– PE is characterized as a two-stage disorder: Stage 1: improper placental vascular development; Stage 2: maternal endothelial activation with evidence of clinical signs of PE (hypertension and proteinuria).
Cigarette smoking in pregnancy, but not smokeless tobacco, reduces the incidence of PE, possibly through increased combustible by-products, such as CO
– Cigarette smoking in pregnancy is associated with a 33% decreased incidence of PE.
– The increased level of CO due to cigarette smoke is hypothesized to lend beneficial properties to decrease the incidence of PE, as outlined in this review.
The effect of smoking on placental development and how it may inhibit the development of PE
– HO attenuates placental apoptosis.
– HO contributes to normal and adaptive placental villous development.
– HO/CO alter placental hemodynamics and contribute to maternal blood flow alterations.
The effects of smoking on maternal adaptation to pregnancy: how it may inhibit the progression of PE
– HO contributes to the maintenance of maternal vascular adhesion molecules.
– HO reduces inflammatory dysfunction.
– HO contributes to an increase in vascular angiogenic molecules and a concomitant decrease in anti angiogenic molecules.
Footnotes
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.