
Abstract
Cardiovascular disease is the leading cause of morbidity and mortality for both men and women in the USA. However, there are differences between the sexes in age-dependent onset, severity, symptoms and outcomes. Basic research into the causes of sex-dependent differences in cardiovascular disease is ongoing and includes investigation into genetic variation in expression and distribution of receptors for the sex steroids; specificity of natural and synthetic ligands that activate the sex steroid receptors; and intracellular mechanisms that are activated by the receptors in all components of the vessel wall and blood elements, which integrate to regulate vascular tone, vascular repair and remodeling in health and disease. In this era of personalized medicine, basic research into mechanisms of sex differences in vascular function will result in improved prevention, detection and treatment of cardiovascular disease in both men and women.
Keywords
There is good news: according to the latest statistics from the American Heart Association (AHA), the mortality from cardiovascular disease in the USA has declined by 29.2% between 1996 and 2006 [1]. However, the bad news is that the death rate from cardiovascular disease for women continues to exceed that of men [1]. What are the reasons for this difference? Does it reflect differences in knowledge and attention to symptoms, access to care, or quality of care delivered to women [1–4]? What are the defining biological factors contributing to sex differences in risk, presentation, and progression of cardiovascular disease? Studies in humans and animals identify important differences in the function of the vascular endothelium and smooth muscle between males and females, including differences in the production of endothelium-derived vasoactive/proliferative factors, responsiveness of the vascular smooth muscle to these endothelium-derived factors, sympathetic adrenergic neurotransmission and activation and adherence of blood elements to the vascular wall [5–19]. Dysregulation of these processes contributes to the development of hypertension, ischemia and atherosclerosis leading to myocardial infarction and stroke. Owing to the fact that the incidence of cardiovascular disease occurs in men at earlier ages than in women, and because the incidence of cardio vascular disease increases in women after menopause and is reduced in postmenopausal women who are using hormone therapy to relieve symptoms of menopause, it was hypothesized that the sex steroid estrogen was protective against progression of cardiovascular disease [20–24]. This article will expand upon this hypothesis by examining the interactions between genetic and hormonal factors contributing to sex differences in vascular function, discuss ongoing research into the effects of hormonal treatment and cardiovascular disease in menopausal women and identify the challenges remaining for cardiovascular research in order to reduce sex disparities in the prevention, diagnosis and treatment of cardiovascular disease.
Genetics of sex differences
What are often defined or categorized as sex differences in vascular functions result from interactions of hormonal milieu with gene expression on the sex chromosomes and autosomes. Therefore, it is important to determine whether a particular characteristic can be attributed to the compliment of sex chromosomes alone or the actions of gonadal steroids (hormonal status) at the time of testing, or hormonal effects on sexual differentiation during development or environmental influences that might impact sexual differentiation and hormonal status [25]. While new animal models are being utilized to investigate these determinants [26], these have not gained widespread usage, nor were they available at the time many studies examining sex differences in vascular function were performed. However, even today, basic science and preclinical studies to investigate causation and examine treatments for cardiovascular disease are not being designed with consideration to the sex, age and hormonal status of the experimental animals. Furthermore, experiments using cells
Sex versus gender
The Institute of Medicine report ‘Exploring the Biological Contributions to Human Health: Does Sex Matter?’ differentiates the term gender from sex by the socio/psychological factors that influence how an individual perceives him or herself within the context of society [31]. Male and female sex, then, is defined genetically by the sex chromosomes (XX or XY) and the development of functional reproductive organs. This distinction is important because every cell has a sex that is defined by the pair of sex chromosomes it contains.
Cardiovascular risk & Y chromosome
Perhaps the most well-studied gene on the Y chromosome associated with cardiovascular risk is the

Integrated actions of sex steroids on the control of vascular function.
Cardiovascular risk & the X chromosome
Any genetic polymorphisms of genes expressed on the somatic region of X chromosome will be expressed in males but will show mosaic expression in females owing to heterogeneous inactivation of one X chromosome [37]. This mosaic expression will include those genes implicated in the development of cardiovascular disease such as those encoding small integrin binding
Hormonal status affects vascular function in part through binding of the sex steroids to specific receptors, which themselves activate or repress transcriptional activity in target cells including the endothelium and vascular smooth muscle. The gene for the androgen receptor is located on the X chromosome. The CAG repeat polymorphism on exon-1 of the gene encodes the transcriptional regulatory domain of the androgen receptor. The length of this domain is inversely correlated with transcriptional activity of the receptor. In adolescent and adult males, shorter repeat segments (associated with greater androgen receptor activity) are associated with lower levels of high-density lipoprotein (HDL), increased intra-abdominal fat, lower estrogen levels, reduced endothelium-dependent vasodilatation, elevated blood pressure and high sympathetic vasomotor tone [40–45]. Although there are inconsistent reports regarding the relationship of these repeat segments with the frequency of coronary artery disease and the incidence of myocardial infarction [40,41], these phenotypes fall within the list of canonical risk factors for cardiovascular disease.
Positive associations related to CAG repeat frequency and free testosterone levels have been identified in women with polycystic ovarian syndrome and in some but not all ethnic groups [46–48]. Women with polycystic ovarian syndrome are at increased risk for cardiovascular disease related to dyslipidemia, glucose intolerance and metabolic syndrome, which is similar to the phenotypic characteristics of men with short CAG repeats [49,50]. Analysis of the CAG repeated regions were not associated with changes in either HDL, blood pressure or sympathetic activation in young, healthy women, which may reflect dilution of repeat effects due to X inactivation as results are expressed using the mean of the two alleles of the polymorphism in the analysis, as it is not possible to determine which allele might be expressed in a particular tissue [45].
Cardiovascular risk & the autosomes
It is now clear that sexually dimorphic expression of genes extends to those located on autosomes; for example in mice, there is sexually dimorphic expression of genes in the liver and adipose tissue affecting steroid and fatty acid metabolism, lipid metabolism and immune responses
Functional categories of genes showing sexually dimorphic expression in liver and adipose tissue of male and female mice.

Gene categories in common between tissues are in bold.
Data taken from [51].
While not all genes may show sexual dimorphic expression affecting cellular processes, there is insufficient information to determine whether or not there is sexual dimorphic expression with aging and changes in hormonal status. Therefore, it is important that scientists who utilize
Relative representation of women in affected patient populations and inclusion in randomized clinical trials of cardiovascular disease prevention.
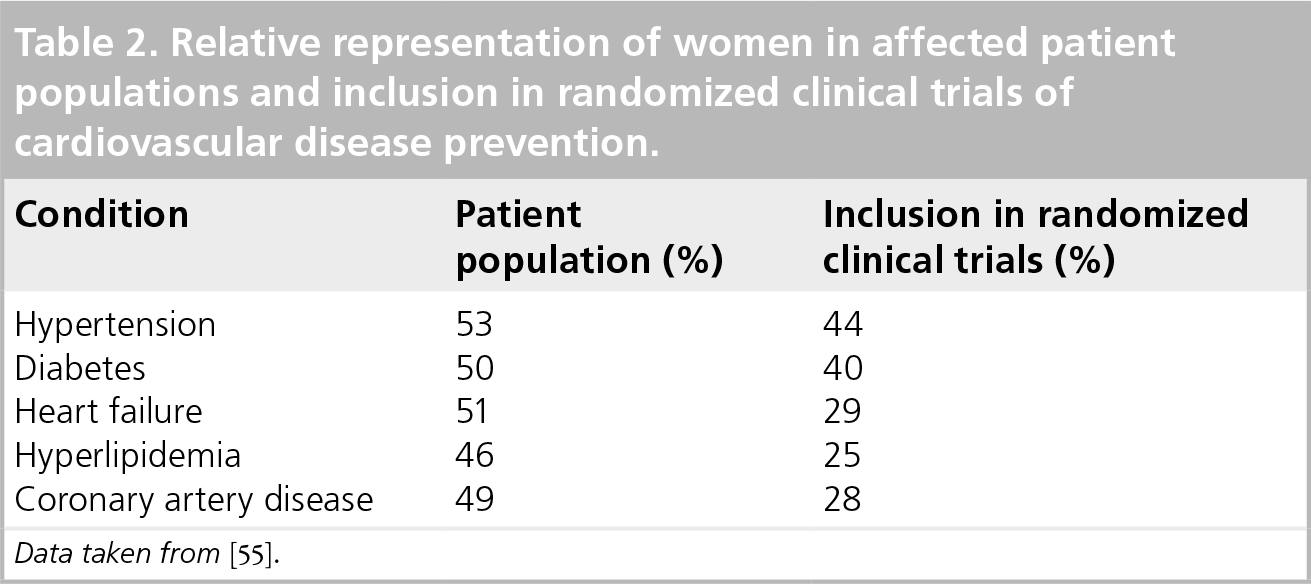
Data taken from [55].
The sex steroids modulate gene expression on the sex chromosomes and autosomes through specific receptors that bind to nuclear binding domains and modulate gene expression in distinct sequences of cascades, which help to regulate vascular function including lipid metabolism, cell proliferation and growth [56].
Genetics of estrogen receptors
Most effects of the female sex steroids (e.g., 17β-estradiol, estrone and estriol) are mediated through the activation of one of two identified estrogen receptors. Genes for these receptors, α and β, are located on autosomes 6q25.1 and 14q22-q24, respectively [57]. Therefore, polymorphisms in these genes, which dictate receptor integrity, have the potential to impact development of a variety of cellular functions implicated in cardiovascular diseases. The first piece of evidence to link estrogen receptors to cardiovascular disease in humans came from a man with a premature stop codon in estrogen receptor-α. Flow-mediated reactive hyperemia of the brachial artery, indicative of endothelial function, was absent in this man; atherosclerosis was accelerated [58,59]. These observations indicated that estrogen receptor-α contributes to maintaining endothelial function associated with flow-mediated dilatation through the release of endothelium-derived relaxing factor nitric oxide, in males. Subsequent studies support expression of estrogen receptor-α and its modulation by estrogen status in women [19,60].
Estrogen receptor-β shares approximately 97% homology with estrogen receptor-α in their DNA binding domains. Both receptors are expressed in the endothelium and vascular smooth muscle. However, the relative distribution of estrogen receptor-β may be correlated with arterial calcification, hypertension and ventricular hypertrophy [61–64].
Genetic polymorphisms in estrogen receptors are associated with various risk factors and the risk of cardiovascular disease. Polymorphisms in estrogen receptor-β are associated with estrogen levels in women [65]. Other associations between single nucleotide polymorphisms (SNPs) in either estrogen receptor-α or- β and cardiovascular risk factors in men and women include insulin resistance, diabetes, BMI, hypertension, plasma lipids and endothelial function [66,67]. Although some SNPs associated with estrogen receptor-α were predictive of adverse events in both men and women, adverse events associated with estrogen receptor-β, so far, have been identified only in women
Cardiovascular consequences associated with single nucleotides polymorphisms in estrogen receptors in men and women.

f: Female; IHD: Ischemic heart disease; m: Male; MI: Myocardial infarction; SNP: Single nucleotide polymorphism.
Genetics of steroid metabolism
Androstenedione is converted to testosterone by 17β-hydroxysteroid dehydrogenase or to 17β-estradiol (estrogen) by aromatase (CYP19A1) in both men and women. Of the two enzymes, most evaluations of genetic polymorphisms related to cardiovascular risk have focused on aromatase. Two SNPs (rs700518 and rs726547) are associated with the relative ratio of testosterone to estrogen in men [65]. In the Study of Women's Health Across the Nation (SWAN), five different SNPs in the aromatase gene were evaluated relative to serum testosterone:estrogen concentrations. Of these, CYP19 rs936306 showed largest allele frequency in African – American women and was associated with a low testosterone to estradiol ratio [75]. Other polymorphisms of CYP19 are associated with insulin insensitivity and diabetes mellitus in women; both conditions are risk factors for cardiovascular disease [76]. How any of these various SNPs directly affects specific function of the vasculature remains to be determined. In addition, whether changes in the relative concentrations of testosterone to estrogen can be defined as either deleterious or protective to the vasculature will depend, in part, upon the integrity of their respective receptors [77].
Testosterone is also metabolized by 5α-reductase to 5α-dihydrotestosterone, which has a higher affinity than testosterone for the androgen receptor. However, no information is available regarding genetic variation in this enzyme and cardiovascular risk in men or women.
Estrogen (17β-estradiol) is metabolized to catechol estrogens. One such compound, 2-hyrdoxyestradiol, binds DNA-promoting development of tumors [78]. Conversion to 2-methyoxyestradiol by
In addition to specific polymorphisms in enzymes metabolizing sex steroids, copy numbers of these genes may also affect phenotypic responses to the hormones, for example, the activity of hydroxysteroid sulfotransferase (SULT1A1), which is responsible for the conversion of 17β-estradiol to estradiol sulfate and estrone to estrone sulfate, was best related to gene copy number [82]. Whether differences in activity of this enzyme affect hormonally driven cardiovascular phenotypes remains to be determined. However, the possibility that genetic polymorphisms and gene copy number affect responses to drugs (or hormones) suggests that a polygenomic approach may be needed to better understand sex differences in vascular responses [81,83].
Distribution of sex steroid receptors
In addition to genetic differences in structure of the sex steroid receptors, their tissue-specific distribution will also influence sex differences in cardiovascular function. However, few studies have examined or compared the distribution of estrogen receptors in vascular tissue between males and females of specified hormonal status. Estrogen receptors are present in all components of the blood vessel wall: endothelium, smooth muscle and adventitia. In arteries and veins collected as surgical waste from patients undergoing vascular repair, mRNA for estrogen receptor-β was greater than mRNA for estrogen receptor-α and averaged approximately 10–20% higher in tissue from females than males [84,85]. These differences in mRNA for the estrogen receptors are consistent with increased expression of estrogen receptor-β in coronary arteries of men and women. Estrogen receptor-β was greatest in the media compared with either the intima or adventia and was also localized in areas of arterial calcification in arteries from males and females [61,86].
The expression of estrogen receptor-α did not correlate with areas of calcified plaque nor was expression altered with hormone treatment of postmenopausal women. However, expression decreased with age only in postmenopausal women using hormonal treatments [61]. The relative expression of either estrogen receptors did not vary with hormonal treatments in postmenopausal women in any layer of the blood vessel wall when examined by immunohistochemistry [61]. This observation differs from changes in the ratio of estrogen receptor-α to -β evaluated by western blot in aortic endothelial cells derived from pigs following ovariectomy or following treatment of ovariectomized animals with either 17β-estradiol, conjugated equine estrogen or raloxifene [87]. Expression of estrogen receptor-α evaluated by quantitative immunofluorescence in endothelial cells of peripheral veins increased with increasing estradiol during the estrus cycle in premenopausal women and decreased in menopause [60]. Regulation of the expression of estrogen receptors by endogenous and synthetic ligands will most likely be influenced by the concentration of ligand, however, systematic studies are needed for comparison of such variation in males [85,88,89].
The expression of androgen receptors was also greater in the media compared with expression in the intima and adventia of coronary arteries of human males. Expression was negatively correlated with plaque area in diseased coronary arteries [86]. As with variation in expression of estrogen receptors by specific ligands, expression of androgen receptors fluctuate in cerebral arteries of rats following manipulation of endogenous testosterone by orchiectomy and testosterone treatment [90]. It is not known how endogenous fluctuations in testosterone affect androgen receptor expression in human vasculature, or how differences in expression may be related to development of cardiovascular disease. Relationships among androgen receptors with estrogen receptors-α, -β or other steroid receptors (i.e., progesterone or corticosteroids – steroid hormones that are not covered in this article) and their competition for cofactors, which might affect receptor binding to nuclear response element requires further investigation [19].
Most studies have evaluated distribution of sex steroid receptors in conduit arteries prone to atherosclerosis. However, no attention has been paid to their distribution and function in the coronary microvasculature. Such analyses might warrant investigation given that coronary microvascular ischemic disease is more prevalent in women than in men [3,4,91].
Ligands & receptor activation
Endogenous ligands for the androgen receptor (testosterone and 5α-dihydrotestosterone, and the estrogen receptors, 17β-estradiol, estrone and estriol) are present in men and women. Testosterone is produced primarily from the testes in males and 17β-estradiol is produced primarily from the ovaries in females. However, in both sexes, the adrenal glands may also be a source of these hormones and testosterone can be aromatized to 17β-estradiol by other nonreproductive tissue such as the vasculature. Therefore, monitoring concentrations of hormones in the blood provides only a partial indication of the potential concentrations at the target tissue.
Dihydrotestosterone has a higher affinity for the androgen receptor than testosterone [44]; when compared to 17β-estradiol, estrone has greater binding affinity for both the estrogen receptors than estriol [92].
Activation of steroid receptors is independent of genetic sex as is evidenced by hormonal treatments with testosterone or estrogenic compounds to individuals undergoing procedures for gender transformation of female-to-male (FM) and male-to-female (MF), respectively. In terms of cardiovascular function, forearm flow-mediated vasodilatation increased following MF but decreased with FM procedures to levels comparable with that observed in females and males, respectively [93,94]. The incidence of venous thrombosis increases in MF persons to a similar degree to what might be expected in women using exogenous hormone treatments [95,96]. Changes in sympathovagal activation and plasma lipids are also reported in transgendered individuals [97,98]. Adverse cardiovascular events such as stroke and myocardial infarction were more frequent in MF than FM individuals [98]. However, it is not possible at this time to determine whether differences in these thrombotic-associated events reflect the type of hormonal product used, since oral estrogenic products are more thrombogenic than transdermal products, or the result of different hormonal modulation of other processes dictated by an XY genotype or epigenic imprinting [99–101]. In FM individuals, decreases in HDL and increases in systolic blood pressure are consistent with patterns of lower HDL and higher systolic pressure in males compared with females [1,98]. However, the quality of long-term outcome data in transgendered individuals are somewhat negated by imprecision in data reporting and heterogeneous study designs [98].
Several synthetic compounds termed selective estrogen receptor modulators have been developed with the idea to target the tissue-specific beneficial effects of estrogen. Tamoxifen and raloxifene are two such compounds approved in the USA for women as adjuvant treatment of breast cancer and osteoporosis, respectively. However, both compounds increase the risk of venous thrombosis and neither are as effective as estrogen in relieving symptoms of menopause, nor are they recommended for the prevention of cardiovascular disease [102].
Sex steroids & vascular function
Receptors for sex steroids are distributed at the membrane surface, throughout the cytosol, in the mitochondria and nucleus of cells

Genomic and nongenomic mechanisms by which estrogen affects functions of the vascular endothelium and smooth muscle.
There is controversy regarding the exact characterization of estrogen membrane receptors [106–108]. However, these receptors seem to be present in cells and tissues derived from both males and females [19,94,109,110]. Indeed, acute infusion of estrogen into arteries or when applied to isolated rings of vascular tissue causes dilatation owing to both an increased release of nitric oxide from the endothelium and the inhibition of ion channels in the smooth muscle [64,111–113–115].
Whether acute administration of testosterone causes dilatation of the arteries and activates the release of nitric oxide directly through activation of androgen receptors or indirectly through aromatization to estrogen remains controversial [116–120]. Details regarding specific intracellular signaling pathways leading to steroidal regulation of nitric oxide have been reviewed elsewhere [19]. However, the net effect of testosterone may not enhance nitric oxide production for sustained time periods as does estrogen. Indeed, serum nitric oxide decreases with increasing concentrations of testosterone with puberty in pigs [121] and with hormonal treatment of FM transgendered individuals (see previous section), which, emphasizes the need to distinguish acute nongenomic actions of the sex steroids from genomic actions.
Other endothelium-derived factors affected by sex steroids include angiotensin converting enzyme, endothelin-1, metabolism of arachidonic acid to prostacyclin (predominately in females) or thromboxane (predominately in males) and endothelium-derived hyperpolarizing factor [104,108]. Sex differences in the production of these factors will also have implications for sex differences in response to some therapies used to treat cardiovascular disease, including angiotensin converting enzyme inhibitors, aspirin, endothelin-1 and angiotensin receptor antagonists [53].
In vascular smooth muscle cells, the predominant nongenomic effects of sex steroids include regulation of membrane potential, intracellular calcium concentration and calcium sensitivity of the contractile proteins
Cytosolic receptors for sex steroids may be involved with post-translational effects, and mitochondrial estrogen receptors have important links to energy production and utilization and production of oxygen-derived free radicals, defining the oxidative stress of cells [90,104,105,124,125].
As discussed in this review, nuclear, genomic effects of the sex steroids are determined by the response elements for receptor binding on specific genes. However, binding of ligand-bound receptors in some cases may require dimerization of estrogen receptors and availability of specific coregulators, which do not show specificity for one steroid receptor compared with another. Therefore, competitive binding for nuclear coregulators will modulate the magnitude of genomic effects in a mixed hormonal environment (e.g., estrogen plus progesterone or testosterone) [19,126–128].
Protein synthesis modulated by the sex steroids includes production and secretion of matrix proteins including matrix metalloproteinases, matrix Gla, osteopontin and bone scialoprotein [88,129–133]. These proteins contribute to arterial stiffening and calcification and plaque vulnerability. Vascular repair and angiogenesis are increased by estrogen in part through increases in proliferation of endothelial progenitor cells [134–139]. In addition, estrogen has anti-inflammatory effects mediated by decreased expression of leukocyte binding sites on the endothelial surface, which limits the attachment and migration of leukocytes during early development of atherosclerotic lesions [140,141].
Metabolic products of 17β-estradiol also have biological activity, which might, in some circumstances act to antagonize the effects of parent compound, for example, the catecholestrogens bind to adrenergic receptors and also compete for COMT that would degrade norepinephrine, thus having the potential to modulate both the magnitude and duration of sympathetic signaling affecting vascular resistance [142,143]. Alternatively, the catecholestrogens are antioxidants, which might potentiate nitric oxide signaling and, along with stimulation of prostacyclin and inhibition of synthesis of endothelin-1 would act to reduce vascular tone. One estrogenic end product of COMT metabolism, 2-methoxyestradiol, inhibits the proliferation of both endothelial and smooth muscle cells. However, it is important to consider that these effects of both the catechol and methoxyestradiols are concentration dependent and affect proliferating cells differently than those that are confluent in an intact vascular wall [144,145].
In order not to oversimplify the actions of the sex steroids, nongenomic and genomic effects of sex steroids are present in all components of the vasculature (e.g., endothelium, smooth muscle, advential fibrobasts and dendritic cells) and blood elements including platelets of males and females.
None of the actions described above occur in isolation and, thus, defining the contribution to testosterone and androgens in the context of vascular reactivity is perhaps more ambiguous than that of estrogen, given the conversion of testosterone to estradiol in males and the differences in reactivity of the androgen receptor based on CAG repeats [43,65,146–149]. These issues may be important when considering potential cardiovascular side effects of various anti-adrogenic compounds in treating men with prostate cancer [150].
The particular intracellular pathway or mechanism activated or dominated by the sex steroids will be tissue-specific depending, in part, upon the expression of receptors, the concentration of specific ligand and the relative activation of competing agonists acting on converging pathways. Attention to these considerations needs to be incorporated into the design of mechanistic studies, particularly in regard to development of tissue-specific selective (estrogen/steroids) receptor modulators.
While the above discussion focused on components of the vascular wall as targets for the sex steroids, it should be emphasized that steroid receptors and cellular actions of hormones also modulate activity of baro- and chemo-receptors, brain nuclei involved with baroreflex and vasomotor control and production of neurotransmitter at the level of the sympathetic nerve endings
The interaction of platelets with the vascular wall contributes to the development of atherosclerotic lesions [152,153]. Although platelets do not contain a nucleus, genomic activity of the sex steroids are present in platelet precursors, megakaryocytes; thus, the proteomic profile of circulating platelets will vary depending upon the hormonal milieu under which they were produced [154–156]. Estrogenic treatments reduce platelet aggregation to specific stimuli and their content of platelet-derived growth factor [154,157,158]. Not only are platelets from male animals more sensitive to aggregation than those of females, they also contain more thromboxane, which, if released at a sight of injury, would increase contraction of the vasoconstriction and proliferation of the smooth muscle [159,160]. These observations need to be validated in humans.
Cell membrane-derived microvesicles (microparticles) released from platelets and other cells during activation and apoptosis act as carriers of biological active molecules among cells and are implicated in the development of atherosclerosis and microvascular disease [161–167]. Although specific studies have not examined differences in production or characteristics of cell-derived microvesicles across the life span in males and females, evidence is beginning to emerge that the number and prothrombic characteristics of microvesicles derived from platelets and endothelial cells change with estrogen status in women [168,169]. Further study is needed to better define how cell-derived microvesicles contribute to sex differences in vascular function.
Sex steroids & prevention of cardiovascular disease
As stated in the initial parts of this review, clinical data provide evidence for earlier development of atherosclerotic disease and hypertension in men compared with women. Owing to the fact that the incidence of both of these conditions increases in women around the time of menopause, it was proposed that female hormones, in particular, estrogen, may be protective against development of atherosclerosis leading to myocardial infarction and stroke. Indeed, results from preclinical, observational and epidemiological studies supported this hypothesis because atherosclerotic-like lesions are decreased in oophorectomized experimental animals treated with estrogen, and adverse cardiovascular events and all causes of mortality are reduced in postmenopausal women who are using menopausal hormonal therapies, women treated with hormones for early ovarian failure and oophorectomized women receiving hormonal treatments [20,170–173]. However, this hypothesis was not supported by the large randomized clinical trial of hormone treatment for the prevention of cardiovascular disease, the Women's Health Initiative (WHI) [174].
Since women in the WHI averaged 63 years of age, approximately 10 years past the average age of menopause when women initiate hormonal treatments for menopausal symptoms, a new hypothesis called the ‘timing hypothesis’ was proposed. The timing hypothesis states that initiation of hormone treatment early in menopause (or oophorectomy) is required for slowing the progression (preventing development) of cardiovascular disease. This hypothesis is supported by preclinical data in primates and sub-analysis of data from the WHI indicating that treatment initiated within 10 years of menopause (or oophorectomy) provides protection against the development of coronary arterial atheroma, coronary arterial calcification and myocardial infarction [170,175,176,177]. In addition, flow-mediated dilatation declines after 5 years past menopause [178]. An ongoing prospective study testing the timing hypothesis is the Kronos Early Estrogen Prevention Study (KEEPS), in which women are randomized to either placebo, oral conjugated equine estrogen or transdermal 17β-estradiol (with pulsed natural oral progesterone) within the first 3 years of menopause and will continue on treatment for 4 years. Primary outcomes for the study are quantifiable changes in disease progression indicated by changes in carotid intima-medial thickening (by B-mode ultrasound) and coronary calcification (by electron beam computer tomography) [179]. KEEPS will conclude in 2012. This study is important because it is the first randomized trial in which both oral and transdermal treatments can be compared on progression of vascular lesions in two major arteries (carotid and coronary) that are implicated in adverse cardiovascular events (stroke and myocardial infarction, respectively). These comparisons are critical for future prescribing practices since adverse thrombotic events may occur less often with transdermal compared with oral estrogenic products, arteries of differing anatomical origins may differ in susceptibilities for stimuli promoting formation of vascular lesions and effects of hormones on those processes may not be the same [180–182].
Current prescribing guidelines for hormonal treatments in women do not recommend their use for prevention of cardiovascular disease but for symptoms of menopause at the lowest effective dose for the shortest duration [102,183]. These guidelines may preclude observing any positive effects on the cardiovascular system, since some data suggest that estrogenic protection against cardiovascular disease is evident only when treatment exceeds 5–6 years [184,185]. This may reflect the time course of physiological processes involved with vascular remodeling and soft tissue calcification.
Whether or not exogenous testosterone products promote or protect against the formation of vascular lesions is unclear in part owing to sex-specific effects, dosing regimes and the aromatization of testosterone to estrogen [110,146,147,186–189]. Furthermore, as mentioned in this review, the integrity of the androgen receptor may determine the overall efficacy of treatment, and perhaps evaluation of this receptor may provide useful information regarding treatment recommendations, for what is now being defined as ‘low T’ in men and the use of testosterone in women at the menopausal transition [190–192].
Future perspective
In the USA, scientific investigation directed toward understanding the factors contributing to this sex-disparity in cardiovascular disease began in the early 1990s with funding initiatives from the US NIH and political mandates for the inclusion of women in clinical cardiovascular trials [193]. Progress has been slow. Even 10 years following the Institute of Medicine's report ‘Exploring the Biological Contributions to Human Health: Does Sex Matter?’ [31], basic and preclinical research does not consistently consider sex and hormonal status to be important biological variables [29,30,194]. Sex disparities in outcomes, treatment and diagnosis of cardiovascular disease remain as evidenced by the latest statistics from the AHA [1]. Some evidence suggests that the classical risk factors (e.g., age, smoking, systolic blood pressure, diabetes and lipids) may not apply to women as they do in men, particularly in regard to coronary calcification, diastolic heart failure and microvascular disease [195–197]. New tests are needed to define risk and guide treatment [168,198].
Organizations such as the Organization for the Study of Sex Differences [301] and The International Society of Gender Medicine are establishing the study of sex/gender differences as a scientific discipline by bringing together outstanding basic, translational and clinical scientists who investigate these differences in a systematic way. Efforts such as these will establish sex/gender as scientific variables defining scientific excellence in study designs, research funding priorities, reporting of data and publication. Initiatives such as the Patience Center Outcomes Effectiveness program in the USA and treatment guidelines cannot continue without sound data based on sex as a variable.
Executive summary
Mortality from cardiovascular disease is greater for women than men.
Study of the biological basis of sex differences in factors contributing to development of cardiovascular disease is in its infancy, but sex/gender based medicine is being established as a scientific discipline.
Both the complement of sex chromosomes as well as sex hormones influences expression of autosomal genes affecting function of all elements of the cardiovascular system. Hormonal status interacts with the genetic complement over the life time to modulate development of cardiovascular disease as well as efficacy of treatments and outcomes.
The current treatment guidelines do not recommend the use of menopausal hormones for prevention of cardiovascular disease in women. However, these recommendations for the lowest dose for the shortest period of time may preclude cardiovascular protection, which may not be manifest until after 5 years of use.
Anti-androgenic treatments to men need to be evaluated with consideration of cardiovascular risk.
Sex and hormonal status are important biological variables that will be used to define scientific excellence in design of basic science and clinical trials, in the reporting of results and in the development of treatment guidelines for cardiovascular disease.
Footnotes