
Abstract
Progesterone is an ovarian steroid hormone that is essential for normal breast development during puberty and in preparation for lactation and breastfeeding. The actions of progesterone are primarily mediated by its high-affinity receptors, which include the classical progesterone receptor (PR)-A and -B isoforms, located in diverse tissues, including the brain, where progesterone controls reproductive behavior, and the breast and reproductive organs. Progestins are frequently prescribed for contraception or during postmenopausal hormone replacement therapy, in which progestins are combined with estrogen as a means to block estrogen-induced endometrial growth. The role of estrogen as a potent breast mitogen is undisputed, and inhibitors of the estrogen receptor and estrogen-producing enzymes (aromatases) are effective first-line cancer therapies. However, PR action in breast cancer is grossly understudied and remains controversial. Herein, we review existing evidence and discuss the challenges to defining a role for progesterone in breast cancer.
Keywords
Several factors contribute to the challenge of demonstrating a clear role for progesterone in breast cancer. Progesterone is difficult to study in isolation from other hormones (e.g., growth factors and prolactin) that also contribute to breast cancer biology. Progesterone receptor (PR) isoforms are primarily expressed in response to estrogen receptor (ER)-α-mediated transcriptional events, but can also occur independently from ER [1]. A subset of mammary epithelial cells (MECs) in the breast that express both PR-A and PR-B also express ERs, and estrogen is usually required in order to induce the expression of PR in these ER+ cells. For this reason, it has been difficult to separate the effects of progesterone alone from estrogen, itself a potent breast mitogen. Indeed, PR isoforms are grossly understudied relative to ER in both normal and cancerous breast cells.
Studies in ER and PR knockout mice have revealed that the concerted actions of estrogen and progesterone are required for normal mammary gland development [2,3]; estrogen/ER promotes the early growth of milk ducts that invade the mammary fat pad emanating from the nipple, while estrogen/ER and progesterone/PR isoforms are responsible for the development of the terminal end-buds (TEBs) or acini located at the ends of ducts that will become the milk-producing structures in the lactating mammary gland. Additional required hormones, EGF and IGF-1, promote the proliferation of terminal end-buds during normal breast development, and augment ductal outgrowth and side branching induced by estrogen plus progesterone [4,5]. In fact, PR isoform expression can not be induced in response to estrogen unless EGF is present [6], suggesting the existence of important cross talk between EGF receptors (EGFR) and/or family members (erbB2) and both steroid hormone receptors.
Another limitation to understanding progesterone/PR action in the context of breast cancer (i.e., uncontrolled proliferation) is that normal proliferating breast epithelial cells are steroid hormone receptor negative [7]. In the normal adult mammary gland, ER and PR are found in a minority luminal epithelial cell population of nondividing cells that lie adjacent to proliferating cells (Figures 1 & 2). These steroid receptor positive (ER+/PR+) cells represent only approximately 7–10% of the epithelial cell population. The most current information suggests that ER+/PR+ cells are capable of proliferating, but are ‘held’ in a nonproliferative or growth-inhibited state by inhibitory molecules, such as TGF-β or high levels of p27, the endogenous inhibitor of cell-cycle-dependent protein kinases (CDKs). Communication between the breast epithelial and stromal compartments mediates the proliferation of neighboring cells by expression and secretion of locally active proproliferative molecules such as Wnts, IGF-II [7], or stroma-derived HGF [8]. Recent evidence suggests that ER+/PR+ cells may serve as feeder cells by providing growth-promoting substances (e.g., Wnts) to nearby progenitor or stem cell populations [9].

Mammary gland architecture.
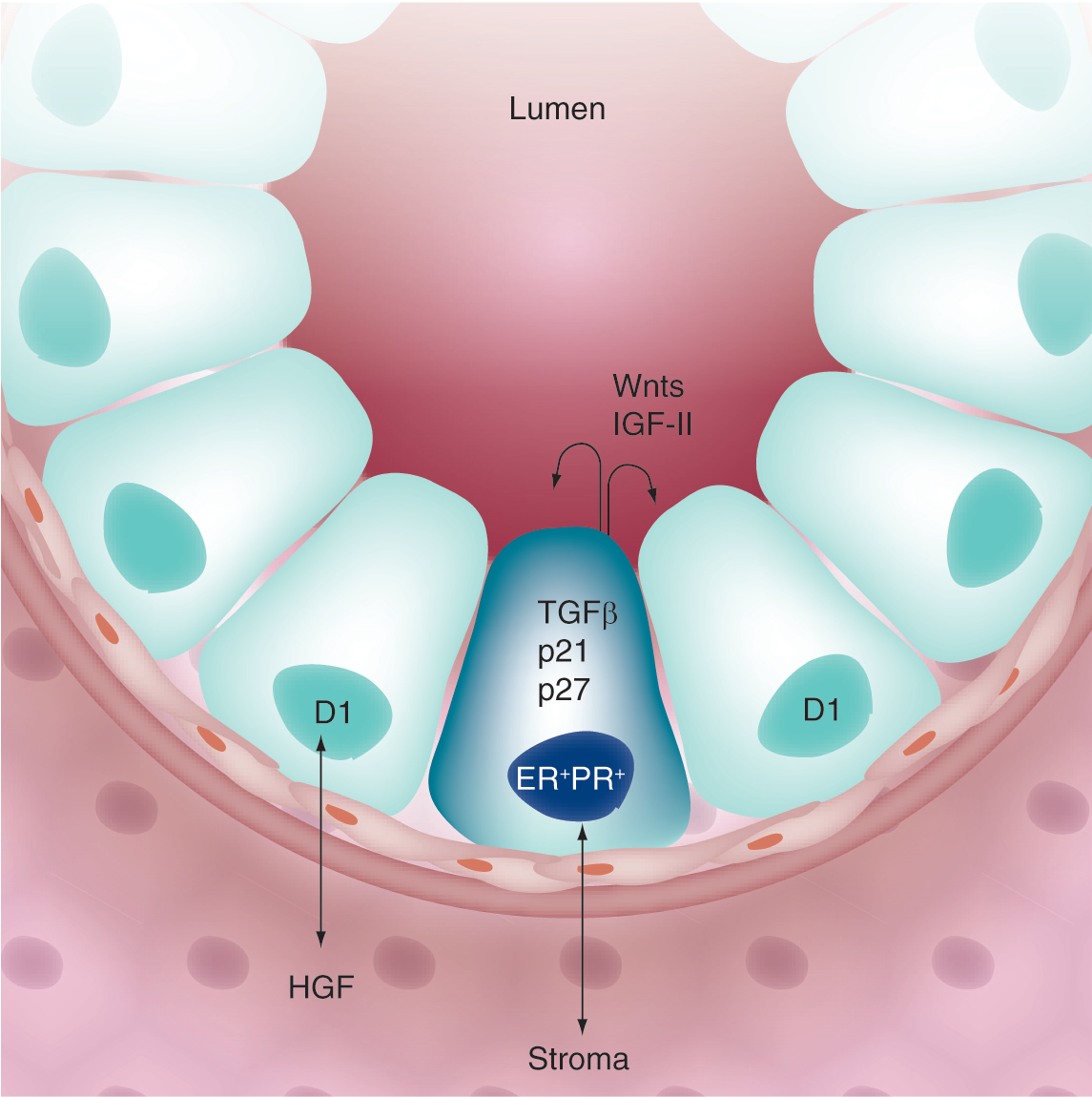
Paracrine signaling in the mammary gland.
In contrast to the normal breast, where proliferating cells are clearly devoid of steroid hormone receptors, the majority of breast cancers (~70%) express ER and PR at the time of diagnosis. Although hormone receptor-positive tumors are often slower growing relative to receptor-negative tumors [10], ER+/PR+ breast epithelial cells may undergo an early switch to autocrine or paracrine signaling mechanisms, whereby negative controls on proliferation are somehow lifted. Another setting where PR-containing cells divide is in the pregnant mammary gland, where PR-B colocalizes with cyclin D1 in BrdU-stained (dividing) cells, [11]. Thus, pathways involved in normal mammary gland growth and development may inappropriately ‘re-assert’ themselves during breast cancer progression. Experimental data in model organisms (primates, mice and rats) and humans suggest a proproliferative role for progestins [12–14]. Herein, we review the evidence regarding progesterone/PR action in breast cancer models, and relate these findings to the potential for future development of PR antagonists for clinical use as part of combined breast cancer therapies.
PR structure & function: classical & membrane-associated rapid signaling
PR isoforms are classically defined as ligand-activated transcription factors and members of a larger family of related steroid hormone receptors (including ER, androgen receptor [AR], glucocorticoid receptor [GR] and mineralocorticoid receptor) that regulate gene expression by binding directly or indirectly to specific sites in DNA (

Progesterone receptor structure.
In the absence of progesterone, PRs are complexed with chaperone molecules including heat shock proteins (HSPs); these interactions allow correct protein folding and assembly of stable PR molecules that are competent to bind hormone [22]. HSPs also function to connect PR to protein trafficking systems. After binding to progesterone, the receptors undergo conformational changes, dimerization and HSP dissociation (
The genomic or classical actions of steroid hormone treatment are delayed by several minutes to hours (i.e., following transcription and translation of target genes). Recently, however, rapidly occurring (within a few minutes) extranuclear or nongenomic effects of cell membrane-localized steroid hormone receptors have become appreciated as a major facet of steroid hormone receptor action. For example, progestin treatment of breast cancer cells causes a rapid and transient (2–10 min) activation of cytoplasmic protein kinases, including mitogen-activated protein kinase (MAPK), PI3K and p60-Src kinase [34–36]. Similar activities have been reported for membrane-associated ER and AR [37]. The mechanism of these effects involves direct binding of steroid hormone receptors to protein–protein interaction domains of signaling molecules located in or near the plasma membrane, in close proximity to growth factor receptors and their immediate effectors. Human PR contains a proline-rich (PXXP) motif that mediates direct binding to the Src-homology 3 (SH3) domains of signaling molecules in the p60-Src kinase family in a ligand-dependent manner [34].

Progesterone receptor function.
In studies using human breast or prostate cancer-cell lines, the rapid actions of AR, PR and/or ER have been shown to contribute to the regulation of cell proliferation in response to their respective hormone ligands [38–40]. While its role in human physiology (i.e., whole organisms) is less clear, steroid hormone receptor-mediated activation of cytoplasmic signaling molecules could theoretically serve to potentiate the nuclear functions of these receptors (Figures 3 & 4). For example, amplification of PR nuclear functions might occur through rapid, direct phosphorylation of PR proteins and/or their coregulators in response to activation of PR-induced cytoplasmic pathways that coincide with ligand binding. Thus, appropriately phosphorylated and activated receptor complexes are directed for efficient regulation of selected target genes. Clearly, such a positive feedback loop would explain the dramatic influence of activated signaling pathways on PR nuclear function. Indeed, several progestin-dependent functions of PR are also MAPK or c-Src-dependent, including upregulation of cyclins D1 and E, CDK2 activation, S-phase entry and anchorage-independent cell growth in soft agar [26,41,42]. C-Src and MAPK-dependent direct phosphorylation of PR is required for PR tethering to SP1 transcription factors on the
How might the extranuclear actions of steroid hormone receptors, including PR, contribute to deregulated breast cancer cell growth and/or increased breast cancer risk? Perhaps by linking steroid hormone action to the regulation of MAPK-regulated genes (i.e., the end point of MAPK signaling is the regulation of transcription factor substrates). In support of this concept, the extranuclear actions of liganded ER are thought to induce a state of ‘adaptive hypersensitivity’ during endocrine therapy in which growth factor signaling pathways are co-opted by upregulated ER [46]. In this model of ER-dependent MAPK activation, liganded ER associated with the cell membrane interacts with the adapter protein Shc and induces its phosphorylation, leading to recruitment of adaptor molecules and activation of Ras and the Raf-1/MEK/MAPK module. MAPK then regulates genes via phosphorylation of ETS factors and/or AP1 components (i.e., independently of ER transcriptional activity). ER activation of MAPK may explain why many tumors respond well to aromatase inhibitors, yet fail to respond to selective ER modulators (SERMs) designed to inhibit ER transcriptional activity in the nucleus, but not ER-dependent MAPK activation in the cytoplasm. Breast cancers often exhibit heightened c-Src and MAPK activities relative to normal breast tissue [47,48]. Steroid hormone receptors including ER, AR, and PR may contribute to the constitutive signaling of these mitogenic kinases via their membrane-associated activities, thereby circumventing endocrine-based therapies (i.e., antiestrogen).
Progesterone action in animal models
Studies in rodents demonstrate that PR-A and PR-B are differentially expressed during pregnancy-induced mammary gland development, with PR-A predominantly expressed during ductal sidebranching, while PR-B expression coincided with the formation of alveoli [49,50]. PR-B but not PR-A was expressed in proliferating cells. Some but not all proliferating cells in both compartments were PR-B+, suggesting that progesterone can induce proliferation through either direct or paracrine mechanisms. By contrast, cells in adult virgin glands were PR and cyclin D1 positive, but failed to proliferate, possibly as a result of high levels of the cyclin-dependent protein kinase inhibitors, p21 and p27 [50]. During the menstrual cycle, cells in the mammary gland undergo sequential waves of proliferation and apoptosis. Notably, in primates (macaques and humans) increased terminal duct lobular unit (TDLU) cell proliferation coincides with peaking serum progesterone levels that occur during the luteal phase [13,51–53], again suggesting a paracrine mechanism for this hormone in adult tissues. Upregulation of local IGF-1 has been suggested as a cooperating factor in this regard [54]. In animal models of postmenopausal hormone-replacement therapy, either parous or nulliparous early and late postmenopausal mice were subjected to estrogen alone (E) or estrogen plus progestin (E+P). In this study, E+P produced a greater proliferative response relative to E regardless of parity or treatment time. E+P acted directly on the mammary gland, rather than via systemic effects [55]. Similar results were reported in surgically postmenopausal macaques [56] and in postmenopausal humans [57]. Although breast cancer development was not modeled in the above animal studies, the results (i.e., proliferation) are in good agreement with human clinical data [58,59], which revealed increased tumor number and size in women taking E+P, while E alone did not significantly alter breast cancer risk or tumor size (discussed below).
Progesterone may act primarily via protooncogenes and growth factors to affect breast cell proliferation and breast cancer etiology. As the majority of early breast cancer lesions express ER and PR and these receptors remain high in 60% of advanced disease, early events may include a switch in the ability of quiescent ER+/PR+ cells to respond directly to steroid hormones and proliferate. Notably, deregulation of the cell cycle appears to be a hallmark of breast cancer. At least 40% of breast cancers overexpress cyclin D1, while up to 30% have lost p27 or p21 and/or contain elevated CDK2 activity [60,61]. Loss or mutation of
Progesterone action in human breast cancer cells
The biochemistry of PR action is well understood, having been largely defined using PR+ human breast cancer cell line models, or PR-null cells into which wild-type or modified PR has been re-expressed. Numerous studies have focused on PR interactions with other regulatory proteins, changes in PR subcellular localization, or post-translational modifications (i.e., phosphorylation, ubiquitinylation or sumoylation) or other conditions that affect PR transcriptional activities, usually measured on artificial gene promoters (reporter genes) that contain one or more tandem progestone responsive elements (PRE) sites [70]. Growth factors, including EGF or heregulin, promote transcriptional synergy with progestins on PR-target genes [45,71,72]. Phosphorylation events primarily serve to augment PR action in a promoter-selective manner [73]. Despite this depth of basic understanding, gene regulation and the associated changes in cell biology in response to PR activation remain elusive. Only a handful of endogenous progesterone-responsive genes have been described in moderate detail [23,29,74]. The majority of genes regulated in response to progesterone lack PR-binding consensus sequences or PREs, and the presence of one or more PREs or PRE half-sites does not predict progesterone-responsive regulation [17]. Numerous genes are regulated upon PR expression, but independently of progesterone [16,75]. Furthermore, many genes are downregulated in response to progesterone/PR-dependent transcriptional repression, largely by unknown mechanisms [16,17]. In most cases, the regulation of particular genes in response to progesterone/PR is only loosely tied (by correlation) to changes in cell biology. For example, many PR-regulated genes have been associated with aspects of tumor progression towards aggressive tumor phenotype. In addition, variation of the PR-A:PR-B ratio is a frequent occurrence in breast tumors relative to normal tissue [76], and is predicted to dramatically alter the genetic program [16,77].
Confounding the role of progesterone in breast cancer is that progesterone has biphasic effects on the proliferation of breast cancer cell lines grown
Notably, progesterone treatment of PR+ cells growing in culture has also been implicated in prosurvival (resistance to chemotherapy-induced apoptosis) [82], and tumor cell differentiation (from luminal to myoepithelial phenotype) without effects on tumor growth [83]; this transition is associated with poor prognosis in breast cancer. Similarly, epithelial to mesenchymal transition (EMT) is an early transitional event that precedes tumor cell invasion and metastasis, which may occur independently of changes in proliferation. During EMT, stationary epithelial cells become more like fibroblast cells and acquire the ability to migrate and invade locally. Interestingly, many of the genes regulated in response to progesterone include those encoding molecules involved in signal transduction and cell adhesion to extracellular matrix (ECM) or other basement membrane components [16,17].
Clinical findings & future perspective
A direct role for PR in breast cancer is perhaps best illustrated by the clinical findings of the Women's Health Initiative (WHI) and Million Women Study, demonstrating that women taking a progestin in combination with estrogen (EPT) as part of hormone replacement therapy, experienced a greater breast cancer risk relative to estrogen alone; tumors were larger and of higher grade [58,59]. The Million Women Study also found that women were more likely to die of breast cancer if they were taking EPT at the time of diagnosis. When the findings of the WHI were announced in mid-2002, the number of prescriptions for EPT dramatically decreased. Population-based studies have shown a decline in breast cancer incidence immediately after these data were made public [86,87]. While it is impossible to prove that the decreased use of EPT caused the decline in breast cancer incidence, this is certainly a plausible explanation. It is possible that discontinuation of EPT suppressed the appearance of breast cancers that would have become evident with continued hormone stimulation. Following the incidence trends will be necessary to determine the magnitude of the effect of EPT in changing breast cancer incidence.
Many aspects of PR action originally discovered in animal or cell-line models of breast cancer have not been well-established in humans. Convincing proof of the role of PR in breast cancer requires demonstration that perturbation of PR function results in important clinical outcomes. For example, we know that high-dose synthetic progestin administration (megestrol and medroxyprogesterone) has activity in advanced breast cancer [88] in a manner analogous to the clinical activity of the synthetic estrogen (diethylstilbesterol) in advanced breast cancer [89]. High-dose estrogen therapy for breast cancer has largely been replaced by selective estrogen receptor modulators (SERMs; tamoxifen, torimifene and fulvestrant) or ligand-deprivation strategies in which aromatase inhibitors are used to prevent the conversion of steroid hormone precursors into estradiol. If progesterone receptor modulators (PRMs) could be shown to alter the growth of PR-expressing breast cancer cells in human clinical trials, then this would clearly validate the findings made
Executive summary
Progesterone is difficult to study in isolation from other hormones and growth factors that similarly influence mammary gland biology and breast cancer (namely, estrogen, prolactin, EGF, IGF and HGF).
Progesterone receptors (PRs) are classically defined as ligand-activated transcription factors, but also function at or near the plasma membrane to directly activate protein kinase pathways (namely, c-Src and the MAP kinase module consisting of Raf-1, MEK1/2 and ERK1/2). This is commonly called ‘rapid’ signaling.
The function of rapid signaling is unknown, but may provide positive regulation of PR-containing transcriptional complexes by direct phosphorylation of PR or coregulatory molecules. Phosphorylation also provides a mechanism for promoter selectivity.
PR-B is required for normal mammary gland development; PR+ cells in the mammary gland may interact primarily with stromal components to mediate proliferative signaling of nearby or neighboring PR-null cells via the action of locally acting growth factors (namely IGF-II, Wnts and/or stromal HGF).
Studies in mice, rats, monkeys and humans support the concept that progesterone/PR contributes to proliferative signaling in the normal adult and neoplastic mammary gland.
Both the rapid actions and the transcriptional activity of PR-B contribute to breast cancer cell proliferation in response to progesterone.
Progesterone/PR act in concert with mitogenic protein kinases and cell-cycle mediators to induce regulation of PR target genes and proliferative responses.
Progesterone acts to sensitize breast cancer cells to the actions of growth factors by upregulation of target genes that include the components of signal transduction pathways (namely, EGFR and EGFR ligands, IRS-2, cyclins D and E, p21).
Understudied areas of progesterone/PR action include the regulation of breast cancer prosurvival and early events in breast cancer cell invasion and metastasis, including preneoplastic transitions during tumor progression (i.e., not necessarily related to altered proliferation).
Selective progesterone receptor modulators (PRMs) have significant activity against breast cancer in clinical trials, but have not been well tolerated by most patients, most likely owing to their high affinity and cross-reactivity to glucocorticoid receptors.
New selective PRMs must be developed before PR may be directly targeted. However, protein kinases that are critical for PR action may also provide effective therapeutic targets; c-Src, CDK2 and/or MAP kinase inhibitors should be considered as part of combination therapies that could be used to complement the use of anitestrogens and antiprogestins.
Several PRMs have been described. Unfortunately, the clinically tested PRMs are not selective and have cross reactivity with GR [90]. Despite this concern, two small trials in breast cancer using these drugs have been reported. Mifepristone has been examined in women with advanced breast cancer and a low response rate was observed. In addition, the side effects of lethargy, nausea and anorexia were noted [91]. Mifepristone has also been tested in meningioma and given for longer periods of time with less toxicity [92]. In this study, glucocorticoids were given during the first 2 weeks of therapy and it is possible that this coadministration of steroids to avoid GR inhibition reduced the side effects. Onapristone, another PRM, has also been tested in breast cancer. This drug was associated with a substantial response rate and sustained stable disease (clinical benefit of 63%), but caused hepatotoxicity that necessitated discontinuation of the trial [93].
In sum, while the bulk of preclinical data suggest an important role for PR function in modulating breast cancer biology, validation of these findings are dependent on a clinical strategy to disrupt PR function in human breast cancers. It will then be important to decipher the contribution of nuclear and membrane PR activities, and target them appropriately with selective PRMs, in addition to targeting the relevant kinases (c-Src, MAPKs and CDK2) required for steroid hormone receptor action. We predict that PR activities will one day be routinely targeted as part of combination therapies aimed at blocking both ER-α and PR-B, along with the associated essential protein kinases.
Footnotes