
Abstract
This manuscript reviews the pharmacodynamics and pharmacokinetics of duloxetine and its efficacy and safety in women with stress urinary incontinence. Duloxetine is a selective inhibitor of neuronal serotonin and norepinephrine uptake which increases urethral striated muscle activity and bladder capacity. Duloxetine is readily absorbed and extensively metabolized; cytochrome P450 1A2 (CYP1A2) inhibiting drugs can markedly increase duloxetine exposure. The clinical efficacy of duloxetine has consistently been demonstrated in several randomized, double-blind studies in women with moderate-to-severe stress urinary incontinence, but the additional benefit relative to placebo was moderate. Duloxetine treatment is frequently associated with adverse events such as nausea, dry mouth, fatigue, insomnia and constipation, but serious adverse events are rare. Therefore, duloxetine appears suitable for the treatment of stress urinary incontinence.
The International Continence Society has defined stress urinary incontinence (SUI) as ‘the complaint of involuntary leakage on effort or exertion, or on sneezing or coughing’ [1]. Of note, this definition does not involve a minimum number of SUI episodes per time interval. Epidemiologic studies on the prevalence of SUI have used various definitions of the condition and, hence, the reported prevalence of SUI varies widely. Nevertheless, all studies agree that it is a highly prevalent condition in adult women of all ages [2–4], particularly in those who have undergone childbirth, including that by cesarean section [5].
Incontinence-specific quality of life instruments have been developed which demonstrate that SUI is a highly bothersome condition with a major adverse impact on quality of life of afflicted patients [6,7]. Urge urinary incontinence is another common form of incontinence, and many patients suffer from a combination of both forms, known as mixed incontinence. While SUI is more frequent than urge or mixed incontinence [2–4,8,9], it appears that urge and mixed incontinence cause greater reductions in quality of life [10].
Overview of the market
Until recently, SUI has largely been a domain of physical therapy [11] or surgery [12]. Existing medical therapies, such as α-adrenergic agonists or estrogens were ineffective and/or had serious side effects, as reviewed by the Cochrane collaboration [13]. All of these earlier medical treatments had been used off-label only. Muscarinic receptor antagonists are the treatment modality most often used for urge incontinence [14] and several studies demonstrate that this class of drug is also effective in mixed incontinence [15–17]. While the effectiveness of muscarinic antagonists in SUI has not been tested systematically, they are sometimes used off-label for this condition.
Introduction to the compound
Duloxetine (Yentreve®, Ariclaim®, formerly known as LY 248,686) has recently been introduced for the treatment of moderate-to-severe female SUI in several European countries. It has also been approved in some countries for the treatment of major depressive disorder [18] and diabetic neuropathic pain [19], but slightly different doses are used in these indications compared with SUI. The molecular basis of the therapeutic effects of duloxetine in all three indications is a selective inhibition of neuronal serotonin and norepinephrine uptake and, hence, an increased availability of these neurotransmitters in the synaptic cleft [20]. This article will review the pharmacodynamic studies on duloxetine as they relate to SUI, its pharmacokinetic profile, its clinical efficacy in the treatment of SUI and its safety in SUI and other patients.
Chemistry
Duloxetine ((+)-(S)-N-methyl-γ-(1-naphtyloxy)-2-thiophenepropylamine hydrochloride, C18H19NOSHCl) is a slightly brownish white solid, which is slightly soluble in water. Since duloxetine is acid labile [20], it is clinically used in capsules containing enteric-coated pellets.
Pharmacodynamics
Several in vitro studies using rat brain synaptosomes, human platelets or heterologously expressed cloned human transporters have demonstrated that duloxetine is a potent inhibitor of the neuronal serotonin (5-hydroxytryptamine [5-HT]) and norepinephrine transporters (Table 1). This was confirmed in radioligand binding studies in which duloxetine potently inhibited the binding of 5-HT and norepinephrine to the cloned human and rat brain 5-HT and norepinephrine transporters (Table 1). Duloxetine only inhibited the neuronal dopamine transporter at considerably higher concentrations, which are not likely to be reached at therapeutic doses (Table 1), and did not exhibit appreciable inhibition of binding to adenosine, choline and γ-aminobutyric acid (GABA) transporters [21]. Moreover, duloxetine does not inhibit another important mechanism of monoamine inactivation, monoamine oxidase [22].
Affinity/potency of duloxetine at amine transporters.
All values are Ki values in nM, except for [23] which are IC50 values.
IC50: Inhibitory concentration 50%; Ki: Binding affinity; ND: Not determined.
Many older uptake inhibitors, particularly tricyclic antidepressants, also act as antagonists at various receptors in therapeutically relevant concentrations. Duloxetine had little, if any, affinity (binding affinity [Ki] > 500 nM) for muscarinic acetylcholine, α1- and α2-adrenergic, dopamine D2, histamine H1 and various subtypes of 5-HT receptors, as well as for adenosine, β-adrenergic, cholecystokinin, GABA-A, glutamate, glycine, melatonin, nicotinic, opiate, neuropeptide Y, neurokinin, neurotensin or somatostatin receptors, Land N-type calcium channels, ATP-sensitive or calcium-activated potassium channels, GABA or glutamate-gated chloride channels, sodium channels, adenyl cyclase, inositol trisphosphate receptors, protein kinase C or nitric oxide (NO) synthase [21]. Therefore, duloxetine is expected to lack the side effects of the older antidepressants which were related to receptor antagonism.
Various approaches have been used to assess the duloxetine-induced inhibition of the neuronal 5-HT and norepinephrine transporters in vivo. One approach is the removal of brain tissue from treated animals with subsequent in vitro measurement of uptake or transporter binding. Such studies in rats have found duloxetine to inhibit 5-HT and norepinephrine transporters at oral doses of 12 and 15 mg/kg, whereas no inhibition of dopamine uptake was observed up to 40 mg/kg [21,23,24].
Another approach is based upon the finding that several compounds that deplete neuronal monoamine stores depend on specific monoamine transporters for their effects. Thus, the in vivo inhibition of monoamine transporters by duloxetine has been tested by studying p-chloramphetamine-induced depletion of 5-HT and 6-hydroxydopamine- or α-methyl-N-tyrosine-induced depletion of norepinephrine stores. Duloxetine inhibited the depletion of 5-HT and norepinephrine stores at intraperitoneal doses of 2.3 and 12 mg/kg [22,25,26]. Similar results were found in studies with mice [22].
A third approach uses in vivo microdialysis to measure drug-induced alterations of extracellular transmitter concentrations. In such studies oral, subcutaneous or intraperitoneal treatment with duloxetine increased the 5-HT and norepinephrine concentrations in the rat frontal and pre-frontal cortex, diencephalon, nucleus accumbens and hippocampus [25,27–33]. Cotreatment with the 5-HT1A receptor antagonists LY 206130 [34], WAY 100,365 [29] and (−)-pindolol [32] enhanced the effect of duloxetine on 5-HT levels, whereas cotreatment with buspirone, a partial agonist at 5-HT1A receptors, partially and transiently inhibited the effect of duloxetine on 5-HT concentrations [30]. Enhancements of the effect of duloxetine on norepinephrine levels were reported inconsistently in these studies. Taken together, these data reflect the role of the 5-HT1A receptor as an autoreceptor modulating neuronal 5-HT release. The α2-adrenergic antagonists atipamezole and 1-PP enhanced the effects of duloxetine on 5-HT levels [31], reflecting the role of these as inhibitory heteroreceptors. Studies in the rat frontal cortex and nucleus accumbens, but not in the hippocampus, also reported duloxetine-induced elevations of dopamine [27–32]. While this may be surprising given the lack of duloxetine effects on dopamine transporters, it is explained by the role of norepinephrine transporters in inactivating dopamine in the frontal cortex and nucleus accumbens.
Electrophysiologic studies, which look at the firing rate of neurons under the control of the respective transmitter, are a fourth approach to assess the inhibition of monoamine uptake. In such studies, duloxetine enhanced the suppression of CA3 pyramidal neuronal firing activity by microiontophoretically applied 5-HT or norepinephrine [23]. Duloxetine alone also suppressed the spontaneous firing rate of dorsal raphe 5-HT and locus ceruleus norepinephrine neurons [23,35,36].
The above data unequivocally demonstrate that duloxetine inhibits the neuronal 5-HT and norepinephrine transporters not only in vitro, but also in various parts of the CNS of experimental animals in vivo. Similar approaches are not applicable to studies in humans for obvious ethical reasons, but alternative techniques, largely reflecting peripheral monoamine transporter activity, have been used in healthy volunteers. Effects on the 5-HT transporter have been studied by measuring its whole blood concentrations during duloxetine treatment, largely reflecting the filling of 5-HT stores in platelets. One placebo-controlled study reported a significant reduction by duloxetine both 20 and 60 mg/day after 7 days and an even greater reduction after 14 days of treatment [37]. Another placebo-controlled study reported a significant reduction in whole blood 5-HT after 7 days of treatment with duloxetine 60 mg twice-daily, whereas treatment with 80 mg/day yielded smaller reductions that failed to reach statistical significance [38].
Studies on the effects of duloxetine on norepinephrine uptake have been able to use a wider range of techniques. In one study, plasma from duloxetine-treated subjects dose-dependently (40–120 mg twice-daily) decreased in vitro radioligand binding to the human norepinephrine transporter [39]. The supine and upright plasma concentrations of norepinephrine were dose-dependently (60–120 mg twice-daily) increased in one study [39], whereas another study, using 60 mg twice-daily or 80 mg/day, did not confirm this finding [37]. The former study also reported reduced plasma concentrations of the norepinephrine metabolite dihydroxyphenylglycol and, hence, markedly reduced dihydroxyphenylglycol/norepinephrine ratios [39]. However, urinary excretion of norepinephrine and its metabolites is considered to be a more reliable indicator of norepinephrine metabolism in humans. This was significantly affected by duloxetine in two studies [38,39].
Elevations of blood pressure and/or heart rate can also be used as indices of sympathetic activity and, hence, of norepinephrine uptake inhibition. Small but statistically significant elevations of blood pressure and/or heart rate were observed in some, but not all, studies with duloxetine [37–39]. A more specific approach is the use of tyramine, an indirect sympathomimetic agent, which acts by releasing endogenous norepinephrine and requires neuronal uptake to be effective. Hence, inhibition of cardiovascular tyramine response can be used as an indicator of uptake inhibition. Attenuation of the cardiovascular tyramine response was detected in one [39] but not two other studies [37,38]. While the above data are not fully conclusive, the overall balance suggests that therapeutically relevant duloxetine doses inhibit both 5-HT and norepinephrine uptake in vivo in humans.
The use of a 5-HT/norepinephrine uptake inhibitor in the treatment of SUI is based upon the concept that both neurotransmitters modulate urogenital function, specifically bladder outlet resistance during the storage phase of the micturition cycle [40,41]. Such modulation is likely to involve Onuf's nucleus in the sacral spinal cord, which controls the activity of the somatic nerve fibers innervating the urethral striated muscle, also known as the external sphincter. Additional parts of the central and peripheral nervous system may also be involved, for example, there may be effects on nerves controlling bladder capacity or a local elevation of norepinephrine concentration in the sympathetically innervated smooth muscle of the urethra, also known as the internal sphincter.
Against this background, the effect of duloxetine on the lower urinary tract has been tested in an anesthetized cat model of acetic acid-induced bladder irritation [42,43]. In this model, intravenous administration of duloxetine dose-dependently increased the activity of the striated external urethral sphincter, as assessed by an electromyogram, and concomitantly increased bladder capacity. Interestingly, duloxetine abolished the acetic acid-induced nonvoiding detrusor contractions during the filling phase, but did not interfere with the voiding contractions; similarly, duloxetine enhanced external sphincter activity during the filling but not the voiding phase [43]. Accordingly, there was no increase in postvoiding residual volume upon duloxetine treatment. Moreover, duloxetine did not affect bladder contractions elicited by peripheral stimulation of the efferent fibers of the pelvic nerve [43]. These data are in line with the concept that 5-HT and norepinephrine act in the lower spinal cord by modulating descending glutamate signals, thereby exerting an indirect effect only [40,41].
The effects of duloxetine on bladder capacity were significantly inhibited by methiothepin but not by LY 53,857, prazosin, idazoxan or propranolol [43], indicating that they are mediated by a 5-HT receptor distinct from the 5-HT2 receptors. On the other hand, the effects of duloxetine upon the external sphincter electromyogram were significantly inhibited by methiotepin, prazosin and idazoxan [43], indicating that it involves not only 5-HT but also α1- and α2-adrenergic receptors.
To determine whether the 5-HT and/or norepinephrine uptake inhibition by duloxetine is involved in its effect on the lower urinary tract, the same model was used for a comparative study of duloxetine, venlafaxine (another mixed 5-HT/norepinephrine uptake inhibitor), S-norfluoxetine (a selective 5-HT uptake inhibitor) and thionisoxetine (a selective norepinephrine uptake inhibitor) [42]. In this study, venlafaxine mimicked the effects of duloxetine and was similarly sensitive to inhibition by the 5-HT receptor antagonist methiothepin. On the other hand, neither S-norfluoxetine nor thionisoxetine mimicked the duloxetine effect, and even their combination failed to do so. While the authors proposed that concomitant inhibition of 5-HT and norepinephrine uptake must be present in a single molecule to modulate bladder and urethral function, a more likely explanation is that S-norfluoxetine and/or thionisoxetine were underdosed. Therefore, this study does not provide conclusive information on whether 5-HT and/or norepinephrine uptake inhibition is required for the urogenital effects of duloxetine.
Given that the above studies come from a single species, an animal model that does not directly reflect SUI and a single group of investigators, additional studies are clearly needed to fully identify the mechanism of action of duloxetine in SUI. For example, it has been shown that duloxetine may also affect vascular tone in the lower urogenital tract [44]. Moreover, the previous studies of duloxetine in the lower urinary tract involved acute treatment only, and studies in the CNS suggest that certain effects of duloxetine may differ upon chronic treatment [36,45,46]. A recent preliminary report on healthy human females shows that a single dose of duloxetine (40 mg) did not affect basal urethral resistance, but increased the pressure amplitude responses to sacral magnetic stimulation [47]. These findings are fully in line with those from the studies in anesthetized cats and corroborate the proposed mechanism of action of duloxetine.
Pharmacokinetics & metabolism
The commercially available duloxetine capsules represent a delayed-release formulation. Its pharmacokinetics have been investigated in healthy subjects and in women with SUI, and its basic pharmacokinetic characteristics are detailed in Table 2. Under steady-state conditions, exposure to duloxetine, judged by its trough plasma concentrations, increases linearly with dose in the range from 20 mg/day to 40 mg twice-daily, and the pharmacokinetics are compatible with a one-compartment model and first order absorption and elimination characteristics [48]. Duloxetine is well absorbed following oral administration [49] with a median lag time of 2–3 h before absorption begins. Maximal plasma concentrations (Cmax) of duloxetine occur after 6 h when administered in the fasted state and after 10 h when taken with a meal [50–55]. However, the concomitant intake of food does not alter the Cmax of duloxetine and has no major effect on the extent of absorption [50]. The ingestion of duloxetine at bedtime rather than in the morning is associated with a later time to Cmax (tmax), reduced Cmax and reduced area under the plasma–concentration curve (AUC) by 29 and 18%, respectively [50].
Pharmacokinetic characteristics of duloxetine.
Data are based upon [48–50,52–55] and information from the regulatory documents of the European and US authorities; for details see text.
tmax: Time to reach maximum plasma levels following oral administration; t1/2: Terminal elimination half-life; Vd: Volume of distribution.
The apparent volume of distribution averages approximately 1640 l [48,52–55]. Duloxetine is highly bound (>90%) to human plasma proteins, binding primarily to albumin and α-acid glycoprotein [49]. The plasma protein binding of duloxetine is not affected by renal or hepatic impairment. An interaction with other highly protein-bound drugs has not been fully evaluated, but the possibility exists that coadministration of duloxetine with another drug that is highly protein-bound may increase the concentration of free duloxetine, potentially resulting in adverse events.
Duloxetine is readily metabolized upon oral ingestion, as indicated by the finding that only 3% of a dose of [14C]-labeled duloxetine is found in plasma as duloxetine [49,52]. Some of the primary duloxetine metabolites, such as 5-hydroxyduloxetine, 6-hydroxyduloxetine or 6-hydroxy-5-methoxyduloxetine, appear to be pharmacologically active [56], whereas the circulating metabolites are considered to be inactive. The major biotransformation pathways for duloxetine involve hepatic oxidation of the naphthyl ring, catalyzed by cytochrome P450 (CYP)2D6 and CYP1A2, followed by conjugation and further oxidation [52]. Thus, duloxetine metabolites found in plasma include 4-hydroxy duloxetine glucuronide and 5-hydroxy, 6-methoxy duloxetine sulfate [52]. The role of CYP2D6 in the metabolism of duloxetine has also been supported by drug–drug-interaction studies (see below). Approximately 70% of an administered duloxetine dose ultimately appears in urine, almost completely in the form of metabolites, and only approximately 20% is excreted in the feces [49,52]. Less than 1% of the administered duloxetine dose is excreted in urine in its unchanged form and numerous metabolites, some representing only minor pathways of elimination, are found there.
Duloxetine has an elimination half-life of approximately 12 h (range 8–17 h) [49–55]. Therefore, steady-state plasma concentrations are typically achieved after 3 days of dosing. While the use of duloxetine in the treatment of SUI has only been approved in women, duloxetine's half-life is similar in men and women. Since SUI may occur in a wide range of age groups [2–4], the pharmacokinetics of duloxetine were compared in healthy elderly (65–77 years) and middle-aged females (32–50 years) [54]. Elderly women had a somewhat slower elimination of duloxetine, but the magnitude of this effect was small relative to the overall interindividual variation. Population pharmacokinetic analyses suggest that the typical values for clearance decrease by approximately 1% for each year of age between 25 and 75 years; but age as a predictive factor only accounts for a small percentage of between-patient variability [54]. A possible effect of ethnicity on the pharmacokinetics of duloxetine has not been studied. Smokers, who tend to have more severe or a greater incidence of SUI due to obstructive pulmonary disease, exhibit a reduced bioavailability of duloxetine by approximately a third, but this has not led to specific dosing recommendations in this subgroup.
The effect of clinically evident hepatic insufficiency on the metabolism and elimination of duloxetine has been studied in six cirrhotic patients with moderate liver impairment (Child–Pugh class B) following a single 20 mg dose [55]; mild and severe impairment have not been studied. Compared with control subjects, patients with moderate hepatic insufficiency had a mean plasma duloxetine clearance approximately 15% that of age- and gender-matched healthy subjects, with a fivefold increase in AUC. Although Cmax was similar in controls and cirrhotic patients, the half-life was approximately three times longer in the latter. Therefore, it is recommended that duloxetine is not administered to patients with any hepatic insufficiency. While it is unlikely that patients with end-stage renal disease would suffer from SUI, it should be noted that limited data indicate that this condition is associated with an approximate doubling of Cmax and AUC. Population pharmacokinetic analyses suggest that mild-to-moderate degrees of renal dysfunction (estimated creatinine clearance of 30–80 ml/min) have no significant effect on the apparent clearance of duloxetine.
In the following section, possible consequences related to the metabolism of duloxetine, particularly with regard to drug–drug-interaction studies are addressed. Duloxetine is a substrate of CYP1A2 and hence, coadministration of fluvoxamine, an inhibitor of CYP1A2, results in an approximately sixfold increase in AUC and a 2.5-fold increase in the Cmax of duloxetine. Other CYP1A2 inhibitors include cimetidine and quinolone antimicrobials such as ciprofloxacin and enoxacin; since these would be expected to have similar effects on the pharmacokinetics of duloxetine, these combinations should be avoided. On the other hand, duloxetine does not inhibit or induce CYP1A2 activity and, accordingly, duloxetine does not affect the metabolism of CYP1A2 substrates such as theophylline [51].
Duloxetine is both a substrate and an inhibitor of CYP2D6 [53], an enzyme involved in the metabolism of approximately 20% of all clinically used drugs. Therefore, coadministration of the CYP2D6 inhibitor paroxetine increased the AUC of duloxetine 1.6-fold [53]. The possible relevance of CYP2D6 inhibition by duloxetine was investigated in an interaction study with the CYP2D6 substrate desipramine, which had a 2.6-fold increase in AUC in the presence of duloxetine [53]. Therefore, coadministration of duloxetine with other drugs that are extensively metabolized by CYP2D6, particularly if they have a narrow therapeutic index, should be approached with caution. These include certain antidepressants (e.g., tricyclic antidepressants such as nortriptyline, amitriptyline and imipramine), phenothiazines and Type 1C antiarrhythmics (e.g., propafenone and flecainide). The plasma concentrations of tricyclic antidepressants may need to be monitored and their dose may need to be reduced when coadministered with duloxetine. Due to the risk of serious ventricular arrhythmias and sudden death potentially associated with elevated plasma levels of thioridazine, duloxetine and thioridazine should not be coadministered. It should also be considered that approximately 1% of Asians and 10% of Caucasians lack CYP2D6 and, hence, are poor metabolizers of duloxetine, with the rest of the population being intermediate or ultrarapid metabolizers [57]. Theoretical considerations predict that poor metabolizers may have markedly enhanced plasma levels of duloxetine, but the authors are not aware of specific studies which have addressed this issue.
Duloxetine is neither a substrate of CYP2C9 nor does it inhibit or induce the activity of this isozyme. Therefore, corresponding drug–drug interactions have not been observed and are not expected. CYP3A, particularly CYP3A4, is one of the quantitatively most important enzymes in drug metabolism [58]. The metabolism of duloxetine occurs largely independently of CYP3A, and duloxetine does not inhibit or induce CYP3A activity. Therefore, an increase or decrease in the metabolism of CYP3A substrates (e.g., oral contraceptives and other steroidal agents) resulting from induction or inhibition is not anticipated, although clinical studies have not been performed.
With regard to other drug–drug interactions, duloxetine does not affect the pharmacokinetics of lorazepam or temazepam, and neither of them alters the pharmacokinetics of duloxetine upon coadministration. Similar to all 5-HT uptake inhibitors, duloxetine should not be combined with monoamine oxidase inhibitors to avoid occurrence of a 5-HT syndrome. On the pharmacodynamic level, duloxetine did not exacerbate the effects of alcohol on psychomotoric tests [59]. However, alcohol consumption may increase the likelihood of hepatic side effects upon duloxetine use.
The commercially available duloxetine formulation has an enteric coating that resists dissolution until it reaches a segment of the gastrointestinal tract where pH exceeds 5.5. Hence, drugs that raise the gastrointestinal pH may theoretically lead to an earlier release of duloxetine. However, coadministration of duloxetine with aluminum- and magnesium-containing antacids (51 mEq) or famotidine had no significant effect on the rate or extent of duloxetine absorption [60]. Whether concomitant administration of proton pump inhibitors affects duloxetine absorption is unknown.
Finally, it should be considered that numerous patients with SUI concomitantly suffer from urinary urgency, in other words, have mixed incontinence [2–4]. The urgency component of mixed incontinence is mostly treated with muscarinic receptor antagonists such as oxybutynin or tolterodine [14,61]. Both the muscarinic receptor antagonist tolterodine [62] and uptake inhibitor duloxetine [63] have been shown to have similar efficacy in mixed incontinence and pure urgency incontinence (muscarinic antagonist) or pure SUI (duloxetine). Due to their differential modes of action, it appears plausible to apply their combination in patients with mixed incontinence, but this has not been tested in clinical studies. Nevertheless, it is important to note that duloxetine does not affect the pharmacokinetic properties of concomitantly administered tolterodine [64].
Clinical efficacy
The clinical efficacy of duloxetine in the treatment of female SUI has primarily been investigated in one Phase II dose-ranging study [65] and three Phase III studies performed in the USA [66], Western Europe and Canada [67] and globally [68]. All four studies had a similar design. Women aged 18 years and older with predominant SUI and at least four (Phase II study) or at least seven (Phase III studies) incontinence episodes/week were eligible to participate. After a 2-week no-drug lead-in period and a 2-week single-blind placebo lead-in, patients were randomized to receive duloxetine or placebo for 12 weeks in a double-blind manner. The Phase II study involved doses of duloxetine 20 mg/day, 20 mg twice daily and 40 mg twice-daily, whereas the Phase III studies used the dose 40 mg twice-daily.
In all studies, the incontinence episode frequency (IEF) was the primary end point [65–68]. Based upon the differential inclusion criteria, the baseline IEF was somewhat smaller in the Phase II study than in the Phase III studies: 1.6–1.9 versus 2.5–2.7/24 h, respectively. The treatment responses varied somewhat between studies and duloxetine reduced the IEF to a significantly greater extent than placebo in three of the four studies (Figure 1). In those cases, the improvements were already significant at the first measured time point, after 4 weeks of treatment. Analyses of voiding frequencies demonstrated that the improvements in the IEF were not due to an increased micturition frequency which, if anything, declined slightly during treatment. The reduction in IEF appeared dose-dependent, with the therapeutically recommended dose of 40 mg twice-daily yielding the largest response among the tested doses [65]. Within each study, a subanalysis of the more severely afflicted patients (those with a basal IEF of at least 14/week) has been carried out. These analyses confirmed the significant efficacy of duloxetine relative to placebo in all four studies, including the one in which the effect of duloxetine had not been found statistically significant for the overall group. Moreover, a secondary analysis of the Phase II study demonstrated that duloxetine was similarly effective in women with pure SUI and in those with mixed incontinence [63]. The cure rate – the percentage of patients without incontinence episodes in the last diary – was numerically greater with duloxetine 40 mg twice-daily than with placebo (18.7 vs 15.2%), but this difference did not reach statistical significance in the only study where it was reported [65].

Effects of duloxetine (40 mg twice-daily) on incontinence episode frequency in Phase II and III studies.
A disease-specific quality of life score (I-QOL) [6] was a coprimary end point in most studies, and a patient global impression index was used as a secondary end point [69]. The I-QOL and patient global impression index were significantly improved in three out of four studies in the intention-to-treat analysis, and at least in the per-protocol analysis in the remaining study [65–68]. The improvements in I-QOL and patient global impression index were also maintained in patients with more severe symptoms at baseline. Alterations of I-QOL were the primary end point of another study, which differed considerably in design [70]. This randomized, placebo-controlled double-blind study investigated 471 incontinent women for 36 weeks. In contrast to the other studies, women with predominant urge incontinence were not excluded (representing 9% of all participants), and a minimum of only one incontinence episode/week was required to participate. Duloxetine did not improve I-QOL to a significantly greater extent than placebo in this patient group.
A subgroup analysis of the pooled data from all four Phase II and III studies showed that relative placebo responses were weakest in women with a high IEF at baseline, those currently using physical therapy and those who had previously undergone incontinence surgery; in contrast, duloxetine responses were very similar in all subgroups of patients [71]. Based upon this experience, an 8-week, randomized, placebo-controlled, double-blind study was performed in 109 women with severe SUI awaiting surgery [72]. Compared with placebo, duloxetine treatment was associated with significant improvements in IEF, I-QOL and pad use. All duloxetine responses were observed within 2 weeks. Most importantly, 10 out of 49 duloxetine-treated women completing the study were no longer interested in surgery, whereas this was not observed in any placebo-treated patient.
A four-armed study has compared the efficacy of duloxetine, pelvic floor muscle training and their combination against placebo in 201 patients [73]. In the intention-to-treat analysis, duloxetine reduced the IEF significantly compared with placebo or muscle training alone, while the combination of duloxetine with muscle training yielded the best treatment results. The adverse-event profile in this study was comparable with that in other studies (Table 3).
Treatment-emergent adverse events observed with duloxetine or placebo in studies with SUI patients.
Data are from the aggregate analysis of the pivotal studies for the use of duloxetine in SUI (n = 958 patients on duloxetine 40 mg twice-daily and n = 955 patients on placebo) as provided in the Summary of Product Characteristics approved by the European Medicines Evaluation Agency.
SUI: Stress urinary incontinence.
Safety & tolerability
Treatment-emergent adverse events with the use of duloxetine in SUI have been described in the reported clinical studies [65–68,70,72] and in meta-analyses thereof [74,75]. The tolerability of duloxetine has also been assessed in the treatment of depression and diabetic peripheral neuropathic pain [19,74–79]. Additional safety information comes from studies in healthy subjects [48,80].
Table 3 shows a combined analysis of the incidence of adverse events during 12 weeks of treatment of SUI with duloxetine (n = 958 patients) or placebo (n = 955 patients) as detailed in the Summary of Product Characteristics approved by the European Medicines Evaluation Agency. While the incidence of adverse events associated with duloxetine treatment within individual studies ranged between 73 and 81%, corresponding incidences with placebo ranged between 50 and 64% (p < 0.05 within each study) [65–68]. Adverse events leading to study discontinuation ranged between 15 and 24% with duloxetine compared with 2–5% with placebo [65–68]. While there was a single death reported during these studies (a 70-year-old woman randomized to duloxetine died from a multifocal cerebrovascular accident not attributed to study medication), the overall incidence of serious adverse events did not differ significantly between duloxetine and placebo; moreover, no serious adverse event resulted in chronic sequelae [81].
The most common side effect of duloxetine was nausea, which occurred in 23.2% of duloxetine compared with 3.7% of placebo-treated patients. Nausea was also the most common side effect with the use of duloxetine in patients with depression [76] or polyneuropathic pain [19]. In studies with depressed patients, which compared duloxetine to other 5-HT uptake inhibitors, the incidence of nausea resembled that seen with paroxetine or fluoxetine [76]. It is assumed that an increased availability of 5-HT acting on 5-HT3 receptors is the reason for nausea [76]. The intensity of duloxetine-associated nausea was mild-to-moderate in 82% of SUI patients; while nausea worsened in one out of 181 patients, it spontaneously resolved within 1 week in 52% and within 1 month in 81% of the afflicted women [66–68,82]. Thus, the prevalence of nausea decreased to approximately 12% after 1 week and 5% after 1 month (Figure 2). Nausea was a reason of study discontinuation in 3.1–6.4% of duloxetine-treated SUI patients [65–68]. A recent study demonstrates that a dose-escalation treatment regimen (20 mg twice-daily for 2 weeks, escalating to the fully effective dose of 40 mg twice-daily) is associated with a significantly reduced incidence of nausea and dizziness compared with the standard treatment (40 mg twice-daily throughout) [83]. Patient counseling regarding the mild-to-moderate intensity and transient nature of duloxetine-associated nausea may also help to increase compliance.
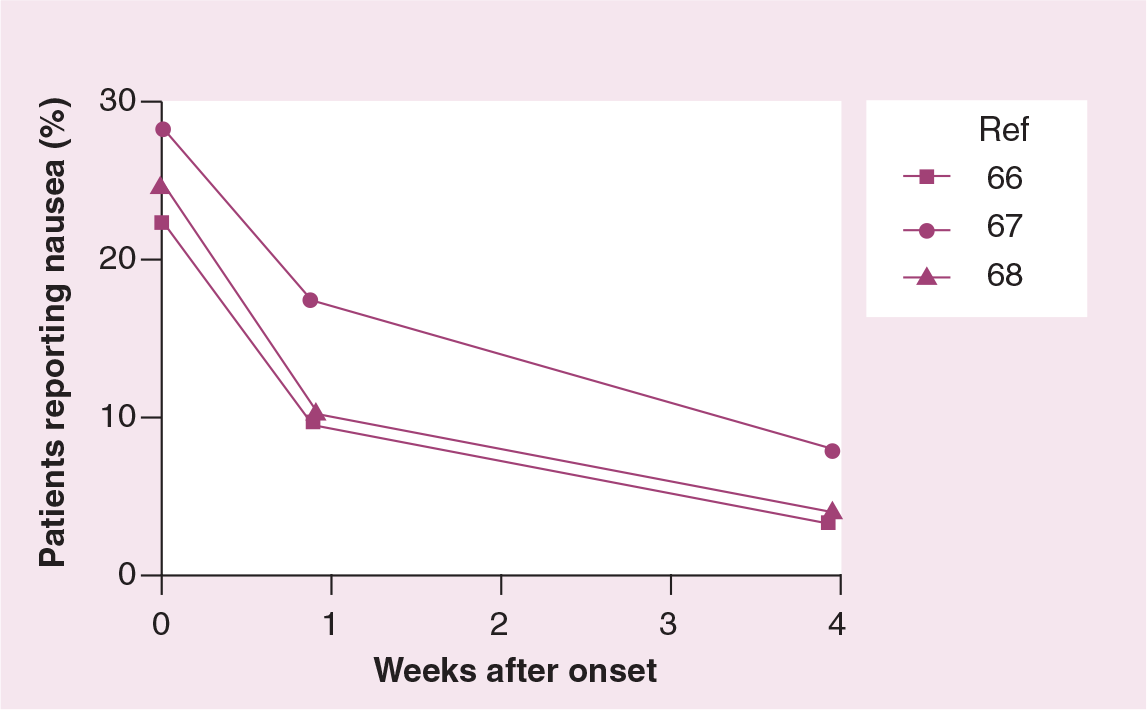
History of nausea in duloxetine-treated SUI patients.
Other frequently reported adverse events of duloxetine in the treatment of SUI include dry mouth (12–19%), fatigue (10–15%), insomnia (13–14%), constipation (10–14%), headache (7–15%) and dizziness (8–12%) (Table 3), the latter also occurring upon withdrawal of duloxetine [83]. Sleep disturbances are typical side effects of amine uptake inhibitors [84]. A study in young, healthy subjects confirmed the clinically observed sleep disturbances with duloxetine treatment by using electroencephalogram (EEG) monitoring; while such disturbances were more frequent with duloxetine than with placebo, the effects of duloxetine (80 mg once-daily or 60 mg twice-daily) were quantitatively similar to those seen with desipramine (50 mg twice-daily) [80].
Since duloxetine is also used to improve mood in depressed patients [18], it has been investigated whether duloxetine may alter mood in SUI patients. An analysis of the placebo-controlled duloxetine studies (involving 1913 patients) and their uncontrolled long-term follow-up (involving 1877 patients) did not provide any evidence for an induction of mania or hypomania [74]. Recent discussions have highlighted the possibility that 5-HT uptake inhibitors used in the treatment of depression may induce suicidal behavior, particularly in children. Suicidal behavior has not been reported in the published studies of duloxetine in the treatment of SUI. Since suicide, fortunately, is a rare event, the available data nevertheless have limited statistical power to firmly exclude such behavior. Therefore, the duloxetine label states ‘A s with other drugs with similar pharmacologic action (e.g., antidepressants), isolated cases of suicidal ideation and suicidal behaviors have been reported during duloxetine therapy or early after treatment discontinuation. Physicians should encourage patients to report any distressing thoughts or feelings at any time.’
Since the desired effects of duloxetine in the treatment of SUI involve an increased bladder outlet resistance, the possibility exists that this may lead to obstructive voiding symptoms or even acute urinary retention. A meta-analysis of nine studies of depressed patients and four studies of SUI patients reported that three (two men and one woman) out of 4719 duloxetine patients discontinued treatment due to obstructive voiding symptoms; however, acute urinary retention requiring catheterization was not observed in any patient [75]. With regard to other genito–urologic side effects, it is well established that 5-HT reuptake inhibitors can have adverse effects on sexual function; some of them are even recommended for the off-label treatment of premature ejaculation. Therefore, it is not surprising that duloxetine was associated with decreased libido, loss of libido or anorgasmia in a small number of patients, but more often than with placebo (Table 3). Similar findings have been obtained in depressed patients treated with duloxetine, whereas direct comparative studies indicate that it may occur less frequently than with drugs such as paroxetine [77].
While the above-mentioned adverse events are unpleasant, most of them are not dangerous. On the other hand, uptake inhibitors, particularly tricyclic antidepressants, have been associated with cardiovascular side effects including arrhythmias. Small clinical pharmacology studies in healthy volunteers inconsistently reported minor elevations of blood pressure and/or heart rate with duloxetine [37–39,48]. Two studies in SUI patients also reported a small (less than 3 bpm) but statistically significant increase in heart rate with duloxetine compared with placebo treatment [67,68]. Duloxetine-associated blood pressure elevations in the treatment of SUI were reported as clinically insignificant [68] or absent [67]. A meta-analysis of studies in depressed patients reported a small but statistically significant increase in heart rate (1.6 vs − 0.6 bpm) and systolic blood pressure (1.0 vs −1.2 mmHg) [79]. Patients with elevated blood pressure prior to duloxetine treatment, if anything, had lowered values upon duloxetine exposure [79]. With regard to an arrhythmogenic potential, no increases of heart rate-corrected QT intervals were observed in SUI patients [68], depressed patients [79] or healthy subjects [48]. Even at clearly supratherapeutic doses of 200 mg twice daily, QT prolongation was not observed [85]. Finally, significant increases in several hepatic enzymes such as alkaline phosphatase, alanine aminotransferase and aspartate aminotransferase were reported during duloxetine treatment, but all increases were within the normal range [81,82].
Regulatory affairs
Duloxetine was approved for the treatment of female SUI by the European Medicines Evaluation Agency and has been introduced into the market for this indication in several European countries. In the USA, duloxetine has been approved by the US Food and Drug Administration (FDA) for the indications of major depressive disorder and polyneuropathic pain; an application for US approval of duloxetine in the treatment of SUI has been withdrawn.
According to its European label information, duloxetine is contraindicated in patients with hypersensitivity to the active substance or to any of the excipients, in patients with liver disease resulting in hepatic impairment and during pregnancy and lactation. Duloxetine should not be used in combination with nonselective, irreversible monoamine oxidase inhibitors such as tranylcypromine, or in combination with CYP1A2 inhibitors such as fluvoxamine, ciprofloxacin or enoxacine.
Future perspective
Until now, duloxetine has mainly been investigated clinically in women with SUI who did not have a major urgency component. Comparisons of women with pure SUI and those with mixed incontinence indicate that duloxetine is similarly effective in both groups [63]. The muscarinic receptor antagonist tolterodine was also found to be effective in mixed incontinence [15–17]. Whether duloxetine or muscarinic receptor antagonists have a better risk–benefit ratio in women with mixed incontinence remains to be determined. While urge incontinence is mostly treated with muscarinic receptor antagonists such as oxybutynin or tolterodine [14,61], it appears plausible that duloxetine may also be effective in such patients, since it increases bladder capacity in animals [43]. Which of the two yields better clinical responses remains to be investigated. Moreover, the different mechanisms of action of muscarinic receptor antagonists and duloxetine make a possible combination attractive in both mixed and urge incontinence. The lack of pharmacokinetic interference between duloxetine and tolterodine [64] facilitates such studies. Finally, it should be considered that men also can suffer from SUI, particularly following prostatectomy. While off-label use of duloxetine in such patients has already started, its efficacy and safety in this group remain to be determined.
Executive summary
Duloxetine is a selective inhibitor of neuronal serotonin and norepinephrine uptake.
This leads to an increase in urethral striated muscle activity and bladder capacity during the filling phase of the micturition cycle, most likely via a site of action in the lower spinal cord (Onuf's nucleus).
Duloxetine in its commercially available formulation is readily absorbed from the gut and has a large volume of distribution.
It is extensively metabolized, mainly by cytochrome P450 (CYP)2D6 and CYP1A2, and CYP1A2 inhibiting drugs can markedly increase drug exposure.
The efficacy of duloxetine in the treatment of stress urinary incontinence (SUI) has been studied in several randomized, double-blind studies in women with moderate-to-severe SUI.
While these studies consistently demonstrated superiority relative to placebo, the additional benefit from active treatment was moderate, reflecting the strong placebo effect in this indication.
The dissociation from placebo appears strongest in women with severe SUI and weakest in those with mild SUI.
Duloxetine treatment is frequently associated with adverse events such as nausea, dry mouth, fatigue, insomnia and constipation, but serious adverse events, particularly with regard to the cardiovascular system, are rare.
Therefore, duloxetine appears to be a suitable option for the treatment of moderate-to-severe SUI, but its role relative to that of behavioral or surgical therapy remains to be determined.
Information resources
European Regulatory authorities
www.emea.eu.int/humandocs/Humans/EPAR/yentreve/yentreve.htm
(Accessed October 2005)
US regulatory authorities
www.accessdata.fda.gov/scripts/cder/drugsa-tfda/index.cfm?fuseaction = Search. Overview&DrugName = CYM-BALTA
(Accessed October 2005)
Manufacturer-sponsored website (German language)
(Accessed October 2005)
Manufacturer-sponsored website
(Accessed October 2005)
Additional information is found at the trial register website of the manufacturer
www.lillytrials.com/results/by_product/results_yentreve.html
(Accessed October 2005)
Disclaimer
Conflict of interest: In recent years the authors have received research grants and/or speaker and consultant honoraria from the following companies in the area of urinary incontinence: 4SC, Astellas, Boehringer Ingelheim, Eli Lilly, Pfizer and Theravance.