
Abstract
Endogenous mechanisms of protection against ischemia can be demonstrated in brain and other organs. The induction of such protection is via a response to sub lethal stress which induces “preconditioning”. The preconditioned organ is then “tolerant” to injury from subsequent severe stress of the same or different etiology. Protection is substantial (70% reduction) but delayed in onset and is transient. Gene expression is unique between brains preconditioned, injured (stroke) or made tolerant. Thus, preconditioning reprograms the response to lethal ischemic stress (stroke), reprogrammed from an injury induction response to a neuroprotective processes. Postconditioning refers to attenuation of injurious processes occurring during reperfusion of ischemic brain. Transient mechanical interruption of reperfusion induces post-conditioning which can attenuate reperfusion injury. Post-conditioning protects ischemic brain by decreasing reperfusion induced oxygen free radical formation. The free radicals produce injury via mitochondrial damage which can be repaired experimentally. Post-conditioning produces neuroprotection as potent as experimental preconditioning. The recognition of broad based gene silencing (suppression of thousands of genes) as the phenotype of the preconditioned, ischemic tolerant brain, may explain failure of all single target drugs for stroke. As risks of reperfusion injury accompany treatment for acute stroke, endogenous neuroprotective and repair mechanisms offer translational stroke therapy.
PRECONDITIONING
Attempts to develop treatments for acute stroke have not fulfilled expectations(O'Collins et al., 2006). Current approaches to neuroprotection in stroke have been mainly those of exogenous drug administration. However there are endogenous approaches to neuroprotection, which have a different logic(Dirnagl et al., 2003). Ischemic brain injury, ischemic injury in other organs and acute brain injury of nonischemic causes (eg seizures(Sasahira et al., 1995) (Dirnagl et al., 2003)), can have injury attenuated and outcome improved by way of preconditioning the organ by exposure to sub lethal stress. The brain, heart and other organs have such endogenous, highly conserved, gene-based neuroprotective programs, the induction of which reduces ischemic injury(Gidday, 2006). Here, brief exposure to sublethal ischemia produces tolerance to a subsequent, severe ischemic challenge. The protection is substantial and dependent on new protein synthesis (Stenzel-Poore et al., 2003, Stenzel-Poore et al., 2004, Barone et al., 1998). The molecular effector of preconditioning is the induction of broad based transcriptional suppression (Stenzel-Poore, Stevens et al. 2003). Such gene silencing is also the effector of neuroprotection against epileptic brain injury where brief, non injurious seizures are used to induce preconditioning stress (Jimenez-Mateos et al., 2008). Although transcriptional suppression characterizes tolerant brain, the protection is protein synthesis dependent. Up regulation of polycomb group proteins have been found as tolerance effectors in brain ischemia and to act as transcriptional suppressors via epigenetic mechanisms (Stapels et al., 2010). Retrospective data support a clinical counterpart of preconditioning in human brain: patients with prodromal transient ischemic attacks (TIA) have milder strokes(Weih et al., 1999).
POST-CONDITIONING
Current clinical treatment for acute stroke is focused on reperfusion of the ischemic region of brain. Here ischemic brain is reperfused by clot dissolution with tissue plasminogen activator (tPA) or via mechanical clot removal (endovascular therapy). Clot dissolution produces reperfusion, and successful reperfusion is associated with better functional outcome (Broderick et al., 2013);(Chimowitz, 2013). However, mechanical clot extraction, while yielding improved reperfusion over tPA clot dissolution, does not show increased clinical benefit over clot dissolution therapy (Broderick et al., 2013);(Ciccone et al., 2013). Thus, an additional restorative approach to ischemic injured but reperfused brain is needed for protection against, or attenuation of, injury that results, in substantial measure, from the burst of oxygen free radicals induced early in reperfusion. Therapy directed against reperfusion injury has been shown effective experimentally in the heart (Zhao et al., 2003) and brain (Zhao et al., 2006) by attenuation of reperfusion using intermittent reocclusion of the artery that perfuses the ischemic vascular bed. This treatment in the heart and brain is known as “postconditioning.” In our experience, postconditioning is effective in brain if implemented within the first 30 minutes of reperfusion, but not later(Pignataro et al., 2008). These observed results of neuroprotection fit with the hypothesis that the neuroprotection of post-conditioning is due to attenuation of mtDNA injury(Chen et al., 2003).
MODELING POST CONDITIONING
We and others modeled postconditioning as a strategy for neuroprotection in brain reperfused after stroke(Zhao et al., 2006, Pignataro et al., 2008). We addressed this issue by modeling reperfusion in the same artery as that affected by focal ischemic stroke. Based on reports in heart, we addressed the reperfusion methodology (time course, time window and permutations and patterns of reperfusion), the potential role of survival kinases and the resultant degree of brain tissue salvage. Preconditioning and postconditioning were also compared. Preconditioning was modeled with transient suture occlusion of the middle cerebral artery for 30 minutes of focal ischemia, followed 72 hours later by 100 minutes focal ischemic challenge (Longa et al., 1989); (Stenzel-Poore et al., 2003). Postconditioning attenuation of reperfusion was modeled (followed injury inducing 100 min focal ischemia) as 2 min reperfusion/2 minutes reocclusion × 5, 3 min reperfusion/3 minutes reocclusion × 3, 10 min reperfusion/10 minutes reocclusion × 1, and 30 min reperfusion/10 minutes occlusion × 1 (Fig. 1). Both preconditioning, and postconditioning with 10 min reperfusion / 10 minute reocclusion (but not with other treatment paradigms - shown in Fig. 1), were highly and equally effective in neuroprotection (Fig. 2). Pre and post conditioning are also equally effective in the heart (Ovize et al., 2010) but multiple brief periods of perfusion / reocclusion were required to precondition heart. In brain, pre and post conditioning do not produce additive neuroprotection (Fig. 2). Further, in brain, pre and post conditioning had very different profiles of effector survival kinases (Fig. 3). Thus we find that post conditioning in brain is distinct from that in heart and has the potential for potent neuroprotection although the time window of effect is narrow.

Schematic diagram showing the various permutations of ischemic challenges. For control ischemia, animals receive 100 mins of MCAO. Infarct volume is determined 24 h after the final ischemic challenge by TTC staining. Preconditioning was carried out as described previously, whereby a 30-min MCAO challenge is paired with 100 mins of MCAO after 72 h of reperfusion. Postconditioning was performed by combining 100 mins of MCAO followed by various brief ischemic durations and reperfusion periods. (From Pignataro et al. 2008.
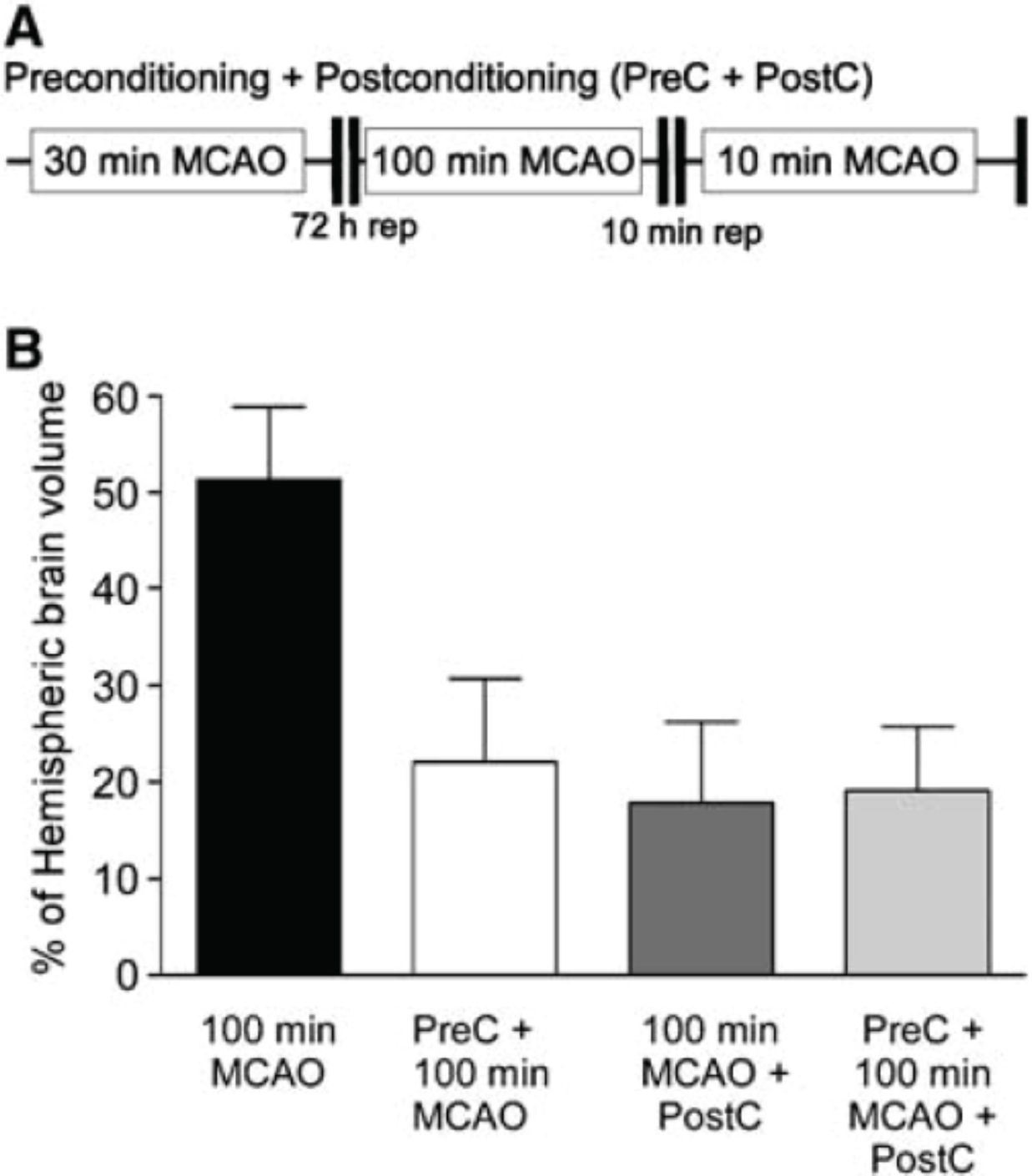
Nonadditive nature of pre- and postconditioning neuroprotection in the brain. (A) Schematic diagram of combined pre- and postconditioning. Animals were subject to 30 mins of MCAO, followed by 72 h reperfusion. Animals were then subject to 100 mins of MCAO, 10 mins of reperfusion, and a subsequent 10 mins of MCAO challenge. Infarct volume was determined 24 h later by TTC staining. (B) Quantification of pre-, postconditioning, and addition of pre- plus postconditioning ischemia. Note the lack of significant further protection by adding pre- and postconditioning. Data shown are mean7 s.d. n=5. Data were analyzed by ANOVA with post hoc Bonferroni's test, *P<0.01 versus 100 mins of MCAO. (From Pignataro et al. 2008.
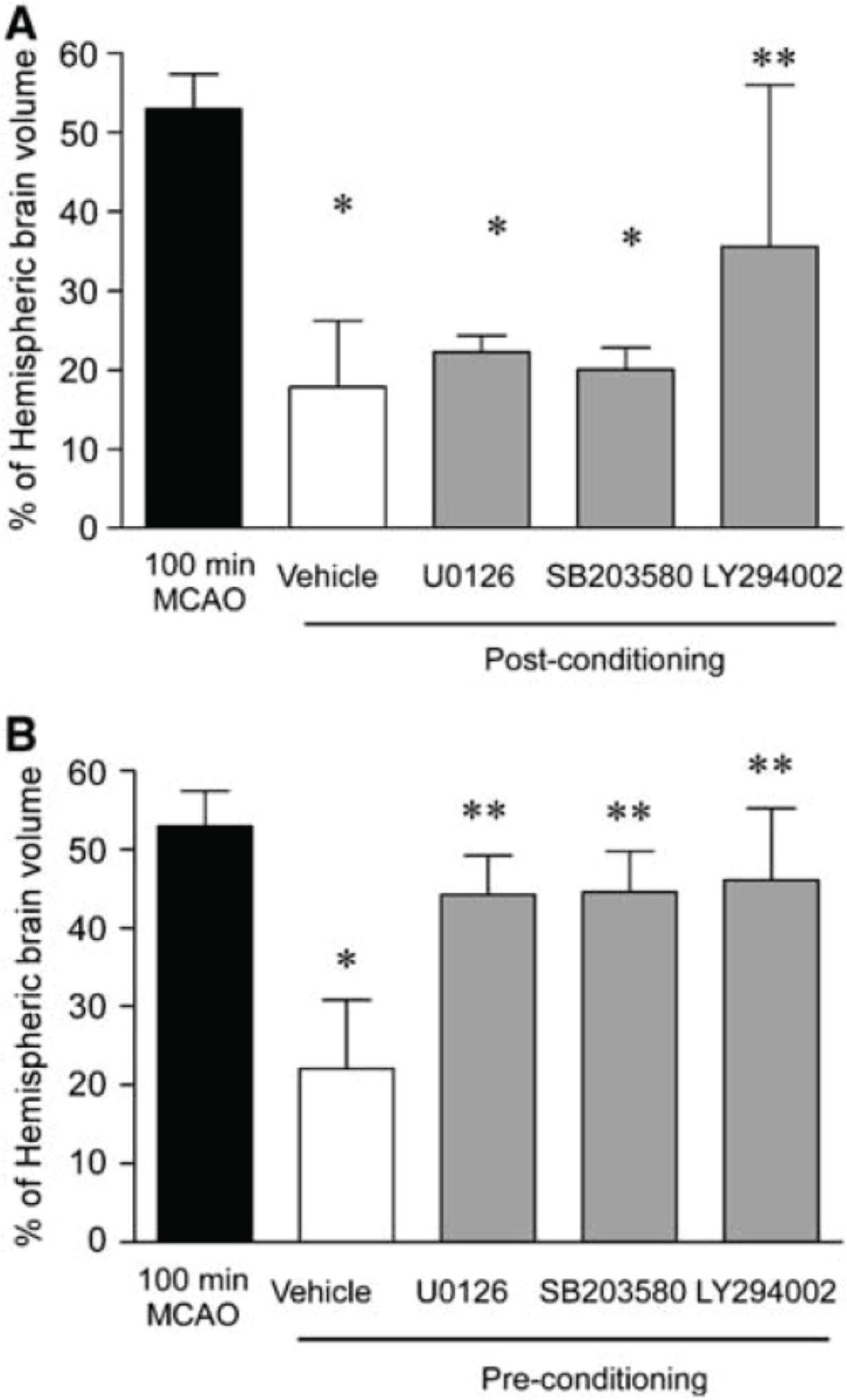
(A) Postconditioning is blocked by LY294002. Animals were administered LY294002, U0126, or SB203580 before ischemia and postconditioning. Infarct volume was determined 24 h later by TTC staining. Data shown are mean7s.d. n=5. Data were analyzed by ANOVA with post hoc Bonferroni's test, *P<0.05 versus control 100 mins of MCAO, **P<0.05 versus vehicle. (B) Effect of SB203580, U0126, and LY294002 on preconditioning-induced neuroprotection. After preconditioning, vehicle or drug was administered. Animals were then subject to 100 mins of MCAO 72 h later and infarct volume determined by TTC staining. Data shown are mean7s.d., n=5. Data were analyzed by ANOVA followed by post hoc Bonferoni's test, *P<0.05 versus 100 mins MCAO and **P<0.05 versus vehicle. (From Pignataro et al. 2008.
REPERFUSION INJURY AND REACTIVE OXYGEN SPECIES (ROS)
The issue of reperfusion induced injury by ROS illustrates the unsatisfactory situation of stroke therapeutic development. Multiple attempts directed at the inhibition of ROS have evolved (Niizuma et al., 2009). While decades of laboratory and clinical studies make it clear that ROS are pathogenically important across the entire spectrum of cerebrovascular disease(Pundik et al., 2012) as well as many other disorders(Ballinger et al., 2000) the beneficial effects of anti-oxidants in clinical trials for stroke have been unimpressive(Lees et al., 2006). These negative outcomes may be attributed in part to the heterogeneous nature of cerebral ischemia, which tends to obfuscate the design and interpretation of clinical trials. There also remains considerable scientific uncertainty about the molecular targets of anti-oxidant drug action. For example, non-selective antioxidants may disrupt ROS signaling required for cell survival and recovery (Hsieh and Yang, 2013). Further, the strategies currently available may not target the key sentinel molecule(s) that integrate the cellular effects of ROS and ROS signaling.
ROS AND MITOCHONDRIAL INJURY
We do know that cellular damage from ROS targets mitochondria. Mitochondrial DNA (mtDNA) is far more sensitive to oxidative damage than the nuclear genome(Grishko et al., 2001);(Yakes and Van Houten, 1997);(Ballinger et al., 2000, Englander et al., 1999). Multiple lines of evidence support the idea that mtDNA serves as a molecular sentinel controlling cell fate in response to oxidant stress. Work in vascular endothelial cells and other cell types shows that mtDNA is highly sensitive to oxidative damage(Ballinger et al., 2000);(Grishko et al., 2001);(Yakes and Van Houten, 1997). There is also a conspicuous association between mtDNA damage and oxidant-induced cell death as the propensity for cytotoxicity is inversely related to the efficiency of mtDNA repair (Grishko et al., 2001);(Harrison et al., 2005). We have shown this relationship in ischemic brain (Chen et al., 2003). Genetic modulation of the first and rate-limiting step in mtDNA repair—mediated by DNA glycosylases that detect and excise oxidatively damaged purine or pyrimidine bases—coordinately regulates ROS-induced mtDNA damage and cell death in all cultured cell populations so far examined(Dobson et al., 2002);(Rachek et al., 2006);(Harrison et al., 2007, Ruchko et al., 2005);(Rachek et al., 2002);(Ruchko et al., 2011). In the nervous system, the DNA glycosylase Ogg1 is neuroprotective in the setting of oxidative DNA damage in vitro and in modeled stroke in vivo (Liu et al., 2011).
We have previously shown the neuroprotective effect of the upregulation of mitochondrial base excision repair (BER) enzymes in ischemic brain as a mechanism of endogenous neuroprotection produced by ischemic tolerance (where brief periods of sublethal ischemia protect against subsequent harmful ischemia) (Chen et al., 2003). The degree of repair potential is injury-dependent. Brief (30min) middle cerebral artery occlusion (MCAO) induced mild oxidative mitochondrial DNA damage which initiated prolonged (up to 72-h) activation of the principal enzymes of the mitochondrial BER pathway. In contrast, prolonged (100-min MCAO) ischemia induced more substantial mitochondrial oxidative DNA damage, associated with transient (approximately 1 h) elevation of BER activity, which then declined below control levels over the course of 4 to 72 h(Fig. 4) (Chen et al., 2003).

Detection of mitochondrial DNA (mtDNA) damage induced by cerebral ischemia assessed by quantitative polymerase chain reaction (QPCR) amplification after 30-minute and 100-minute middle cerebral artery occlusion (MCAO) or sham (S). Repair of mtDNA damage occurs after 30-minute (but not 100-minute) MCAO. (From Chen et al. 2003).
NOVEL NEUROPROTECTIVE STRATEGY FOR REPERFUSION INJURY
With this background, we sought to extend postconditoning neuroprotection to the organelle level. Accordingly we developed mtDNA repair as a new intervention in tissue ischemia. We devised a novel fusion protein construct that targets DNA repair glycosylases to mitochondria (Koczor et al., 2009). The constructs, consist of the TAT sequence from HIV (to facilitate cellular uptake of the protein), the mitochondrial targeting sequence from Mn-SOD, and one of two DNA glycosylases, either the mammalian Ogg1, which recognizes and cleaves oxidized purine base products, or the bacterial Endonuclease (Endo) III, which recognizes and cleaves oxidized pyrimidines. Both fusion proteins accelerate the first and rate-limiting step in mitochondrial BER, although they target different oxidized base products. Most importantly, both constructs prevent, and in some instances reverse, tissue injury evoked by multiple different injurious stimuli in intact animal models (Chouteau et al., 2011). Our studies demonstrate in clinically-relevant rodent models (hydrogen peroxide-(Chouteau et al., 2011), ventilator-induced lung injury(Hashizume et al., 2013) and hyperoxiainduced fetal lung injury and dysmorphogenesis (Gebb et al., 2013) that mt-targeted DNA repair enzymes exert no off-target effects, prevent oxidative mtDNA damage, and suppress end organ injury / mortality. We also demonstrate the effectiveness of mt-targeted DNA repair “drugs” in ischemia-reperfusion injury in brain and in the setting of brain reperfusion following modeled tPA clot lysis or clot extraction as emergency stroke therapy. Accordingly, using a model of middle cerebral artery occlusion/reperfusion in the mouse, enhancement of mtDNA repair attenuates the degree of brain infarction following reperfusion induced as an acute treatment for stroke. The time window of effect is at least three hours following reperfusion onset.
SUMMARY
Thus there is potent neuroprotection for stroke which is non pharmacologic but rather induced by endogenous mechanisms. Preconditioning brain (by exposure to brief ischemia of a duration less than that inducing injury) is mediated by transcriptional suppression. Potential clinical indications include cardiovascular surgical procedures with high risk of intraoperative / post operative stroke or neurovascular procedures with significant ischemic potential. Postconditioning (transient interruption of blood flow during therapeutic reperfusion) is protective in experimental systems. The reperfusion injury caused by tPA thrombolysis or endovascular clot removal permits reactive oxygen species induced mitochondrial injury. Such reperfusion induced injury can be corrected experimentally using base excision repair techniques. Such additive therapy holds promise for acute revascularization treatment of acute stroke.