
Abstract
Synthetic oleanane triterpenoids are multifunctional drugs being developed for the prevention and treatment of a variety of chronic diseases driven by inflammation and oxidative stress. Low nanomolar concentrations of triterpenoids inhibit the induction of inflammatory cytokines, and these drugs are potent activators of the Nrf2 cytoprotective pathway. In contrast, low micromolar concentrations of triterpenoids increased the production of ROS and induced apoptosis in a dose-dependent manner in malignant MCF10 CA1a breast cancer cells. Because cancer cells respond differently to ROS than normal cells, it should be possible to exploit these differences therapeutically. In an experimental model of lung cancer, the triterpenoids activated the Nrf2 pathway, as seen by induction of the cytoprotective enzyme NQO1, and reduced the toxicity of carboplatin and paclitaxel. The induction of the Nrf2 pathway in the lung did not suppress the efficacy of treatment with carboplatin and paclitaxel, as the average tumor burden in the group treated with the combination of CDDO-Me and carboplatin/paclitaxel decreased by 90% (P < 0.05 vs. the controls and both single treatment groups). Understanding the dose response of triterpenoids and related drugs will help provide the proper context for optimizing their potential clinical utility.
INTRODUCTION
More than 6000 pentacyclic triterpenoids can be found in nature, and useful biological activities have been described for triterpenoids such as oleanolic acid, ursolic acid and betulinic acid (Hill and Connolly 2012). Because so many triterpenoids are found in plants, many triterpenoids have been safely ingested by humans for centuries. Oleanolic acid itself has weak anti-inflammatory activity and is readily available in kilogram quantities. In order to enhance the potency of oleanolic acid, a new chemical synthesis program was started. The rationale was to select a natural product with a good safety profile and known biological activity as a scaffold, make chemical modifications to improve potency, and then screen for anti-inflammatory activity (Sporn et al. 2011).
Over 300 semi-synthetic oleanane triterpenoids have been synthesized and tested for their ability to suppress inflammation. Three of the most potent of these new triterpenoids include CDDO-Imidazolide (CDDO-Im), CDDO-methyl ester (CDDO-Me), and CDDO-ethyl amide (CDDO-Ea), which differ only in the functional group at the C28 position (Sporn et al. 2011). Notably, the biological response to these compounds is highly dependent on dose (Liby and Sporn 2012). The triterpenoids are among the most potent known activators of the cytoprotective Nrf2 pathway, and thus low nanomolar concentrations of these drugs inhibit both inflammation and oxidative stress (Dinkova-Kostova et al. 2005; Liby et al. 2005). Higher nanomolar concentrations of these triterpenoids induce differentiation of adipocytes and immature cancer cells and block proliferation of cancer cells (Suh et al. 1999; Ito et al. 2001). Increasing the dose of drug to low micromolar concentrations, which can be achieved in vivo, can initiate cell death in cancer cells without harming normal tissues (Ito et al. 2000; Chauhan et al. 2004; Ikeda et al. 2004).
The multifunctional nature of the synthetic oleanane triterpenoids has also been observed in humans. In a phase 1 trial for treatment of patients with advanced cancers, CDDO-Me was well-tolerated in a dose-escalation study when given orally at 5–900 mg/day and provided some objective improvements in the size of the tumors (Hong et al. 2012). Unexpectedly, the drug also improved biomarkers of renal filtration in the oncology patients. In subsequent clinical trials, CDDO-Me, or bardoxolone methyl, significantly improved kidney function in diabetic patients with chronic kidney disease (Pergola et al. 2011). However, the clinical development of the synthetic triterpenoids is currently on hold because of unexpected complications in patients with stage 4 chronic kidney disease.
Because bardoxolone methyl was well-tolerated in other clinical trials, the unexpected toxicity may be unique to patients with advanced kidney disease (Ruiz et al. 2013). The ability of high doses of triterpenoids to selectively induce cell death in cancer cells implies that these drugs could still be used for the treatment of cancer, especially if combined with standard chemotherapy. An intriguing hypothesis in the field of cancer research suggests that it may be possible to selectively kill cancer cells without harming normal cells by exploiting the altered redox balance found in cancer cells (Trachootham et al. 2006; Trachootham et al. 2009; Raj et al. 2011; Watson 2013). Because of their higher endogenous levels of reactive oxygen species (ROS), cancer cells are dependent on the Nrf2 antioxidative and cytoprotective pathway to maintain redox balance. They are thus highly vulnerable to cell death from drugs that increase ROS, while normal cells, which have low endogenous levels of ROS can more easily tolerate additional insults. In order to test this idea experimentally, we first examined the ability of the triterpenoids to induce ROS and apoptosis in the MCF10 model of progressive malignancy and then used the A/J mouse model of lung cancer to investigate the efficacy of the triterpenoids in combination with carboplatin and paclitaxel for the treatment of established lung tumors.
MATERIALS AND METHODS
Reagents
CDDO-Me and CDDO-EA were synthesized as previously described (Honda et al. 1999; Honda et al. 2002; Yates et al. 2007) and provided by Reata Pharmaceuticals. Carboplatin and paclitaxel were provided by the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis of the National Cancer Institute. All other reagents were from Sigma unless otherwise indicated.
Tissue culture, ROS assays and annexin experiments
The various MCF10 cell lines (Miller 2000; Strickland et al. 2000; Santner et al. 2001) were kindly provided by Fred Miller (Wayne State University School of Medicine) and were grown in DMEM/F12 supplemented with 5% horse serum, 29 mM sodium bicarbonate, 10 mM HEPES, 20 ng/ml EGF (R & D Systems), 10 μ/ml insulin, 0.5 μ/ml hydrocortisone, and 100 ng/ml cholera toxin in 5% CO2 at 37°C. Nrf2 wild-type (+/+) and knockout (−/−) mouse embryo fibroblasts, generously provided by Jeff Chan UC Irvine, were grown in DMEM/F12, 10% serum, 1% non-essential amino acids and 0.8% 2-mercaptoethanol. In order to measure ROS, cells were plated and allowed to attach overnight. Cells were treated either with triterpenoids alone for 4 hrs or with drug for 16 hrs prior to being challenged with tert-butyl hydroperoxide (tBHP) for 15 min. In all of these ROS experiments, cells were incubated with 10 μM of H2DCFDA (Molecular Probes) for 1–2 hrs prior to harvesting. The production of intracellular ROS was measured by flow cytometry using an excitation wavelength of 480 nm and an emission wavelength of 525 nm. Apoptosis was measured using the TACS annexin V kit (R & D Systems) and flow cytometry, and the percentage of apoptotic cells included both early (annexin positive) and late (PI positive) apoptotic cells.
Lung cancer studies
All animal studies were done in accordance with protocols approved by the Institutional Animal Care and Use Committee at Dartmouth. Seven week-old female A/J mice (Jackson Laboratory) were injected i.p. with a single 0.32 mg dose of vinyl carbamate (Toronto Research Chemicals) in acidified saline (pH 5). Mice were fed semisynthetic AIN-93G diet (Harlan Teklad), beginning one week before initiation with the carcinogen and continuing for the duration of the experiment. Twelve weeks after initiation, the mice were randomized and fed control AIN-93G diet or triterpenoids mixed into this diet for an additional 12 weeks. One week after the treatment diets started, mice were also injected with six doses of carboplatin (50 mg/kg ip) and paclitaxel (15 mg/kg ip), given every other week. At the end of the experiment, lungs were harvested, inflated with formalin, step-sectioned (6 sections per lung, 100 microns apart) and stained with H&E. The number, size, and histopathology of the tumors were evaluated in a blinded fashion as described previously (Liby et al. 2007; Liby et al. 2009). In order to detect activation of the Nrf2 pathway, 12 female A/J mice were fed triterpenoids in AIN-93G diet for 1 wk at the same concentrations used for the lung cancer experiments. Tissues were harvested, and the induction of NQO1 mRNA in peripheral blood mononuclear cells (PBMC) and NQO1 enzyme activity in liver and lungs were determined as described (Yates et al. 2007; Pitha-Rowe et al. 2009).
Statistical analysis
All in vitro experiments were repeated 2–3 times, and means ± SD are shown and were analyzed by t-test or one-way ANOVA. Results for the in vivo experiments are described by the mean ± SE and were analyzed by one-way ANOVA and a Tukey test or by one-way ANOVA on ranks (Kruskal-Wallis) and Dunn's method, if the data did not fit a normal distribution (SigmaStat3.5). All p values are two-sided.
RESULTS AND DISCUSSION
Increasing malignancy in various MCF10 cell lines correlates with enhanced production of intracellular ROS
The development of a series of human breast epithelial cell lines with a common genetic background but with various degrees of malignancy provides a unique model to study the progression of cancer (Pauley et al. 1993). The MCF10A cells were derived from human breast epithelium obtained from a mastectomy, and the cells spontaneously immortalized. Neither the original tissue nor the resulting cell line displayed symptoms of malignancy, and these cells do not grow in immunodeficient mice. Transfection with an activating T24 H-Ras sequence into MCF10A cells yielded new cells (MCF10 AT1) that have lost their anchorage dependence and are invasive in vitro. The MCF10 AT1 cells form palpable premalignant lesions that resemble hyperplasia or carcinoma in situ when injected into athymic mice with some lesions progressing to invasive cancer (Miller 2000). By serially passing the MCF10 AT1 lesions between nude mice and tissue culture plates, the highly tumorigenic, undifferentiated, and metastatic MCF10 CA1a cells were created (Strickland et al. 2000; Santner et al. 2001). A brief summary of these cells lines, representing a spectrum of progression from the relatively normal MCF10A breast epithelial cells to fully malignant, poorly differentiated MCF10 CA1a cells is shown in Fig. 1.
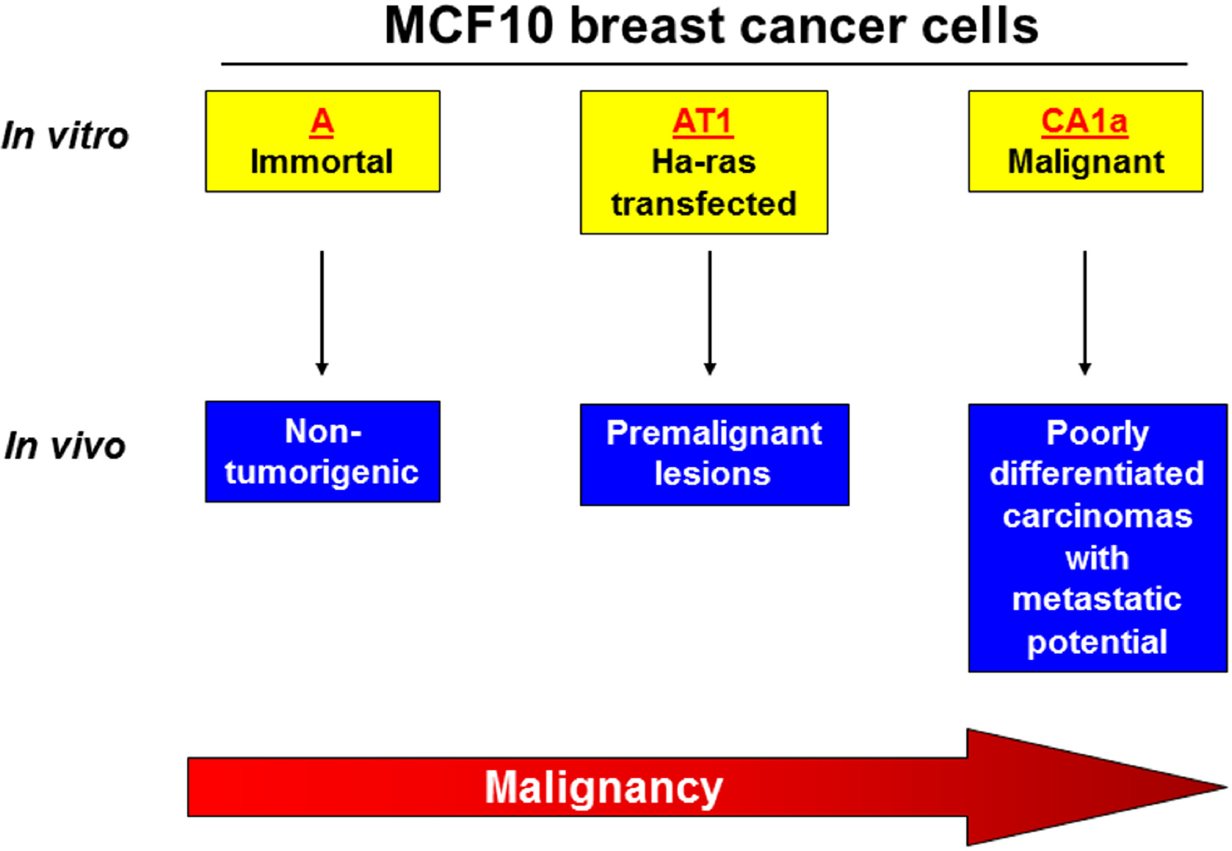
The progressive MCF10 model of breast cancer. The MCF10A spontaneously immortalized cell line was derived from non-malignant human breast epithelium and does not grow in immunodeficient mice. The introduction of an activating Ha-Ras mutation into MCF10A cells resulted in a new cell line (AT1) that forms palpable premalignant lesions when injected in vivo. Repeated passing of MCF10 AT1 premalignant lesions into nude mice created highly tumorigenic and invasive MCF10 CA1a cells. These 3 cells lines thus represent a spectrum of progression from the relatively normal MCF10A breast epithelial cells to fully malignant, poorly differentiated MCF10-CA1a breast cancer cells capable of metastasizing.
We first used these cell lines to test the promising idea that the high metabolic activity of cancer cells produces higher endogenous levels of ROS in malignant cells than in normal cells (Trachootham et al. 2009). MCF10A and AT-1 cells were incubated with the nonfluorescent indicator H2DCFDA, which passively diffuses into cells. In the presence of ROS, the probe is deacetylated and oxidized to fluorescent H2DCF, which can be detected by flow cytometry. As shown in Fig. 2, the endogenous levels of ROS were slightly, but reproducibly, higher in the untreated premalignant AT-1 cells than in the normal MCF10A breast epithelial cells. When challenged with the oxidant tert-butyl hydroperoxide (tBHP), ROS levels increased in both cell types in a dose-dependent manner, but were markedly higher in the Ras transformed MCF10 AT-1cells than in the normal cells (Fig. 2A). These studies were extended to include the fully malignant MCF10 CA1a cells. In these experiments, the levels of intracellular ROS were increased in a dose-dependent manner in response to treatment with tBHP, with strikingly higher levels of ROS in the most malignant CA1a cancer cells (Fig. 2B) compared to the other two cell lines.

Transformation of MCF10 cells with activated Ras enhances intracellular production of ROS. MCF10A immortalized breast epithelial cells, MCF10 AT-1 premalignant cells containing an activating mutation in H-Ras (A and B) and fully malignant MCF10-CA1a cells (B) were challenged with the oxidant tert-butyl hydroperoxide (tBHP) for 15 min, and reactive oxygen species (ROS) were measured by flow cytometry. These results are representative of 3–4 independent experiments. P < 0.05 vs. control, # P < 0.05 vs. control and vs. MCF10A cells.
The effects of triterpenoids on ROS production are dependent on dose
Synthetic oleanane triterpenoids are among the most potent known activators of the Nrf2 cytoprotective pathway (Dinkova-Kostova et al. 2005; Liby and Sporn 2012). Low nanomolar concentrations of triterpenoids decrease ROS levels induced by tBHP, but this protective effect is lost in cells that lack functioning Nrf2 protein (Liby et al. 2005). As shown in Fig. 3, pretreatment for 16 hrs with 0.1 μM of CDDO-Im reduced ROS production by 80% in response to a challenge with tBHP in the MCF10A breast epithelial cells and by 67% in the AT1 premalignant cells. In contrast, increasing the concentration of triterpenoid to 0.3 μM increased ROS production in both cell types, albeit to a higher level in the AT1 cells. Notably, cell death was observed primarily in the MCF10 CA1a cells treated with the higher concentration of CDDO-Im, and this observation was quantified in later experiments. The Nrf2-mediated upregulation of glutathione is the main cytoprotective mechanism used by sulforaphane to protect against hydroperoxides and other electrophiles (Higgins et al. 2009), and CDDO-Im does increase glutathione levels (Ikeda et al. 2003), which we have confirmed by flow cytometry. Ongoing experiments will determine whether the induction of glutathione by the triterpenoids is responsible for the protection against tBHP.
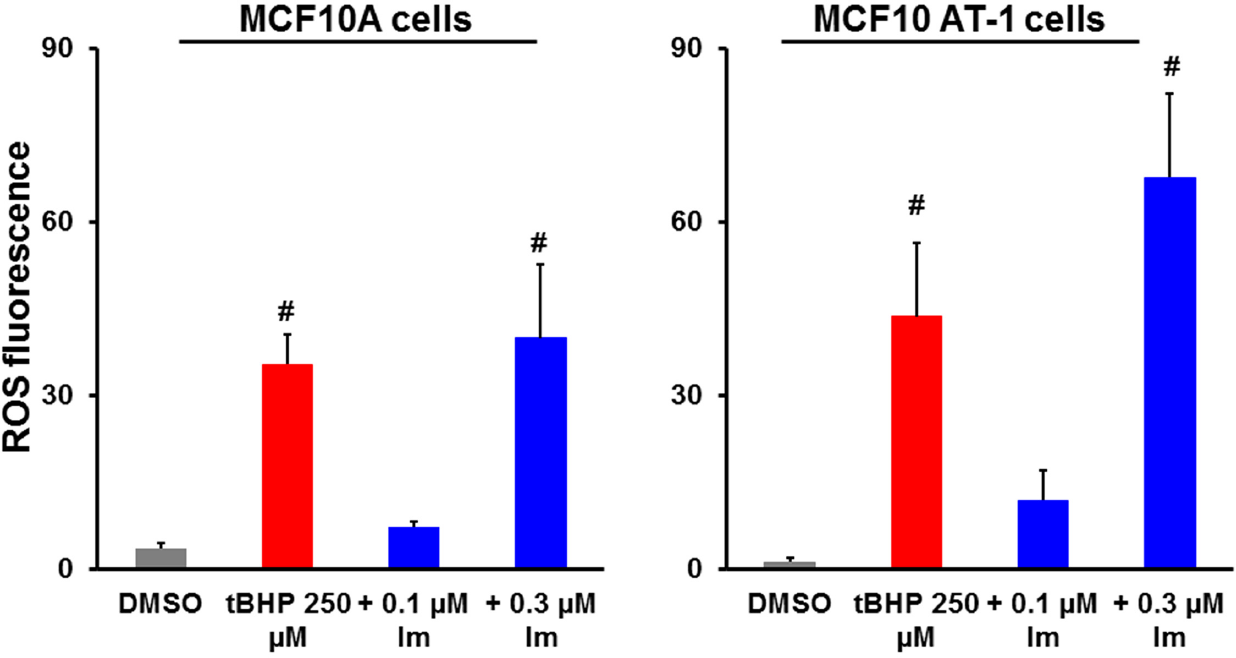
Different doses of the triterpenoid CDDO-Im reduce or amplify the production of ROS in MCF10 cells. MCF10 A and MCF10 AT-1 cells were treated with CDDO-Im for 16 hrs, incubated with H2DCFDA for 2 hrs, and challenged with tBHP for 15 min. The fluorescent ROS was detected by flow cytometry, and the results are representative of 3 independent experiments. P < 0.05 vs. control and 0.1 μM CDDO-Im.
Triterpenoids alone can increase ROS production and induce apoptosis
Because cancer cells produce more ROS, it may be possible to exploit this abnormality by using drugs to further enhance the production of intracellular ROS to toxic levels, triggering cell death specifically in malignant cells (Trachootham et al. 2009). When cells were treated with 0.3–0.6 μM CDDO-Im alone for 4 hr, ROS levels increased more than 2 fold in the CA1a cells and 5 fold in the AT1 cells, with higher levels observed in the CA1a cells than in the AT-1 cells (Fig. 4). When cells were incubated with the same concentrations of triterpenoids that increased ROS but for longer periods, these higher concentrations induced cell death (Fig. 5). Notably, the 0.1 μM concentration of CDDO-Im that diminished ROS production did not cause apoptosis, but the doses of CDDO-Im that augmented ROS production also increased apoptosis in a dose dependent manner, with higher levels in the CA1a cells than in the AT1 cells.

Higher concentrations of the triterpenoid CDDO-Im alone increase the production of ROS in MCF10 cells. MCF10 AT-1 and CA1a cells were treated with CDDO-Im for 4 hrs, incubated with H2DCFDA for 1 hr, and ROS were detected by flow cytometry. The results represent 4 independent experiments.
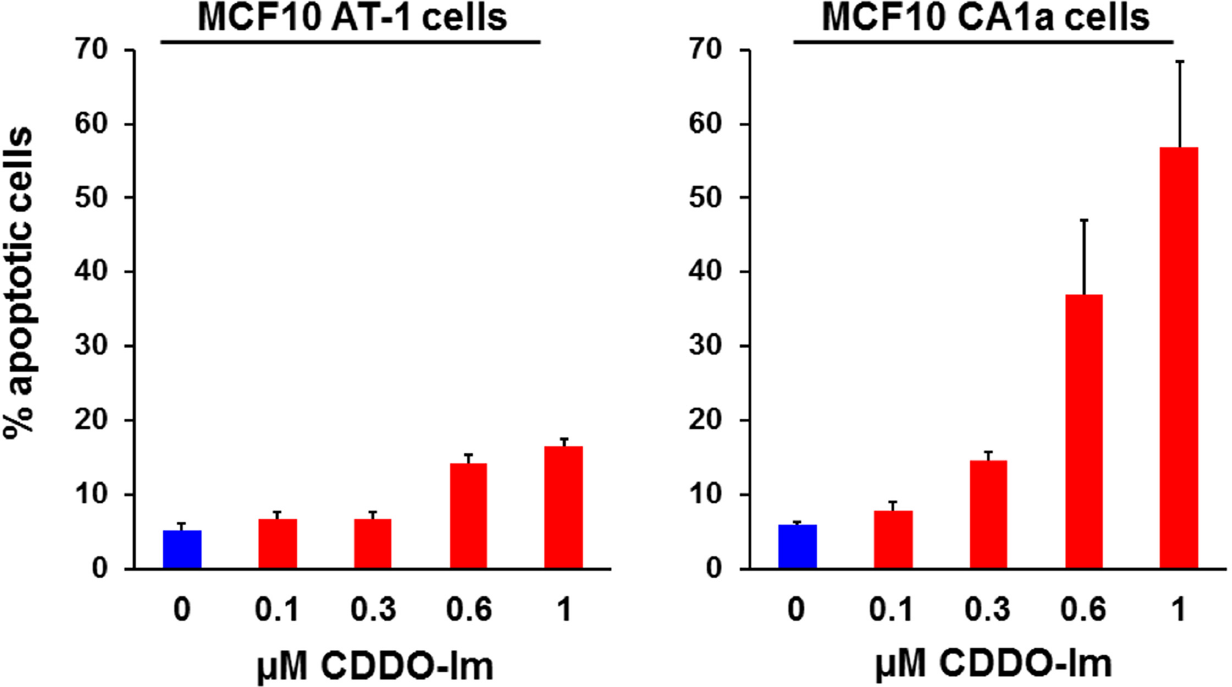
The same concentrations of CDDO-Im that increase ROS also induce apoptosis in malignant MCF10 CA1a cells. MCF10 CA1a cells were treated with various concentrations of CDDO-Im for 6 hrs, and annexin V and propidium iodide staining were detected by flow cytometry. The results represent 2 independent experiments.
Nrf2 knockout cells are more sensitive to treatment with CDDO-Im than wild-type cells
Because the Nrf2 pathway regulates a number of anti-oxidative genes, inactivating Nrf2 should make cells more responsive to agents that induce ROS. As shown in Fig. 6A, treatment with 250 μM of the oxidant tBHP for 15 min induced higher levels of ROS in Nrf2−/− knockout (KO) cells than in wild-type+/+ (WT) cells. Treatment with CDDO-Im alone for 4 hrs also enhanced ROS production, with markedly higher levels in the Nrf2 KO cells than in the WT cells. Pretreating cells with 0.1 μM of CDDO-Im eliminated ROS induced by tBHP in WT cells but had no effect in the KO cells (Fig. 6B). This protective effect of the triterpenoid was lost in the WT cells if the concentration of CDDO-Im was increased to 0.3 μM, but the levels of ROS were consistently higher in the KO cells than in WT cells in all of the samples. As shown in Fig. 6C, the enhanced production of ROS in Nrf2 KO cells treated with the oxidant tBHP or the triterpenoid CDDO-Im is also evident in an apoptosis assay, as 0.6 μM CDDO-Im induced more cell death in the Nrf2 KO cells than in the WT cells.
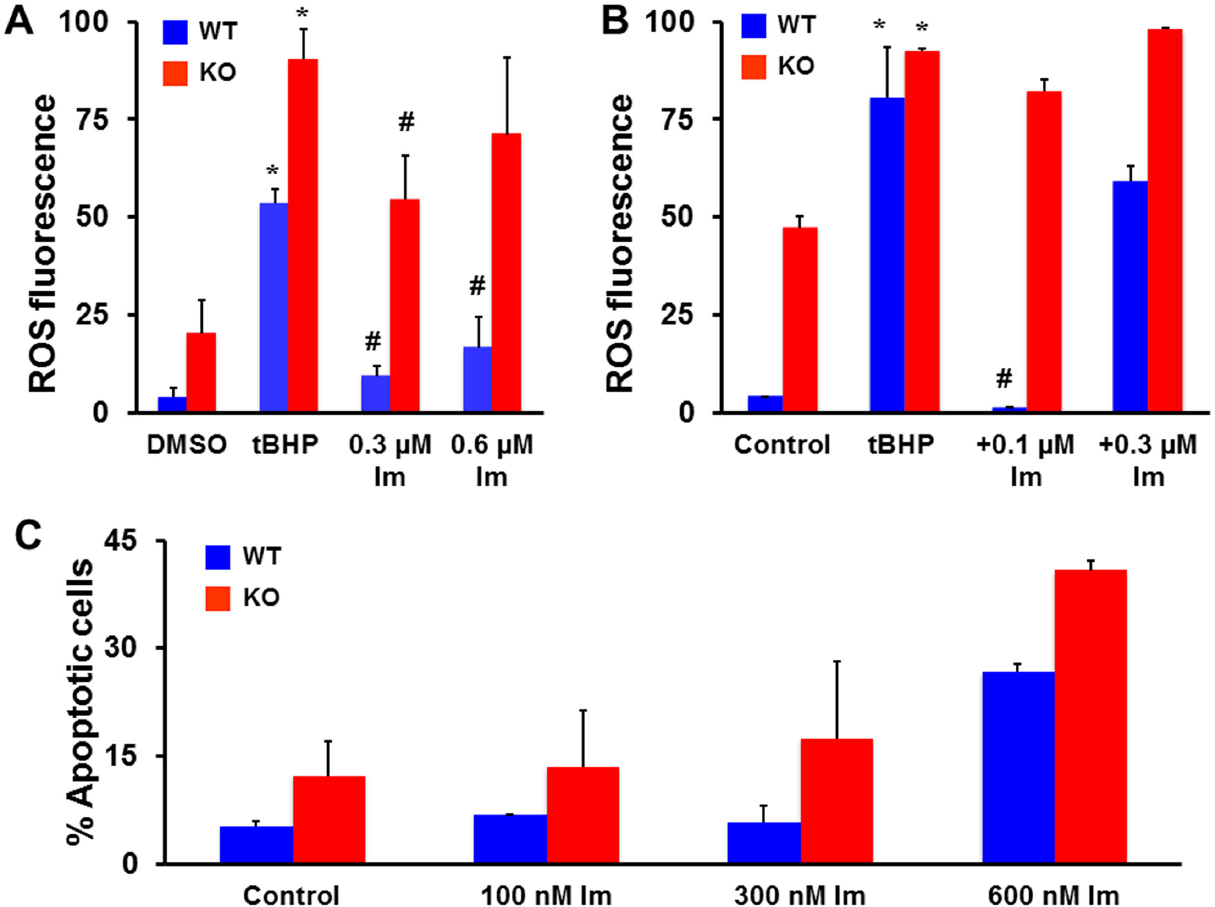
ROS production and apoptosis are higher in Nrf2 knockout fibroblasts than in wild-type cells treated with CDDO-Im. Nrf2−/− knockout (KO) and wildtype+/+ (WT) fibroblasts were treated with CDDO-Im or tBHP (250 μM) alone for 4 hrs (A) or were incubated with CDDO-Im for 15 hrs and then challenged with tBHP for 15 min (B) and ROS were detected by flow cytometry. Cells were also treated with CDDO-Im for 14 hr, and the percentage of apoptotic cells determined by annexin V and propidium iodide staining followed by flow cytometry (C). * P < 0.05 vs. control and # P < 0.05 vs. tBHP.
A/J mouse model of lung cancer
The MCF10 series of cell lines and Nrf2 wild-type and knockout fibroblasts are useful models for studying ROS production and apoptosis, especially with the correlation of genetic mutation and malignancy in the MCF10 cells. However, the need to use tumor xenografts in nude mice, which lack an appropriate tumor microenvironment and functioning immune system, make this model less appropriate for in vivo studies. Instead, we have used the highly relevant A/J mouse model (Liby et al. 2007). Strain A mice can develop spontaneous lung tumors after a long latency, and a variety of carcinogens, including several found in cigarette smoke, have been used to induce adenomas in these mice. When vinyl carbamate is used as a carcinogen, highly reproducible invasive adenocarcinomas develop with a shorter latency and at a higher frequency than with other carcinogens. Vinyl carbamate is activated to an epoxide that forms DNA adducts and subsequent mutations in K-ras (You et al. 1989), which are found at high frequency in lung and other cancers (Pylayeva-Gupta et al. 2011; Prior et al. 2012), and are similar to the H-ras mutations found in the MCF10 AT1 cells.
We have previously shown that triterpenoids are effective for both prevention and treatment of experimental lung cancer (Liby et al. 2007; Liby et al. 2009). However, genetic analysis of lung cancers and other types of tumors has raised concerns that either mutations that cause constitutive activation of the Nrf2 cytoprotective pathway or drugs that induce this pathway may interfere with chemotherapy and cause drug resistance (Kensler and Wakabayashi 2010; Sporn and Liby 2012). Two standard chemotherapeutic agents used to treat lung cancer are carboplatin, which crosslinks to DNA and interferes with DNA replication, and paclitaxel, which targets microtubules by inhibiting depolymerization. Based on the differential effects observed in the various MCF10 cell lines, we hypothesized that the triterpenoids could simultaneously enhance the efficacy of treatment with carboplatin and paclitaxel and reduce the toxicity of these chemotherapeutic agents.
Triterpenoids activate the Nrf2 pathway in vivo and reduce the toxicity of carboplatin and paclitaxel treatments
Because the effects of the triterpenoids on the Nrf2 cytoprotective pathway at doses used for treatment of established tumors (Liby et al. 2009) were not known, A/J mice were fed CDDO-Me at 80 mg/kg diet and CDDO-Ea at 800 mg/kg diet for one week. Although CDDO-Im is one of the most potent and active triterpenoids for in vitro studies, its relative instability and poor pharmacokinetics makes it more challenging to use for long-term animal studies so CDDO-Me were used in these studies. For many chemotherapeutic agents, including C/P, myelosuppression is the dose-limiting toxicity so peripheral blood mononuclear cells (PBMC) were harvested from the mice and pooled. As shown in Fig. 7A, both triterpenoids upregulated expression of NQO1 mRNA in PBMCs by 9–14 fold; NQO1 is a prototypical Nrf2 target widely used to screen for potential cytoprotective and chemopreventive activity (Dinkova-Kostova et al. 2005; Baird and Dinkova-Kostova 2011). NQO1 enzyme activity was also markedly increased in both the liver and lung of the A/J mice (Fig. 7B).
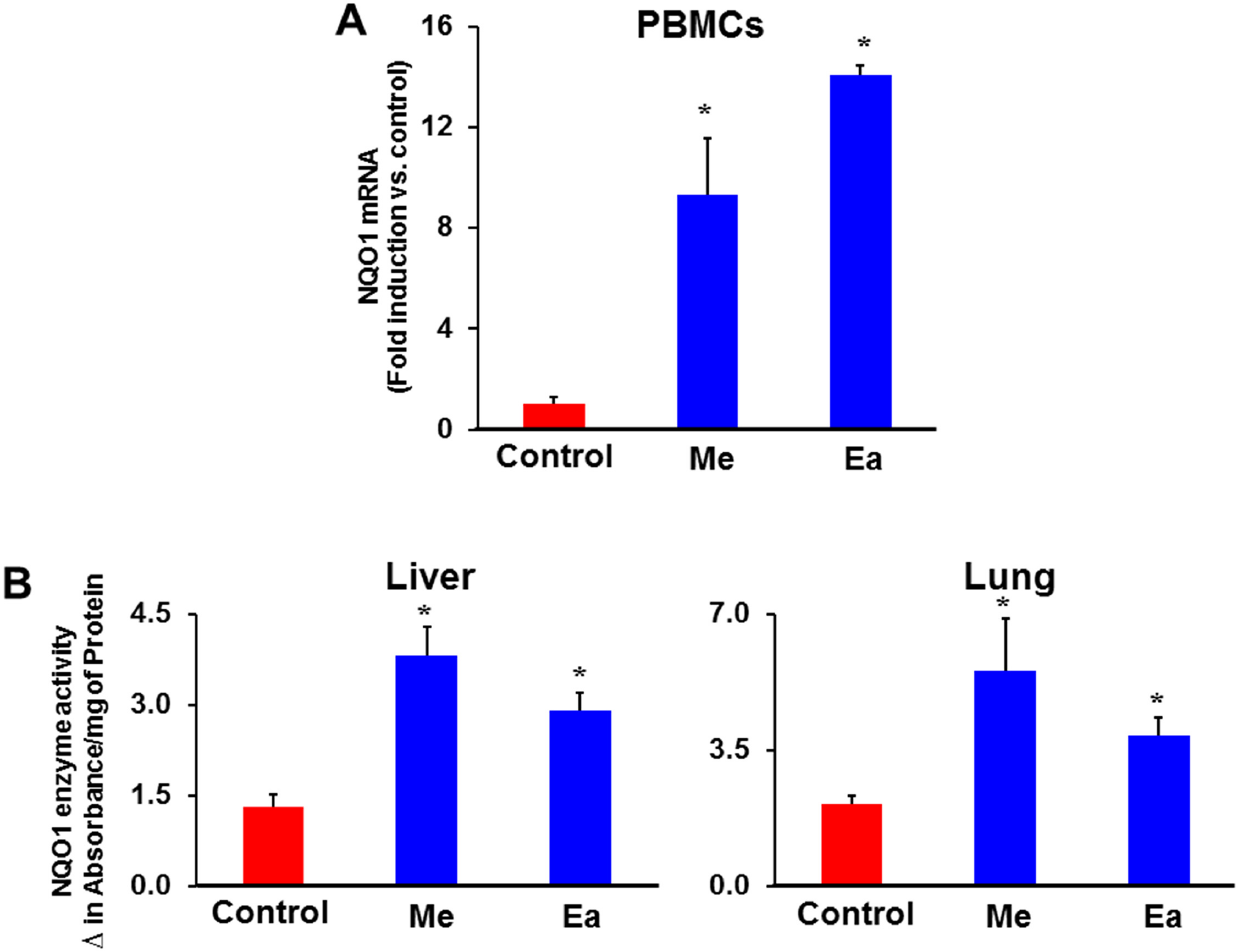
Triterpenoids induce NQO1 mRNA (A) and enzyme activity (B) in vivo. Twelve female A/J female mice were fed triterpenoid (CDDO-Me 80 mg/kg diet or CDDO-Ea 800 mg/kg diet) for 1 week. Induction of NQO1 mRNA was measured in peripheral blood mononuclear cells (PBMCs), and NQO1 enzyme activity was analyzed in livers and lungs. * P < 0.05 vs. control.
For a first pilot study with C/P, lung carcinogenesis was initiated with vinyl carbamate. Eight weeks after initiation, mice were randomized and fed triterpenoids mixed into powdered diet. By this time, the mice had developed lung adenocarcinomas (Liby et al. 2008), which was confirmed by the histopathology on sections from the lungs of the mice in this study. In addition to treatment with triterpenoids, the mice also received three weekly injections of carboplatin (C − 50 mg/kg) and paclitaxel (P − 15 mg/kg) and then two additional doses of C/P two weeks apart (Fig. 8A). This regimen proved too toxic, as only 38% (3 of 8) of mice injected with C/P survived this protocol. In contrast, 88% (14/16; P < 0.05 vs. C/P) of the mice on a triterpenoid diet and injected with C/P survived. In order to reduce the toxicity observed in the pilot experiment, the protocol was modified. Treatment with the triterpenoids was delayed until 12 weeks after injection with vinyl carbamate, and five C/P injections were given every other week (Fig. 8B). This altered regimen was considerably less toxic, as 91% (21 of 23) of the C/P group and 97% (31/32) of the triterpenoid groups survived.

Protocol for lung cancer studies. A. In a pilot study (A), A/J mice were injected with vinyl carbamate (VC). Eight weeks later, the mice were fed triterpenoids in diet. One week later, the mice were injected with 5 doses of carboplatin and paclitaxel (C/P), the first three doses once a week and the last two doses two weeks apart. In a second experiment (B), triterpenoid diets were not started until 12 weeks after initiation with VC, and the 5 C/P injections were given every two weeks.
Triterpenoids do not reduce the efficacy of treatment with carboplatin and paclitaxel on established lung tumors
When fed in diet for 12 weeks, the triterpenoid CDDO-Me alone significantly (P < 0.05) decreased the average tumor size and total tumor burden on lung sections. As shown in Table 1, the average tumor burden (ATB) was reduced by 75%, from 15.9 ± 2.7 mm3 in the control group to 3.9 ± 0.6 mm3 in mice fed CDDO-Me. Surprisingly, treatment with CDDO-Ea alone did not significantly alter the ATB in this study. In previous studies, the same dose of CDDO-Ea lowered ATB by 75% (Liby et al. 2009), but treatment was started 8 weeks after initiation and continued for 16 weeks. In contrast, treatment with the triterpenoids was delayed until 12 weeks after injection with vinyl carbamate and continued for 12 weeks in the current study. Because of the numerous mutations (Pleasance et al. 2010) and heterogeneity (Gerlinger et al. 2012) found in advanced cancers, it is easier to treat early lesions rather than end-stage disease (Sporn 2006). CDDO-Ea is also less potent than CDDO-Me in vitro and in vivo (Liby and Sporn 2012), and the lack of response to this single agent when treatment was started later and for a shorter period is additional evidence to support the benefits of early intervention in carcinogenesis.
Treatment with a triterpenoid and carboplatin/paclitaxel inhibits lung carcinogenesis in A/J mice.

Female A/J mice were injected i.p. with vinyl carbamate (0.32 mg/mouse). Twelve weeks after injection with the carcinogen, mice were fed triterpenoids in diet for 12 weeks and half of the groups also were injected with six doses of carboplatin (50 mg/kg ip) and paclitaxel (15 g/kg ip) every other week. Values are mean ± SEM.
P < 0.05 vs. control
P < 0.05 vs CDDO-Me alone
P < 0.05 vs. CDDO-EA alone
P < 0.05 vs. triterpenoid and C/P alone.
The use of the standard chemotherapeutic agents carboplatin and paclitaxel (C/P) for treatment of established lung tumors significantly (P < 0.05) reduced both the number and size of tumors, and the ATB in the C/P group (3.3 ± 0.5 mm3) was only 21% of the control group (15.9 ± 2.7 mm3). Notably, the average number and size of the tumors was lower with the combination of a triterpenoid and C/P in comparison to all individual treatments, and the ATB declined by 90% for the two combination groups (1.4 ± 0.4 mm3) compared to the control group (15.9 ± 2.7 mm3; P < 0.05 vs. control, triterpenoid alone, and C/P alone). Even though CDDO-Ea was ineffective as a single agent in this study, it did not interfere with the activity of C/P alone, and indeed the combination of CDDO-Ea and C/P was as effective as CDDO-Me and C/P. Moreover, the combination of either triterpenoid with C/P improved the histopathology of the tumors. As described previously (Liby et al. 2007; Liby et al. 2009), tumors are classified based on objective criteria and in a blinded fashion as low, medium, or high grade adenocarcinomas. By the end of the study at 24 weeks after initiation, 70% of the control tumors were high grade adenocarcinomas, 29% were medium grade, and only 1% were low grade tumors. CDDO-Me alone reduced the severity of these tumors as only 45% were high grade and 55% were medium grade (P < 0.05 vs. control), and similar effects were observed with C/P as 43% of tumors were high grade and 51% were medium grade in this group. In groups treated with the combination of a triterpenoid and C/P, however, only 33–36% of the tumors were high grade, and 58–60% were medium grade (P < 0.05 vs control).
In conclusion, these studies show that high levels of triterpenoids induce ROS and apoptosis of MCF10 CA1a malignant breast cancer cells. Although other studies also describe the induction of ROS by low micromolar concentrations of triterpenoids (Ikeda et al. 2003; Ikeda et al. 2004; Brookes et al. 2007; Kim et al. 2011), the current studies are the first to use a series of cells on a common genetic background but with variable degrees of malignancy to confirm that the sensitivity of cells to the induction of ROS and apoptosis in response to treatment with the triterpenoids increases with malignancy. Because low nanmolar concentrations of triterpenoids activate the Nrf2 cytoprotective pathway and thus reduce ROS (Dinkova-Kostova et al. 2005; Liby et al. 2005), it is possible that the triterpenoids could cause drug resistance to chemotherapy in tumors (Hayes and McMahon 2009; Kensler and Wakabayashi 2010). However, the triterpenoid CDDO-Me alone, at concentrations that activate the Nrf2 pathway in vivo, reduces the average tumor burden by 75% in an animal model of lung cancer. Furthermore, CDDO-Me enhances the efficacy of standard of care chemotherapy with carboplatin and paclitaxel so that the tumor burden in this combination group is only 10% of the tumor burden found in the lungs of the control group. Understanding the dose response of the triterpenoids will aid in the optimization of their use in the clinic, but their ability to alter redox balance in cancer cells without harming normal tissue may provide an attractive strategy for treating advanced malignancies (Trachootham et al. 2009).
Footnotes
ACKNOWLEDGEMENTS
The synthetic triterpenoids were originally developed by Michael B. Sporn in collaboration with Tadashi Honda and Gordon W. Gribble. The expert technical assistance of Renee Risingsong, Ryan Collins, Darlene Royce, and Charlotte R. Williams for these studies is greatly appreciated. This work was supported by the NIH (R01 CA78814) and Reata Pharmaceuticals, Inc.
Patents issued for synthetic triterpenoids and unrestricted research grant from Reata Pharmaceuticals, Inc.