
Abstract
The peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptors that function as transcription factors regulating the expression of genes involved in cellular differentiation, development, metabolism and also tumorigenesis. Three PPAR isotypes (α, β/δ and γ) have been identified, among which PPARβ/δ (PPARD) is the most difficult to functionally examine due to its tissue-specific diversity in cell fate determination, energy metabolism and housekeeping activities. PPARβ/δ acts both in a ligand-dependent and -independent manner. The specific type of regulation, activation or repression, is determined by many factors, among which the type of ligand, the presence/absence of PPARβ/δ-interacting corepressor or coactivator complexes and PPARβ/δ protein post-translational modifications play major roles. Recently, new global approaches to the study of nuclear receptors have made it possible to evaluate their molecular activity in a more systemic fashion, rather than deeply digging into a single pathway/function. This systemic approach is ideally suited for studying PPARβ/δ, due to its ubiquitous expression in various organs and its overlapping and tissue-specific transcriptomic signatures. The aim of the present review is to present in detail the diversity of PPARβ/δ function, focusing on the different information gained at the systemic level, and describing the global and unbiased approaches that combine a systems view with molecular understanding.
Introduction
Systems biology aims at deciphering complex biological units by aggregating biological information of various natures (gene, RNA, protein) in order to gain the most comprehensive view of the events that shape life. Another way of saying this is that systems biology aims to understand how a biological system, made up of multiple interacting linear or non-linear pathways, behaves, and explains the global pattern observable at the level of the organism. The present review compiles a diversity of details regarding the activity of peroxisome proliferator-activated receptor β/δ (PPARβ/δ), viewed at the system level.
Several qualities make PPARβ/δ an interesting molecule to explore using system approaches: its ubiquitous expression, the rather broad and poorly specific nature of its ligands (mainly unsaturated free fatty acids) and the fundamental functions that PPARβ/δ controls, such as energy metabolism or cell survival. Herein, we will start with a chapter concerning the gene, its structure and evolutionary history, and the main classical properties of PPARβ/δ as a transcription factor. We will then proceed with the systemic and integrative views that mouse models carrying alterations of PPARβ/δ activities provide. The third part will describe the global and unbiased approaches that bridge a system view and molecular mechanisms. These approaches encompass microarray and genome-wide Chromatin Immunoprecipitation (ChIP). This review will end with the description of polymorphisms in the human PPARD gene that are associated with various pathologies.
Molecular properties of PPARβ/δ
PPARβ/δ (PPARD) gene
Shortly after the characterization of a new nuclear receptor (NR) involved in the peroxisome proliferation response to some chemicals in mice, which was accordingly referred to as peroxisome-proliferator activated receptor (PPAR) [Issemann and Green, 1990], three PPAR genes, α (PPARA), β (PPARD) and γ (PPARG), were identified in Xenopus laevi [Krey et al., 1993]. When characterized in mouse, rat or human, the mammalian PPARα and PPARγ genes were easily identified, while the third gene was less clearly homologous to the Xenopus PPARβ and was alternatively called NUC-1 in human [Schmidt et al., 1992], fatty acid activated receptor (FAAR) [Amri et al., 1995] in mice, and lastly PPARδ [Evans et al., 2004]. It now appears clear that they are indeed homologous to the Xenopus PPARβ [Germain et al.,2006; Laudet, 1997]. These laborious identification efforts explain the different names for PPARβ/δ in the literature.
PPARs belong to a subfamily of the NR superfamily, together with the thyroid hormone receptors, retinoic acid (RA), vitamin D, ecdysone, and the orphan receptors Rev-ErbAα (5ear1;NR1D1) and E75 (NR1D3, from Drosophila). The latter two represent the closest relatives of the PPARs [Laudet et al., 1992]. The ancestral genes in this subfamily appeared more than 500 million years ago [Knoll, 1992], followed by further duplication. The ancestral thyroid hormone receptor (TR) gene duplicated into two genes, TRα (NR1A1) and TRβ (NR1A2), and the ancestral retinoic acid receptor (RAR) gene duplicated into three genes, RARα (NR1B1), RARβ (NR1B2), and RARγ (NR1B3). Similarly, the three PPAR loci, α, β, and γ, appeared during this second period [Laudet et al., 1992], with the duplication events likely being contemporaneous with the appearance of the early vertebrates [Keese and Gibbs, 1992]. Interestingly, PPAR genes have evolved 2–3 times faster than the RAR and TR genes according to the amino acid sequence differences observed between the Xenopus and mammalian homologs. Among the PPAR subtypes, PPARβ/δ exhibits an even higher rate of evolution. This relatively rapid evolution emphasizes the need for careful evaluation when studying PPAR activities in various species. PPARD has been assigned to chromosome 6, at position 6p21.1–p21.2 in human [Yoshikawa et al., 1996], and Ppard has been assigned to chromosome 17 in mouse. The six exons in the 3’ part encode the full PPARβ/δ protein.
PPARβ/δ protein structure
Like most NRs, the PPAR protein structure consists of four main domains: the unstructured N-terminal A/B-domain, the C-domain folded in two zinc fingers which corresponds to the DNA-binding domain (DBD), the D-domain or hinge region, and finally the E-domain forming a bundle of helices and stranded beta-sheet, which accommodates a ligand binding pocket. While the A/B-and D-domains are only poorly conserved between the PPAR isotypes, the C- and E-domains share a high degree of sequence and structural homology (reviewed in [Escher and Wahli, 2000]).
Whether and by which mechanism the N-terminal activator domain (AF1) of PPARs may regulate transcriptional activity remains debated. This domain appears to be a determinant for PPAR subtype-specific activity [Bugge et al.,2009; Castillo et al.,1999; Hummasti and Tontonoz, 2006], also restricting the number of genes transcriptionally regulated by each of the PPAR subtypes [Hummasti and Tontonoz, 2006].
The ligand binding domain (LBD) in all three PPARs is a very large Y-shaped cavity (about 1400 cubic angstroms) as compared to other NRs, and this characteristic allows the PPARs to interact with numerous structurally-distinct ligands [Nolte et al., 1998]. However, a novel aspect of the PPARβ/δ pocket is the narrowness of one of the Y arms, which cannot accommodate bulky polar heads [Xu et al.,2001; Zoete et al., 2007] such as Thiazolidinediones (TZDs) and L-tyrosine-based agonists. When the human PPARβ/δ LBD was first crystallized in absence of ligand, the crystals indicated the presence of vaccenic acid, of bacterial origin, in the ligand pocket [Fyffe et al., 2006]. Crystallization in the presence of the potent and selective PPARβ/δ ligand GW0742 was later performed [Batista et al., 2012] and both studies suggest that two residues in the hormone binding pocket (Val312 and Ile328) are important for ligand-selective binding to PPARβ/δ. Finally, in the true absence of ligand, the structure of PPARβ/δ seems to correspond to an equilibrium of different conformations, among which are those favoring coregulator recruitment.
PPARs bind to DNA as obligate heterodimers with members of the retinoid X receptor (RXR) subfamily of NRs. The PPAR:RXR complex behaves as a permissive heterodimer since it can regulate gene expression upon activation by either RXR or PPAR ligands. DNA binding properties of PPARβ/δ are similar to those of PPARα and PPARγ, with a common consensus sequence (PPAR-responsive element; PPRE), which consists of a direct repeat of a close derivative of the AGGTCA consensus hexamer, separated by one base-pair, the PPAR moiety occupying the 5’ half-site [IJpenberg et al., 1997]. The only difference that contributes to the strength and specificity of PPAR isotype binding lies in the 5’ flanking region, though it seems mainly important for PPARα and not for PPARβ/δ [Juge-Aubry et al., 1997].
Patterns of PPARβ/δ expression
PPARβ/δ gene expression is truly ubiquitous [Braissant et al.,1996; Kliewer et al., 1994], suggesting the importance of this receptor in both systemic activity and/or for basic cellular functions. Different methods such as in situ hybridization, qPCR and tissue microarray-based immunochemistry have been used to provide a large survey of PPARβ/δ-expressing cells in the body, the most exhaustive one being that of Higashiyama et al. [Higashiyama et al., 2007]. This study confirmed the wide distribution of PPARβ/δ protein in mouse tissues. A brief summary with key references is given below.
PPARβ/δ is expressed in organs/cells highly associated with fatty acid catabolism such as hepatocytes in the liver [Sanderson et al.,2010; Sanderson et al.,2009; Shan et al., 2008], adipocytes in the brown and white adipose tissue (BAT and WAT) [Leibowitz et al.,2000; Mottillo et al.,2012; Pan et al.,2009; Reilly and Lee,2008; Roberts et al.,2011; Wang et al., 2003] and skeletal muscle cells [Giordano et al., 2009].
PPARβ/δ is also widely observed in the nucleus of epithelial lineage cells from keratinocytes [Kim et al.,2006; Schmuth et al.,2004; Tan et al.,2001; Westergaard et al., 2001] to enterocytes [Girroir et al.,2008; Gupta et al.,2000; Gupta et al.,2004; He et al.,1999; Park et al., 2001]. In the nervous system, PPARβ/δ is found in both axons and dendrites of neurons residing in different brain areas and in microglia cells [Higashiyama et al.,2007; Xiao et al., 2010] of the central nervous system, as well as in astrocytes [Chistyakov et al., 2014] and in the neurofibers of the peripheral nerves and spinal cord [Jana et al.,2012; Peters et al., 2000].
In the immune system, PPARβ/δ expression has been particularly characterized in macrophages [Bouhlel et al.,2009; Chawla,2010; Desvergne,2008; Lee et al., 2003]. In the cardiovascular system, PPARβ/δ immunostaining is present in the nucleus of cardiomyocytes and vascular smooth muscle cells in the aorta [Cheng et al.,2004a; Cheng et al.,2004b; Finck, 2007] and other vascular districts [Zhang et al., 2002]. In the endocrine system, PPARβ/δ is seen in the delta cells of the Langerhans islet and in the secretory cells of the adrenal cortex [Lee et al.,2006; Takahashi et al., 2006]. In the reproductive organs, the nucleus of both spermatid/spermatocytes in the testis and follicular epithelial cells in the ovary are positively stained for PPARβ/δ [Higashiyama et al., 2007]. Finally, PPARβ/δ has been also found in cartilage and bone compartment [Scholtysek et al., 2013].
However, there are some divergent aspects of studies PPARβ/δ expression patterns that cannot be listed herein. These relate to experimental conditions, mouse strain and environment at the time of measurement, as PPARβ/δ expression level is dictated by both exogenous and endogenous signals. The use of different approaches (in situ hybridization, qPCR for RNA levels, various antibodies and methods for protein levels) to evaluate PPARβ/δ expression also likely contributes to some of the divergencies found in the literature. Of particular note, questions remain regarding how truly specific commercial antibodies are, suggesting that off-target activity may contribute to false positive signals. Despite these difficulties, the ubiquitous expression of PPARβ/δ - with the basal highest expression in gastrointestinal tract and skeletal muscle [Braissant et al., 1996] - likely reflects its role in multiple biological functions.
Transcriptional activity of PPARβ/δ
The classical view of the prototypic activity of PPARβ/δ, as for other NRs, is to activate transcription in a ligand-dependent manner, via binding to specific response element in the promoter of target genes and recruitment of coactivator complexes. The somewhat dogmatic concept is that agonist ligands induce a conformational change in PPARβ/δ:RXR that favors the dissociation of corepressors and the association with coactivators [Yu and Reddy, 2007]. A large number of coactivator complexes interacting with transcription factors have been identified and it is hypothesized that combinatorial usage of these complexes provides the basis for cell type-specific, gene-specific, and signal-specific transcriptional responses. Where and with which complexes active PPARβ/δ interacts is, however, not yet clear [Khozoie et al.,2012; Ricote and Glass, 2007].
One intriguing question concerns ligand-dependent repression of gene expression, particularly with respect to PPARβ/δ, as it may represent an important activity of this receptor. The best-documented mechanism was shown in macrophages where, in absence of ligand stimulation, PPARβ/δ sequesters the transcriptional repressor B-cell lymphoma 6 protein (BCL6). Upon ligand stimulation, PPARβ/δ releases BCL6, thereby triggering BCL6-dependent repression of NF-kB target genes such as Monocyte chemotactic protein 1 (MCP1) [Lee et al., 2003]. Other mechanisms are likely involved, depending on cellular context, but still remain to be elucidated.
In absence of ligand, PPARβ/δ may also exert transcriptional repression. This was proposed to explain the regulation of a set of genes whose expression increased upon deletion of PPARβ/δ [Adhikary et al.,2011; Khozoie et al., 2012]. Previous studies had shown that by occupying PPREs while interacting in a ligand-independent manner with silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) and Histone deacetylase 1 HDAC1, PPARβ/δ could down-regulate PPARα-and PPARγ-dependent transactivation [Shi et al., 2002]. Corepressor interactions with PPARβ/δ are indeed particularly strong and might even impair its classical ligand-dependent positive transcriptional activity when either Nuclear receptor corepressor (NCoR) or SMRT is overexpressed [Krogsdam et al., 2002], thereby contributing to the complexity of the system. Since these studies, little if any progress has been made with regard to assessing the mechanism by which PPARβ/δ could act as a bona fide repressor.
In addition to LBD conformational changes upon ligand binding, there are some important post-translational modifications that may also alter the activity of PPARβ/δ in the absence/presence of ligand. Phosphorylation is one of the most common modifications and several consensus phosphorylation sites can be mapped along the PPARβ/δ sequence. Along these lines, both cAMP and PKA increased the basal and ligand-dependent activity of PPARβ/δ [Burns and Vanden Heuvel, 2007], while Wortmannin, an inhibitor of the PI3K, could inhibit the positive transcriptional activity of PPARβ/δ on prostaglandin E2 receptor EP4 gene expression [Han et al., 2005]. However, at the molecular level, the phosphorylation events of the PPARβ/δ protein and their functional consequences remain to be directly explored.
Ligand-induced proteolysis through the ubiquitin-proteasome system is a common mechanism to end transcriptional activity promoted by the ligand-activated receptors. However, this mechanism seems to be reversed for PPARβ/δ, as in the absence of ligand, PPARβ/δ has a very short half-life due to efficient ubiquitination and proteosomal degradation. Ligand binding inhibits this ubiquitination thereby increasing its half-life [Genini and Catapano,2007; Rieck et al.,2007; Wadosky and Willis, 2012], albeit this phenomenon may depend on the level of PPARβ/δ expression at the time of ligand stimulation [Rieck et al., 2007]. Interestingly, ubiquitin-C is itself a target gene of PPARβ/δ [Kim et al., 2004]. Finally, even though PPARβ/δ presents a potential Small Ubiquitin-like Modifier (SUMO)ylation site in its D region (K185) [Wadosky and Willis, 2012], no experimental data to date has confirmed a role for SUMOylation in regulating PPARβ/δ activity.
PPARβ/δ ligands
Natural ligands
As for PPARα and PPARγ, many unsaturated fatty acids can bind to PPARβ/δ, in a pattern closely resembling that of binding to PPARα [Desvergne and Wahli, 1999]. One proposed model of fatty acids delivery to PPARβ/δ is via the very large density lipoprotein (VLDL), through release of their triglycerides (TG), as shown in macrophages [Chawla et al.,2003; Ziouzenkova and Plutzky, 2004].
However, it is still unresolved which particular fatty acids act as endogenous ligand specific for PPARβ/δ. Arachidonic acid derivatives were the first to be pointed at, starting from those obtained upon cyclooxygenase 2 (COX2) activation, such as prostacyclin (PGI2) and other prostaglandins [Yu et al., 1995], but also metabolite derivatives obtained through the 12/15 lipoxygenase activity, such as 9-HODE,13-HODE, 12-HETE and 15-HETE. While they are known as low affinity PPARγ activators [Nagy et al., 1998], these metabolites activate PPARβ/δ with some intriguing results since 13-s HODE is reported to be an inhibitor of PPARβ/δ in colon epithelial cells [Shureiqi et al., 2003], but an agonist in pre-adipocytes [Coleman et al., 2007]. Such observations point to the concept of tissue-specific response, due for example to presence or absence of other coregulators. Along these lines, a report suggested that retinoic acid (RA) could be a ligand for either PPARβ/δ or RARs, depending on the relative expression of CRABPII (delivering RA to RARs) and FABP5 (delivering RA to PPARβ/δ) [Schug et al., 2007]. Further studies from the same group highlighted some of the biological outcomes of this crosstalk between RA and PPARβ/δ [Schug et al., 2008], albeit discrepancies concerning the effectiveness of RA stimulation on PPARβ/δ have been reported [Rieck et al., 2008].
Exogenous fatty acids are also studied, such as ombuin-3-O-beta-D-glucopyranoside (ombuine), a flavonoid from Gynostemma pentaphyllum, activating both PPARβ/δ and PPARα [Malek et al., 2013]. This example underscores the fact that most of the natural ligands known to interact with PPARβ/δ also interact with the other PPAR subtypes. It is an interesting feature when searching for dual agonists, albeit it may also lead to off-target effects.
Synthetic ligands
Tremendous effort has been devoted towards optimizing synthetic ligand binding selectivity to PPARβ/δ, thereby avoiding off-target effects. Recently a high-throughput screening (HTS) assay [Xia et al., 2013] has been established in order to select PPAR agonists with low toxicity and high efficiency. Below are a few, and by no means exhaustive, illustrations of synthetic PPARβ/δ agonists and antagonists that have been used or are presently being tested in in vivo studies.
Agonists
The very first synthetic compound, called L165041 [Berger et al., 1999], was an established leukotriene antagonist, which activates human PPARβ/δ, but also triggers PPARγ activation at high doses [Forman et al., 1997]. The compound GW501516 [Sznaidman et al., 2003], which was more potent and more specific, subsequently took over in most published experimental work and became the reference compound as a PPARβ/δ agonist [Pelton, 2006]. However, while being investigated for its potential activity in metabolic disorders, as reviewed in Lamers et al. [Lamers et al., 2012], its uncontrolled use in human as a doping substance and subsequent high risks of abuse contributed to stop all further development of the molecule. Today, GW0742, which was developed together with GW501516, is a highly selective PPARβ/δ agonist that is commercially available for non-human research purposes [Sznaidman et al., 2003]. The most recent agonists developed to target PPARβ/δ in clinical settings were MBX-8025/RWJ800025 (MBX-8025 presently in phase II trials, generated by Metabolex) [Billin, 2008] and KD-3010 developed by Kalypsys [Iwaisako et al., 2012].
Antagonists and Inverse Agonists
The irreversible PPARγ antagonist GW9662 [Leesnitzer et al., 2002] was initially used since it also exerted a potent PPARβ/δ antagonistic activity [Seimandi et al., 2005]. The first selective PPARβ/δ antagonist was GSK0660, which was identified and characterized in 2008 [Shearer et al., 2008], although its poor bioavailability has impaired its use in in vivo studies. The same was true for SR13904, which in addition to PPARβ/δ also antagonizes PPARγ transactivation, albeit with much weaker potency [Zaveri et al., 2009]. Finally, GSK3787 is a potent PPARβ/δ antagonist with good pharmacokinetic properties. It has a good bioavailability and can be used in animal studies [Palkar et al.,2010; Shearer et al., 2010]. However, this compound is also an irreversible antagonist, which covalently binds PPARβ/δ and it is not as selective as hoped. These antagonists can also be referred to as inverse agonists, as they bind PPARβ/δ as an agonist, but induce an opposite pharmacological response, decreasing the basal expression level of PPARβ/δ target genes [Shearer et al., 2008] and increasing the recruitment of corepressors [Palkar et al., 2010]. More recently two novel compounds have been described in the class of PPARβ/δ antagonists: DG172 and PT-S58. DG172 exhibits high binding affinity and potent inverse agonistic properties, enhancing transcriptional corepressor recruitment and down-regulating transcription of PPARβ/δ target genes [Lieber et al., 2012]. Moreover, DG172 is bioavailable after oral treatment in mice. PT-S58, a cell-permeable diaryl-sulfonamide, acts as a pure competitive PPARβ/δ subtype-specific antagonist targeting the ligand binding site of PPARβ/δ while not allowing coregulator interactions [Levi et al.,2013; Naruhn et al., 2011].
Altogether, these molecules represent important tools to study the systemic impact of PPARβ/δ activities in animal models, together with the generation of genetically-modified mouse models.
Integrative physiology of PPARβ/δ
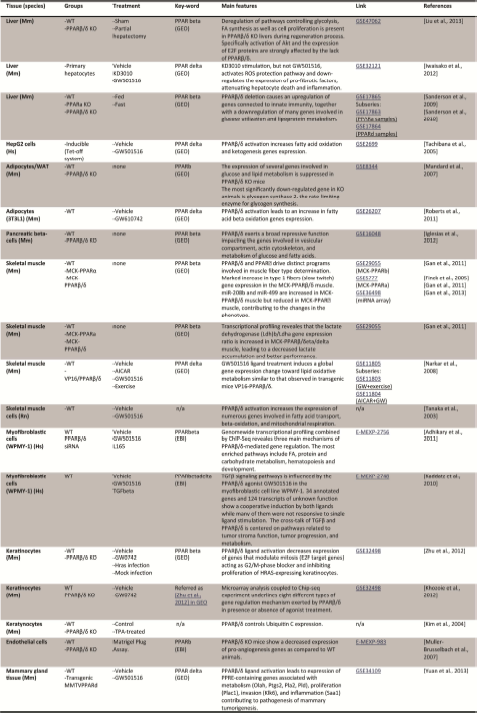
In the context of this review on PPARβ/δ and systems biology, the present section focuses on knowledge gained with a systemic view from animal models, either through gene modification or through animal treatment with agonist or antagonist ligands. Although these reported studies have deeply contributed in shaping our present understanding of PPARβ/δ, they also present several caveats. It indeed remains difficult when using transgenesis approaches (total body or tissue-specific) activating or deleting PPARβ/δ, to disentangle the truly specific effects from the secondary and compensatory effects. The use of constitutively active PPARβ/δ via its fusion with trans-activator domains, such as VP16, can alter the secondary/tertiary structure of PPARβ/δ, leading to different interaction surfaces and in turn diverse transactivation functions. Agonist and antagonist compounds are also not exempt from off-target activities. These few examples underline the need to cautiously interpret observations made in such models.
Generation of PPARβ/δ null mice
Four models of PPARβ/δ total body Knock Out (KO) have been reported. In the first published model [Peters et al., 2000], Ppard was disrupted via insertion of phosphoribosyltransferase II expression cassette in the last exon, encoding the ligand binding domain. Mice for this modification presented significantly lower survival rate in mixed background but this phenotype was back to normal in C57BL/6N background, in striking contrast with the three other models, described below. It must be noted that the strategy of the gene alteration has likely driven the generation of a hypomorph allele rather than a null allele, since theoretically, PPARβ/δ obtained from this gene could be expressed, albeit truncated of its C-terminus 60 amino-acids, but left with intact N terminus and DNA binding domains. Another strategy consisted of framing the exon 4 encoding the first moiety of the DNA-binding domain of PPARβ/δ with two LoxP sites, allowing the generation of either a conditional knockout allele, or a constitutive deletion of the exon 4, with a subsequent frame shift of the encoding sequence [Barak et al., 2002]. Using the latter, a PPARβ/δ null mouse model was generated, although this gene alteration resulted in severe placental defects and frequent mid-gestation lethality. Notable changes in the structure of the placenta of PPARβ/δ KO mice include reduced size of the entire tissue with loosening of its attachment to decidua and hemorrhages at various locations due to flooding of maternal blood. Replacing exon 4 with the β-galactosidase gene resulted in placental defects and lethality similar to those previously reported [Barak et al., 2008]. Using a similar strategy, Nadra et al. [Nadra et al., 2006] replaced exon 4 and part of exon 5 with a PGK-neo cassette deleting the two zinc fingers of the DNA-binding domain. Here again, deletion of PPARβ/δ severely affected placental development, leading to embryonic lethality at embryonic day 9.5 (E9.5) to E10.5 of most, but not all, PPARβ/δ-null mutant embryos. The observed placental alterations mainly reside in the Giant cell layer. In fact, both molecular and cellular studies highlighted the role of PPARβ/δ in the differentiation pathway of these particular cells through activation of Akt signaling and inhibition of Id-2 [Nadra et al., 2006].
Further analyses demonstrated that the lethality observed is 100% in pure background (C57BL/6 and SV129, B. Yaacov and B. Desvergne personal communication, respectively), whereas it is partial on mixed backgrounds (C57Bl6/FVB or C57Bl6/SV129, Barak Yaacov and B. Desvergne personal communication, respectively). The viability and maintenance of PPARβ/δ null mice is achieved by the interbreeding of survivors on mixed genetic backgrounds. The fact that mixed background increases chances to obtain null mutant pups, in addition to a “founder” effect rather than a specific genetic background, underline the complexity of the genetic network in which PPARβ/δ is involved with respect to placenta development.
The development of the surviving mice was grossly normal and numerous studies could benefit from this model, as seen in the next section. In parallel, tissue-specific knockout mice and transgenic mice expressing normal, mutated or strongly-active forms of PPARβ/δ have been generated in order to study tissue-specific functions exerted by this receptor without interference from the PPARβ/δ systemic deletion (see Table 1).
PPARβ/δ mutant phenotypes.

PPARβ/δ as a potent regulator of metabolism and inflammation
Energy metabolism in muscle
While initially overshadowed by PPARα and PPARγ, interest in PPARβ/δ rose significantly when its role in muscle energy metabolism was identified. The first indications that PPARβ/δ might play a role in muscle metabolism came from the observations that PPARβ/δ expression in skeletal muscle is increased upon fasting [Holst et al., 2003] and upon exercise [Russell et al., 2005], suggesting a role for PPARβ/δ in the adaptive response of skeletal muscle to increased demand for fatty acid oxidation. Skeletal muscle indeed represents an important consumer of fatty acids. In a transgenic mouse model, the overexpression of PPARβ/δ in skeletal muscle provokes a shift towards more oxidative fibers and promotes a general decrease of body fat content [Luquet et al., 2003]. Similar results were obtained by expressing overactive PPARβ/δ fusion protein (PPARβ/δ-VP16) under the control of the alpha-actin promoter [Wang et al., 2004], which resulted in an average 2-fold increase in type I muscle fibers, and a subsequent increase in muscle oxidative capacity, leading to a remarkable increase in running distance and time on a treadmill [Wang et al., 2004]. Because of this phenotype, the latter model was also called the “marathon mice”.
Conversely, when PPARβ/δ is specifically deleted in skeletal muscle, using the CRE-recombinase system under the control of alpha-actin promoter, the muscle fibers exhibited lower oxidative activity, while the body fat mass increased and led to insulin resistance [Schuler et al., 2006]. Altogether, these observations suggest PPARβ/δ activation plays a role in skeletal muscle adaptation to physical stress, although the molecular mechanisms underlying this effect are not yet clearly understood.
To further study the PPAR-dependent energy substrate usage in muscle and to characterize it at the molecular level, Gan et al. [Gan et al., 2011] generated transgenic mouse lines expressing either PPARβ/δ or PPARα under the control of skeletal muscle-specific promoter (MCK-PPARβ/δ or MCK-PPARα constructs). Comparing these two mouse models, the authors demonstrated that PPARβ/δ interacts with the exercise-inducible kinase AMP-activated protein kinase (AMPK), which in turn promotes glucose uptake, fatty acid oxidation, mitochondrial biogenesis, and insulin sensitivity. In addition, PPARβ/δ-mediated activation of AMPK leads to a synergistic activation of lactate dehydrogenase b (Ldhb) gene transcription, increasing the ratio Ldhb/Ldha and diminishing the accumulation of lactate. The high glycogen stores and increased levels of GLUT4 in mouse muscles with MCK-driven PPARβ/δ expression further suggested a broad reprogramming of glucose utilization pathways mediated by PPARβ/δ [Gan et al., 2011]. In parallel, slow-twitch genes are increased in MCK-PPARβ/δ soleus muscle, via an indirect mechanism involving a complex NR/microRNA circuitry [Gan et al., 2013].
Altogether, these results clearly confirm that PPARβ/δ in skeletal muscle controls different phases of the adaptive response to training (i.e., the number of oxidative myofibers), and metabolic switch (i.e., the increase in fat burning capability), by reprogramming the expression of glucose utilization genes. We might also ask whether these observations could be of any use for improving structural muscular disorders. Miura et al., [Miura et al., 2009] found that GW501516 systemic treatment stimulated utrophin A mRNA levels in muscle cells derived from the Duchenne muscular dystrophy mdx mouse model. Over a 4-week trial period, the agonist treatment augmented the percentage of muscle fibers expressing slower myosin heavy chain isoforms and stimulated transcription of utrophin A and its expression in the sarcolemma. The mdx sarcolemma integrity was improved, together with a limitation of the eccentric contraction-induced damage of mdx skeletal muscles [Miura et al., 2009]. More generally, a better knowledge of the metabolic role of PPARβ/δ in skeletal muscle, where it contributes to enhanced muscle endurance, might be useful in designing therapeutic strategies for muscular degenerative diseases such as muscular dystrophy.
Adult cardiomyocytes express a high level of PPARβ/δ, and also utilize fatty acid as their main energy source. With respect to PPARβ/δ functional delineation in cardiac muscle, three different mouse models have been generated. The cre-loxP-mediated deletion of PPARβ/δ restricted to cardiomyocytes presented the most dramatic phenotype due to the loss of this receptor, with congestive heart failure and reduced mouse survival [Cheng et al.,2004a]. This was associated with down-regulation of the constitutive expression of key fatty acid oxidation genes, and a consequent decrease of basal myocardial fatty acid oxidation with lipid accumulation. In a second model, PPARβ/δ cardiac-specific deletion was induced at 2 months of age. This “adult” model underscored the overall alteration of energy substrate usage, with decreases of both fatty acid and glucose oxidation rates concomitant with a reduction in the expression of genes in both pathways [Wang et al., 2010]. It also suggests that the severity of the cardiac failure in the first model might be due to a role of PPARβ/δ in cardiac development. In the third model, mice overexpressed PPARβ/δ under the control of the alpha myosin heavy chain (αMyHC) promoter.
Mirroring the PPARβ/δ deletion phenotype, these mice presented increased myocardial glucose utilization, with upregulated expression of Glut4 and glycolytic genes, and were resistant to diet-induced lipid accumulation in cardiomyocytes and subsequent lipotoxic cardiomyopathy [Burkart et al., 2007]. More recently, a fourth mouse model overexpressing PPARβ/δ specifically in cardiac muscle has been generated. The constitutively active form of PPARβ/δ-VP16 was placed under the control of the promoter αMyHC for cardiac-muscle-restricted expression, and was constructed to be inducible by Tamoxifen administration. This report mainly describes how this methodology may be used to efficiently generate transgenic mouse models expressing a constitutively active form of PPARβ/δ upon Tamoxifen administration in a tissue-specific manner [Kim et al., 2013]. However, the overexpression of such constitutively activated PPARβ/δ cannot take into account subtle activities depending on expression levels and ligand availability, and this may lead to overestimates of the importance of certain regulatory activities.
PPARβ/δ in adipose tissue
The role of PPARβ/δ in fatty acid catabolism is also effective in white and brown adipose tissue (WAT and BAT, respectively). Transgenic mice expressing, in the WAT, a hyper-active form of PPARβ/δ (PPARβ/δ-VP16) are resistant to both high-fat diet-induced and genetically predisposed obesity, together with decreased lipid accumulation in adipose tissue and diminished lipidemia, in line with an increased fatty acid consumption [Wang et al., 2003]. The effect of PPARβ/δ over-activation was even more dramatic in metabolically-active BAT, with an increased expression of genes involved in fatty acid hydrolysis, oxidation, and uncoupling of oxidative phosphorylation [Wang et al., 2003]. This over-activation may, however, mask more subtle regulatory activity, such as the PPARβ/δ-mediated induction of Twist1, which acts as a negative regulator of energy dissipation, in part via a decrease of UCP1 [Pan et al., 2009].
Lessons learned from PPARβ/δ deletion in mice are painting a more complex picture. Total body PPARβ/δ null mice exhibit a paradoxically leaner phenotype with a significant reduction of both WAT and BAT mass, an effect possibly resulting from an alteration of fatty acid transport [Barak et al.,2002; Peters et al., 2000]. However, PPARβ/δ null mice fed with high fat diet displayed an increased susceptibility to weight gain, coupled with blunted BAT UCP1 expression [Wang et al., 2003]. Finally, mice carrying an aP2-driven adipose-specific deletion of PPARβ/δ did not show any altered fat mass content [Barak et al., 2002] (reviewed in [Christodoulides and Vidal-Puig, 2010]). These results suggest that most effects on adipose tissue may be the consequence of systemic and muscle metabolism rather than reflecting a direct activity of PPARβ/δ in the adipose tissue.
PPARβ/δ-specific roles in liver metabolism
Liver is one of the most important organs in controlling energy homeostasis, via regulation of energy availability and storage. PPARβ/δ is expressed in all the main cell types present in the liver: hepatocytes, Kupffer cells and hepatic stellate cells. The exploration of PPARβ/δ-mediated response in the liver suggested the dual implication of PPARβ/δ not only in fatty acid metabolism, but also directly in glucose metabolism. In particular, Lee et al. [Lee et al., 2006] demonstrated that PPARβ/δ enhances glucose utilization in the liver, via activation of the pentose-phosphate pathway, and promotes liver lipogenesis, recapitulating an insulin-sensitizing effect. Conversely, PPARβ/δ null mice suffer from glucose intolerance [Lee et al., 2006]. Deciphering what is due to a direct activity of PPARβ/δ in the liver from a combined effect on liver, muscle, and adipose tissue remains difficult. In an effort to address this question, Liu et al. [Liu et al., 2011] achieved a liver-specific PPARβ/δ activation by employing an adenoviral-mediated gene delivery system and used, as a control, liver-specific PPARβ/δ null mice. The comparison between mice overexpressing PPARβ/δ in liver with those bearing a liver-specific deletion confirmed its role as an insulin sensitizer. In fact, overexpressing PPARβ/δ in liver causes a genetic reprogramming that leads to an increased glucose utilization and increased lipogenesis. However, no specific alterations have been pointed out regarding the liver-specific deletion of PPARβ/δ, suggesting a moderate activity under normal conditions.
These effects of PPARβ/δ in controlling hepatic lipid metabolism may also have consequences on its role in the resident macrophages in liver, the Kupffer cells, that have also been implicated in fatty liver disease and insulin resistance [Lanthier et al., 2010]. More specifically, PPARβ/δ is a key regulator of the alternative activation of Kupffer cells towards antiinflammatory activity (macrophage M2 subtype) in the presence of IL4 and IL13 stimulation [Odegaard et al., 2008]. An imbalance of inflammatory mediators in liver can also affect hepatic stellate cells where, regardless of their activation status, PPARβ/δ is expressed at high levels [Hellemans et al., 2003]. However, no in vivo mouse studies using PPARβ/δ activation or inactivation mouse models have yet pushed these cellular studies forward.
These observations confirmed a key role of PPARβ/δ in tissues and circumstances where PPARα is also a key regulator of fatty acid oxidation, thereby raising questions regarding their respective roles. Microarray analyses comparing the liver transcriptome of PPARα null mice versus PPARβ/δ null mice revealed only minor overlap between PPARα- and PPARβ/δ-dependent gene regulation, and further reinforced the observations that PPARβ/δ governs glucose utilization and lipoprotein metabolism and has an important anti-inflammatory role in liver [Sanderson et al., 2010].
PPARβ/δ-specific roles in the pancreas
Pancreas-specific deletion of PPARβ/δ, obtained through PDX1-mediated PPARβ/δ gene deletion, caused an increased number of islets and, more importantly, enhanced insulin secretion, leading to hyperinsulinemia and lower glycemia in mutant mice. This was due to alterations in the machinery of exocytosis, from Golgi functions to routing of granules and vesicles to the cell periphery, rather than an altered metabolic response [Iglesias et al., 2012]. In contrast, systemic treatment with PPARβ/δ agonists resulted in increasing glucose-stimulated insulin secretion (GSIS) and normalizing pancreatic islet hypertrophy in ob/ob mice [Tanaka et al., 2003] and db/db mice [Winzell et al., 2010]. Treatment with the PPARβ/δ agonist GW501516 also restored the impaired GSIS observed in mice carrying a pancreas-selective deletion of Desnutrin (also called ATGL/PNPLA2) [Tang et al., 2013]. This study also suggested that in homeostatic conditions, the ATGL-mediated lipolysis would provide ligand for PPARβ/δ, whose activity on mitochondrial oxidation would contribute to islet β cell GSIS.
Finally, and more indirectly, PPARβ/δ agonists increase the production of glucagon-like peptide-1 (GLP-1), an intestinal incretin that can preserve the morphology and function of pancreatic β-cells [Daoudi et al., 2011]. This mechanism might in part explain the apparent contradiction between the observations made in the model of pancreas-specific deletion of PPARβ/δ and those made with systemic PPARβ/δ agonist treatment. However, and as mentioned above, we cannot ignore the caveats that each model carries (compensatory effects and off-target activities for example).
Systemic metabolic effects of PPARβ/δ
The tissue-specific deletions of PPARβ/δ allowed the evaluation of its tissue-specific activity. Systemic treatments of mice with specific PPARβ/δ agonists allowed an integrated analysis of these responses, mainly affecting the overall metabolic homeostasis. For example, the treatment of obese animals with specific PPARβ/δ agonists results in normalizing the metabolic parameters with decrease of circulating triglycerides and reduction of adiposity [Tanaka et al.,2003; Wang et al.,2003; Wang et al., 2004].
Circulating insulin levels also declined, whereas the improvement in glucose tolerance and insulin sensitivity, as determined by the glucose and insulin tolerance tests (GTT and ITT), was moderate [Tanaka et al., 2003]. This PPARβ/δ-mediated systemic improvement in lipid homeostasis also led to decreased high-fat-diet-induced liver steatosis [Wu et al., 2011]. Such PPARβ/δ-driven treatment recapitulates the improved carbohydrate catabolism in the liver, the promotion of fatty acid oxidation in the muscle and the inhibition of free fatty acid release from adipocytes, thereby explaining the overall enhanced insulin sensitivity [Lee et al., 2006]. Similar observations were made in OLETF rats, an animal model of type II diabetes with obesity, whose treatment with the highly PPARβ/δ-specific agonist GW0742 attenuates hepatic fat accumulation and improves insulin signaling [Lee et al., 2012].
PPARβ/δ also participates in regulating lipid systemic transport through lipoproteins. Treating obese rhesus monkeys, an animal model for human obesity and associated metabolic disorders, as well as diabetic db/db mice, with a selective PPARβ/δ agonist caused a beneficial increase in serum HDL cholesterol and a decrease in small-dense LDL [Leibowitz et al.,2000; Oliver et al., 2001], together with a reduced cholesterol absorption in the intestine, associated with a decrease in Niemannn-Pick C1 like 1 protein [van der Veen et al., 2005]. Conversely, PPARβ/δ deficient mice exhibit LDL hypertriglyceridemia, due to increased hepatic production of VLDL and decreased LPL-mediated catabolism [Akiyama et al., 2004].
Altogether, these observations emphasize the intertissular, reciprocal, metabolic regulation that PPARβ/δ coordinates. It explains the difficulties, particularly in the adipose tissue, in discriminating tissue-specific from systemic regulation. It also permits insistence on the potential therapeutic activities of PPARβ/δ in obesity and/or in type 2 diabetes. Indeed, the implication of PPARβ/δ in energy consumption rendered this receptor an attractive therapeutic target, especially for its anti-obesity activity. The first human clinical trial using a PPARβ/δ agonist has been concluded by Sprecher et al [Sprecher et al., 2007], in which GW501516 treatment has been tested in a small cohort of healthy volunteers. Several successive clinical trials have been conducted, or have recently been completed, as reviewed in [Lamers et al., 2012] to treat hyperlipidemia, insulin resistance and obesity. However, despite its promising potential as a treatment for obesity and dyslipidemia, the use of PPARβ/δ agonist, in particular GW501516, in clinical trials was limited, due to its possible use as a doping substance (http://www.independent.co.uk/life-style/health-and-wellbeing/health-news/warning-to-beijing-olympics-over-pills-that-mimic-exercise-882608.html). In addition, the ubiquitous expression of PPARβ/δ generates some concerns regarding the possible onset of adverse side-effects, due to the activation of PPARβ/δ in tissues not related to the therapeutic effects.
Inflammation
Macrophages are key players in inflammation. The first report on PPARβ/δ acting on inflammation in macrophages described the association and dissociation of PPARβ/δ with the transcriptional corepressor BCL6, in absence and presence of ligand, respectively. This results in ligand-dependent PPARβ/δ-mediated inhibition of NFkB target genes, thereby limiting the inflammation [Lee et al., 2003]. Macrophages, however, differentiate from tissue resident monocytes either toward a pro-inflammatory (M1 or classically activated macrophage, e.g. by IFNγ, TNFα or bacterial LPS) or an anti-inflammatory (M2 or alternatively activated, e.g. by IL-4 or IL-13) phenotype according to the specific stimuli of the environment. Specific deletion of PPARβ/δ in macrophages (Lys-Cre-driven specific deletion; [Kang et al., 2008]) and irradiated wild-type mice subjected to PPARβ/δ-/- bone marrow transplantation [Odegaard et al., 2008] both demonstrated that alternative activation of resident macrophages in liver and adipose tissue depends highly on PPARβ/δ activity, which contributes to the M2a-specific gene expression program. Of interest is the fact that M2a macrophages not only limit inflammation but also affect metabolic regulation, in a variable manner depending on the animal model used [Desvergne, 2008]. Finally, PPARβ/δ also acts as a transcriptional sensor of dying cells, facilitating the engulfment of apoptotic cells by macrophages. Mice bearing a macrophage-specific deletion of PPARβ/δ exhibit a decreased expression of opsonins, impairing the capability of phagocytes to recognize apoptotic bodies and to clear them from the environment. This leads to a progressive formation of autoantibodies that predispose PPARβ/δ KO mice to develop autoimmune disease [Mukundan et al., 2009].
The vascular compartment and its endothelial cells also play an important role in inflammation, which is associated with increased local vasodilation, vascular permeability, and leukocyte recruitment due to augmented expression of adhesion molecules on endothelial cells. Selective PPARβ/δ ligand GW501516 treatment inhibits the expression of major endothelial cell inflammatory responses (V-CAM-1, E-selectine, ICAM-1) involved in leukocyte recruitment in vivo [Piqueras et al., 2009]. PPARβ/δ also protects endothelial cells from tissue damaging oxidative stress, which is produced by immune cell activity and may contribute to increased inflammatory response [Fan et al.,2008; Liou et al., 2006] [Jiang et al.,2009a] [d'Uscio et al., 2012]. Other observations include the anti-inflammatory effect of PPARβ/δ ligand administration, which limited the development of atherosclerosis in models using LDL receptor knockout mice [Barish et al.,2008; Graham et al.,2005; Lee et al.,2003; Takata et al., 2008].
These studies likely explained the positive effects of PPARβ/δ agonist treatment in a variety of models of induced inflammation in mice, such as LPS-induced pulmonary inflammation [Haskova et al., 2008], chronic inflammation in the adipose tissue [Rodriguez-Calvo et al., 2008], experimental colitis [Hollingshead et al., 2007], systemic septic shock [Kapoor et al., 2010], TPA-induced hyperplasia and inflammation in the skin [Peters et al., 2000] and hepatotoxicity [Shan et al., 2008] (reviewed in [Bishop-Bailey and Bystrom, 2009]). The anti-inflammatory activity of PPARβ/δ might also be beneficial in the brain. For instance, activation of PPARβ/δ with GW0742 in a murine model of experimental autoimmune encephalomyelitis limits the appearance of cortical lesions and decreases IL-1β expression, and reduces the incidence of clinical symptoms [Polak et al., 2005]. PPARβ/δ may also contribute to the anti-inflammatory activity of Palmitoylethanolamide (PEA) in spinal cord injury [Paterniti et al., 2013]. As can be seen from this paragraph, the diverse importance of the mechanisms proposed is difficult to establish when studying the integrated model represented by the mouse. In particular, the reciprocal link between inflammation and metabolism, along with the attendant question of which starts first, is very intriguing and is clearly highlighted when phenotyping the activity of PPARβ/δ in the full organism.
PPARβ/δ in cell fate
PPARβ/δ in cell differentiation
Mouse observations have identified a certain number of cell types sensitive to PPARβ/δ activation in their differentiation process, as we described in the preceding section concerning placental Giant Trophoblastic cells.
In the skin, PPARβ/δ promotes keratinocyte differentiation [Kim et al.,2005; Schmuth et al.,2004; Tan et al., 2001]. Particularly, in the context of wound and inflammation, which triggers increased PPARβ expression and promotes the synthesis of a ligand so far unidentified [Tan et al., 2001]. The end of the wound healing process is marked by increased activity of the TGF-β/Smad pathway that terminates PPARβ/δ-induced expression and activation [Tan et al., 2004]. This is consistent with the high expression of PPARβ/δ in the mouse epidermis during fetal development, which progressively disappears from the interfollicular epithelium after birth, but is reactivated in the presence of various stimuli such as tetradecanoylphorbol acetate topical application, hair plucking, or skin wound healing, with a demonstrated role in the timing and efficiency of keratinocyte differentiation [Michalik et al., 2001].
The role in keratinocyte differentiation and the high constitutive expression of PPARβ/δ in the epithelial compartment of the gastrointestinal tract raised the question as to whether PPARβ/δ would similarly affect homeostatic regulation of intestinal cell proliferation/differentiation. Indeed, in PPARβ/δ null mice, Varnat et al. demonstrated that PPARβ/δ contributes to Paneth cell terminal differentiation [Varnat et al., 2006], via a PPARβ/δ-dependent down-regulation of Hedgehog signaling, suggesting that PPARβ/δ is downstream of the Wnt-β-catenin/TCF4 pathway.
In the brain, PPARβ/δ is highly expressed during the development of the central nervous system [Braissant and Wahli,1998; Cullingford et al.,1998; Moreno et al.,2004; Woods et al., 2003]. PPARβ/δ null mice exhibit myelination defects [Peters et al., 2000] suggesting a role in oligodendrocyte differentiation and maturation, further confirmed in studies using cells in culture [Saluja et al.,2001; Vittoria Simonini et al., 2010].
PPARβ/δ importantly contributes to the bone differentiation/remodeling cell program in mice [Scholtysek et al., 2013]. First of all, PPARβ/δ is the most abundantly expressed PPAR isotype in bone. Its activation decreases osteoclastogenesis and reinforces osteoblast differentiation via increased WNT signaling to mesenchymal cells. A mirror image is observed in PPARβ/δ null mice that present a higher number of osteoclasts and progressive osteopenia, particularly aggravated upon ovariectomy in females [Scholtysek et al., 2013].
PPARβ/δ is the predominant PPAR isotype expressed in hematopoietic stem cells (HSCs) [Ito et al., 2012]. Bone marrow transplantation using PPARβ/δ-deleted KitposScaposLinneg cells revealed that PPARβ/δ loss did not affect homing of HSCs but profoundly impacted long-term repopulating capacity, whereas treatment with PPARβ/δ agonists improved HSC maintenance in vivo [Ito et al., 2012].
Finally, as discussed above, the role of PPARβ/δ in adipose tissue is highly complex and intimately linked with the systemic metabolic consequences of PPARβ/δ activation, and it seems that PPARβ/δ has little or no specific activity on adipocyte differentiation. However, while total body KO mice have reduced adipose stores, mice with selective adipocyte PPARβ/δ KO have normal adipose tissue. Considering that the aP2 promoter which drives PPARβ/δ deletion is activated at a late stage of adipocyte differentiation, we cannot exclude a possible role at an earlier stage, as suggested by studies using cell culture [Bastie et al.,2000; Hansen et al.,2001; Matsusue et al., 2004].
In conclusion, these in vivo observations are quite heterogenous. PPARβ/δ seems to act in many different cell types and in different tissue contexts to modulate cell fate. Is there a unifying mechanism for all these observations? It is indeed tempting to hypothesize that these activities come from a PPARβ/δ role in cellular basal metabolism. However, this remains to be demonstrated.
PPARβ/δ in cell survival
The study of Di Poi et al. [Di-Poi et al., 2002], discussed in the previous section, highlighted another important PPARβ/δ-mediated response to the inflammatory insult: cell survival. This occurs via PPARβ/δ-dependent up-regulation of integrin-linked kinase (ILK) and 3-phosphoinositide-dependent kinase-1 (PDK1), which activate Akt1 via phosphorylation, and through down-regulation of PTEN [Di-Poi et al., 2002]. This is in turn accompanied by an increased response to chemotactic signals, in particular through cytoskeleton remodeling [Tan et al., 2007].
Accordingly, different models of ischemia-reperfusion have been studied to evaluate the importance of this activity in various tissues. Ischemia-reperfusion is characterized by a first step of adaptation to the lack of nutrients and oxygen creating a cellular stress with acidosis, swelling, changes in the expression of adhesion molecules and other modifications due to adaptation of the cell to a forced anaerobic condition. Reperfusion in this context results in an oxidative insult with induction of oxidative stress in cells rather than restoration of normal function. Such a model is for example used for mimicking ischemic acute renal failure, where the major cellular damage is acute tubular necrosis. In such a mouse model, preconditioning with PPARβ/δ agonist dramatically protected against the damage, via PPARβ/δ-dependent increased Akt activity, whereas PPARβ/δ heterozygous and PPARβ/δ null mice were very sensitive to kidney failure [Letavernier et al., 2005]. These results, reproduced in a diabetic rat model [Collino et al., 2011] and in endothelial cells in culture [Jiang et al.,2009b], point to PPARβ/δ as a remarkable target for preconditioning strategies. The same protective properties are found in the heart, where in vivo activation of PPARβ/δ via agonist treatment preserves the heart from ischemia/reperfusion injury in Zucker fatty rats [Yue et al., 2008], as well as in brain undergoing ischemia [Iwashita et al., 2007]. Finally, treatment with the PPARβ/δ agonist L-165041 protects the testis from ischemia and reperfusion damage, reducing TNFα, IL-6 along with tissue injury via inhibition of extracellular-regulated kinase 1/2 phosphorylation [Minutoli et al., 2009].
PPARβ/δ in cell proliferation and tumor biology
The role of PPARβ/δ in cell proliferation is more ambiguous. Different groups observed that PPARβ/δ has a negative role towards proliferation of keratinocytes [Tan et al., 2001] [Burdick et al.,2007; Burdick et al.,2006; Kim et al.,2006; Kim et al., 2005], at least in part through down-regulation of cyclin A [Tan et al., 2001], through reduced ubiquitination of PKCα [Kim et al., 2005], or via Cdkn1c gene encoding the cell cycle inhibitor p57(Kip2) [Muller-Brusselbach et al., 2007]. However, other studies have shown that PPARβ/δ can promote keratinocyte proliferation [Di-Poi et al.,2002; Michalik et al.,2001; Romanowska et al., 2010]. For example, overexpression of PPARβ/δ in the skin, driven by the human CYP1A1 promoter, provoked hyper-proliferation of keratinocytes, dendritic cell accumulation, and endothelial activation, closely mimicking a psoriasis lesion [Romanowska et al., 2010]. Moreover, under UV stimulation, PPARβ/δ activates Src protein, that in turn promotes the Epithelial to Mesenchymal Transition. This complex process leads to a progressive de-differentiation of keratinocytes, thereby contributing to a higher proliferation rate and, in turn, to tumor progression [Montagner et al., 2014]. A potential and likely only partial explanation for these discrepancies may involve in vivo an intricate combination of PPARβ/δ activation not only in keratinocytes but also in the adjacent fibroblasts, regulating the expression of various cytokines, a hypothesis verified at least in a system of organotypic culture [Chong et al., 2009].
At first glance, due to its role in cell differentiation and the limited evidence supporting its pro-proliferative activity to date, PPARβ/δ should not be thought of as a pro-tumorigenic molecule. However, two main features link PPARβ/δ to tumorigenesis in the intestine: i) PPARβ/δ is a Wnt target gene in this tissue [He et al., 1999], a feature often associated with pro-tumorigenic activity, and ii) PPARβ/δ has a strong anti-apoptotic activity, which is particularly remarkable under conditions of stress (see above). Moreover, the analyses of PPARβ/δ expression, performed in various contexts, underscore its high expression in cancer cells.
The discussion below will only present examples of contradictory results obtained in in vivo studies, as numerous exhaustive reviews have been written on the subject [Michalik and Wahli,2008; Muller-Brusselbach et al.,2007; Muller et al.,2008; Peters et al., 2011]. Most of the in vivo studies evaluate the role of PPARβ/δ in intestinal cancers and can be divided in two principal conditions: PPARβ/δ deficiency and PPARβ/δ activation.
In the context of ApcMin/+ genetic background, PPARβ/δ deletion enhances the number of colon polyps formed in the intestine [Harman et al.,2004; Reed et al., 2004]. In the same manner, the treatment of PPARβ/δ-deficient mice with a potent carcinogen (azoxymethane, AOM) causes an increased number of polyps formed within this tissue [Hollingshead et al.,2008; Marin et al., 2006], suggesting a protective effect of PPARβ/δ in tumorigenesis. However, other studies showed that PPARβ/δ disruption (total body KO or intestinal-specific PPARβ/δ deletion), either combined with ApcMin/+ genetic background [Wang et al., 2006] or with AOM treatment [Zuo et al., 2009], led to reduction of polyp formation and to a marked resistance against carcinogen action. Finally, two other reports stated that total body deletion of PPARβ/δ in ApcMin/+ genetic background or intestinal-specific PPARβ/δ deletion combined with carcinogenic treatment did not result in any changes in colon tumor formation [Barak et al.,2002; Monk et al., 2012].
Some contradictions were shown in the case of PPARβ/δ activation via treatment of mice with PPARβ/δ agonist. GW501516 administration in ApcMin/+ mice leads to an increased number and size of polyps in intestine [Gupta et al., 2004] [Wang et al., 2006], whereas GW0742 administered to
ApcMin/+ mice did not affect size or number of polyps [Marin et al., 2006]. In contrast, treatment with high doses of GW0742 agonist in the AOM-induced model of carcinogenesis leads to a significant decrease in tumor incidence [Hollingshead et al., 2008] [Marin et al., 2006].
A partial explanation for the controversial findings reported in the in vivo studies may come from PPARβ/δ activity in the tumor microenvironment. For example, PPARβ/δ up-regulates VEGF expression in epithelial cells [Piqueras et al.,2007; Wang et al., 2006], a critical pro-angiogenic factor, also affecting vascular permeability, and immune cell activity and their response to tumor. An elegant demonstration comes from the study of Muller-Brusselbach et al. [Muller-Brusselbach et al., 2007], who showed that growth of syngeneic PPARβ/δ wild-type tumors is impaired in PPARβ/δ null mice, due to a deficit in microvessel formation and maturation. This is also consistent with the correlation found in human tumors between high levels of PPARβ/δ and cyclooxygenase 2 expression on the one hand, and the microvascular density and venous vessel tumor invasion on the other hand [Yoshinaga et al., 2009], thereby confirming its role in affecting the tumor microenvironment.
This brief description is by no means exhaustive, as numerous papers continue to feed the controversy, but fail to provide definitive reasons for such discrepancies. The controversies are not over [Peters et al.,2008; Xu et al., 2013], and further studies need to be done in order to establish a role for PPARβ/δ in cancer, and in doing so to ensure the safety of PPARβ/δ activation through synthetic agonists.
Genome-wide analyses of PPARβ/δ transcriptional activities
Mouse models are crucial for gaining a systemic view of PPARβ/δ functions relevant to the full organism. To gain insights into the mechanisms by which the transcription factor PPARβ/δ may act, whole genome studies allow a broad and unbiased view of the primary targets it affects.
Microarray studies
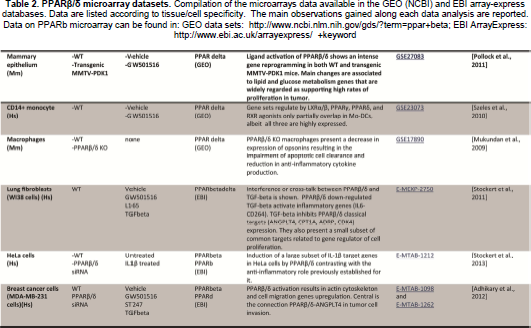
Due to the fact that PPARβ/δ is ubiquitously expressed and controls many different biological pathways, the compilation of microarray data, in different contexts, could be helpful in order to highlight some “core” functions and tissue- or cell-specific activity on gene expression from a wider point of view. Table 2 recapitulates the microarrays available in the GEO (NCBI) and EBI array-express databases at the following links: http://www.ncbi.nlm.nih.gov/gds and http://www.ebi.ac.uk/arrayexpress/+keyword. The keywords PPAR beta, PPARβ/δ, PPARdelta and PPAR beta/delta enable you to find all the available microarray data. This search is likely not complete, but it provides a good handle on conducting a deeper search. Doing a rigorous meta-analysis across all data obtained in microarrays remains very challenging, due to different initial conditions, different platforms used, and most importantly, to the experimental design aimed at answering a different and specific question for each study. The summary presented below only aims at presenting the main observations and leads that these microarray studies highlighted, albeit many of them have been previously stressed (see section on Integrative Physiology).
PPARβ/δ microarray datasets.
Firstly, microarray represents a powerful technique for assessment of gene expression regulation in understanding the impact of PPARβ/δ deletion/activation on lipid and glucose metabolism. The main observations were achieved studying liver, adipose tissue, and pancreas comparing both PPARβ/δ wild type and KO mice, or agonist-treated versus vehicle control mice expression profiles. In particular, PPARβ/δ deletion caused suppression of the expression of several genes involved in glucose and lipid catabolism [Mandard et al.,2007; Sanderson et al., 2010], serving to confirm the role of PPARβ/δ as a key regulator of oxidative metabolism. The global gene expression pattern in PPARβ/δ-depleted pancreatic cells also highlighted its positive regulatory activity on a combination of genes involved in insulin secretion and its repressive role on genes involved in pancreatic cell proliferation [Iglesias et al., 2012]. Genetic reprogramming of oxidative metabolism genes is also involved in PPARβ/δ regulation of muscle response to physical stress and muscle endurance. Activation of PPARβ/δ by ligand treatment or transgenic expression leads to a general upregulation of genes involved in fatty acid transport, beta-oxidation, and mitochondrial respiration [Tanaka et al., 2003], which further increases when coupled with exercise [Narkar et al., 2008]. Microarray also identified Lactate b dehydrogenase and AMPK as two of the main enzymes involved in the endurance program [Gan et al., 2011]. Moreover, the classical switch toward fiber type I observed in PPARβ/δ-activated genetic reprogramming goes through a distinct muscle microRNA (miRNA) network and the signaling of estrogen-related receptor gamma (ERRgamma) [Gan et al., 2013].
Another aspect of PPARβ/δ-mediated gene regulation relates to this receptor's role as a potent anti-inflammatory player [Iwaisako et al.,2012; Kaddatz et al.,2010; Mukundan et al.,2009; Stockert et al.,2011; Stockert et al., 2013]. In fact, PPARβ/δ deletion triggered a reduced expression of anti-inflammatory cytokine and opsonins in macrophages [Mukundan et al., 2009]. As a mirror image, PPARβ/δ activated by ligand stimulation is able in fibroblasts to counteract TGFβ-activated genes and to activate TGFβ-repressed genes [Kaddatz et al.,2010; Stockert et al., 2011]. Importantly, different agonists can exert different activation of genetic regulation by PPARβ/δ. For example in liver, KD3010 but not GW501516, attenuates the gene expression signature consistent with inflammation and hepatocyte cell death, stimulates ROS protection pathways and down-regulates pro-fibrotic factor expression [Iwaisako et al., 2012].
Genome-wide regulation of gene expression mediated by PPARβ/δ in tumor development also reflects the controversies discussed above. In mammary epithelium, PPARβ/δ acts as an oncogene affecting cell invasion, motility, but also inflammation-related pathways [Yuan et al., 2013]. Actin cytoskeleton and cell migration genes were deregulated after PPARβ/δ agonist treatment and ANGPLT4 has been highlighted as a central step in tumor cell invasion [Adhikary et al., 2012]. In contrast, activation of PPARβ/δ in keratinocytes seems to be protective against tumor formation by controlling the expression of members of the ubiquitin-proteasome degradation pathway [Kim et al., 2004] or by repressing E2F target genes through the interaction with p130/p107 [Zhu et al., 2012]. These discrepancies may be due to the fact that the analysis has been performed in isolated tissues/cells, thereby losing the contribution of the paracrine activity exerted by other cell types residing in the tumor environment (tumor stroma).
Particular attention should be given to two microarray studies, performed in myofibroblasts [Adhikary et al., 2011] and in keratinocytes [Khozoie et al., 2012], where all the collected data comparing the absence and presence of a PPARβ/δ activator have been filtered by PPARβ/δ silenced or PPARβ/δ KO derived gene expression profiles, in cells treated with the same experimental conditions in order to exclude non-specific signals. By combining microarray profile with ChIP-seq data, the authors described different types of PPARβ/δ-mediated gene regulation (summarized in the next section “ChIP and ChIP-seq with PPARβ/δ antibody”), from repression in presence of ligand to activation in absence of ligand. Interestingly, genes belonging to a given regulatory pattern were directly associated with particular functional pathways, thereby confirming the biological relevance of these findings.
In conclusion, microarray analysis of PPARβ/δ activities contributed to a better knowledge of the molecular properties and biological functions of this ubiquitously-expressed receptor, highlighting both pan-tissue and tissue-specific genetic regulation. Nonetheless, a rigorous meta-analysis across all data obtained in microarrays remains to be done.
ChIP and ChIP-seq with PPARβ/δ antibodies
The analysis of the binding landscape of a given transcription factor is an important approach currently used to identify its possible target genes in the genome. However, binding of a TF in the vicinity of a gene is not sufficient, but only increases the probability that this gene is regulated by this TF. Therefore, combining the binding analysis above with a gene expression analysis greatly facilitates interpretation of the data. Irrespective of transcriptional regulation, the whole genome binding landscape may also give some insights into general DNA-binding properties.
Chromatin Immunoprecipitation (ChIP) is a technique used to study the interaction between DNA and proteins within the cellular context, rather than using a synthetic piece of DNA in in vitro experiments such as the gel electro-mobility shift assay. It is clear that the quality (specificity and affinity) of the antibody is a crucial determinant of the effectiveness of such an experiment. This is indeed a critical issue for PPARβ/δ, as well as for many other NRs, because of the relatively poor efficiency and specificity of NR monoclonal or polyclonal antibodies available commercially. However, we present below first a series of papers demonstrating PPARβ/δ interaction on known gene sequences, and second two papers analyzing genome-wide binding sites of PPARβ/δ.
ChIP has successfully elucidated PPARβ/δ interaction with the promoter of proteolipid protein (PLP) when cells are treated with the fibrate Gemfibrosil [Jana et al., 2012]. PPRE sequences binding to PPARβ/δ were also identified by ChIP in the promoter region of the calreticulin gene [Riahi et al., 2010]. Animal studies demonstrated that oral administration of GSK3787 antagonizes the GW0742-induced PPARβ/δ promoter occupancy of Angptl4 and ADRP genes. This correlates with a reduced ADRP and Angptl4 mRNA expression in WT but not in PPARβ/δ null mice colon, which is consistent with the ChIP results [Palkar et al., 2010]. The PPARβ/δ antibody used in this study was developed by Girroir et al. [Girroir et al., 2008].
ChIP experiments were also very useful in suggesting indirect mechanisms of PPARβ/δ-mediated transcriptional regulation. Indeed, the enhanced expression of the SIRT1 gene after PPARβ/δ ligand activation is not associated with PPARβ/δ binding to its 5’ flanking region, but is mediated instead by a canonical Sp1 binding site. Consistently, this potent trans-activating effect of PPARβ/δ/GW501506 was completely abolished in the presence of Mithramycin, an inhibitor of Sp1, suggesting that Sp1 could act as an ancillary factor for PPARβ/δ [Okazaki et al., 2010]. Finally, in HCT116 colorectal carcinoma cells, VEGFA transcription is regulated by PPARβ/δ via β-catenin-mediated chromatin loops. The authors show that chromatin loops around VEGFA are released upon PPARβ/δ activation. The model predicts that β-catenin mediates repressive looping and that PPARβ/δ-specific ligands release the loops, creating an active transcription unit [Hwang et al., 2012].
In concert, this limited number of examples (this presentation is not exhaustive) demonstrate that ChIP of PPARβ/δ can be obtained in certain contexts. Other contexts were not so successful and required, for example, the use of a V5- PPARβ/δ tagged protein [Yamamoto et al., 2011]. Recently, however, two groups have succeeded in performing genome-wide analyses of PPARβ/δ binding. Adhikary et al., [Adhikary et al., 2011] performed genome-wide analyses of human myofibroblasts (WPMY-1 cell line), treated with or without agonist (GW501516), in control cells and in PPARβ/δ-depleted cells, to identify PPARβ/δ-mediated gene regulation. ChIP-Seq analyses of PPARβ/δ, RNA POL II, and H3K4me3 have been performed, using for PPARβ/δ a Santa Cruz antibody. The sequencing data identified a total of 4542 enriched peaks for PPARβ/δ, most of them inside transcribed genomic regions or less than 25Kb upstream. Among the 4542 enriched peaks for PPARβ/δ, the high confidence peak set defined as having a false discovery rate (FDR) <0.05 led to the identification of 443 peaks. This high confidence peak set was then filtered for H3K4m3 and RNA POL II enrichment as markers for active and inducible proximal promoter. The identified enriched sites were subsequently combined with transcriptional profiling derived from both agonist GW501516-treated or PPARβ/δ-silenced myofibroblasts. This additional analysis enabled the definition of 112 bona fide PPARβ/δ target genes classified according to three distinct types of transcriptional response: ligand-independent repression by PPARβ/δ; ligand-induced activation and/or derepression by PPARβ/δ; and ligand-independent activation by PPARβ/δ. The majority of enriched pathways were associated with carbohydrate and lipid metabolism. However, the analysis highlighted PPARβ/δ involvement in non-metabolic functions including hematopoiesis and muscle/heart development. No analysis for the presence of other transcription factor sites coupled to PPARβ/δ genome-specific peaks was proposed. Sequencing data were deposited at EBI ArrayExpress (E-MTAB-371) [Adhikary et al., 2011].
In 2012, a second study coupling the expression profiles with ChIP-seq allowed examination of PPARβ/δ-dependent regulation of gene expression in primary culture of mouse keratinocytes, [Khozoie et al., 2012]. The study was carried out on primary keratinocytes derived from both PPARβ/δ wild type and PPARβ/δ KO mice, in the presence and absence of the selective agonist GW0742. An initial microarray analysis identified a total of 612 target genes for PPARβ/δ. These target genes were then classified according to the type of transcriptional regulation, resulting in a total of eight different types of regulation ranging from repression to activation with or without ligand stimulation, plus combinations of these effects.
ChIP-seq was subsequently performed to examine the molecular mechanism by which PPARβ/δ differentially regulates these target genes. The authors used the polyclonal antibody anti-PPARβ/δ described by Girroir et al. [Girroir et al., 2008]. ChIP-seq data revealed 6839 sites occupied by PPARβ/δ in chromatin prepared from control cells, while more than twice as many sites were identified in chromatin prepared from agonist-treated cells, the specificity of which was controlled by comparing with chromatin prepared from PPARβ/δ null mice. Only 203 out of the 612 PPARβ/δ-regulated genes displayed in the microarray have been found to be occupied by PPARβ/δ at the chromatin level within 10Kb of the transcriptional start site. The search for DNA binding motifs proximal to PPARβ/δ binding regions identified two main phylogenic families of transcription factors, the ETS and the CREB/ATF/AP1, associated with the PPARβ/δ binding site in various combinations depending on the type of regulation. More particularly, ATF4 is required for ligand-dependent induction of PPARβ/δ target genes for some of the types of gene regulation identified in the study [Khozoie et al., 2012].
In conclusion, these studies offer an important glimpse into the complexity of PPARβ/δ-mediated transcriptional regulation. Not only ligand stimulation, but also the DNA binding of other transcription factors influences the transcriptional activity of PPARβ/δ itself [Khozoie et al., 2012]. However, it would be of major interest to complete the picture with genome-wide analyses in different cellular and treatment contexts. This would require having access to a fully reliable antibody. Alternatively, developing a flag-tagged knock-in PPARβ/δ mouse model might help to overcome the lack of a highly specific and efficient PPARβ/δ antibody.
Human PPARβ/δ (PPARD) gene polymorphisms
The search for human polymorphisms and their consequences on human physiology might reveal important activities of PPARβ/δ in human. 90 single nucleotide polymorphisms have been identified and listed in the NCBI reference assembly, among which 21 SNPs have been cited in the literature. We briefly describe below a few polymorphisms that have been associated with a phenotype (see Table 3). It must be noted that none of the polymorphisms identified to date change the amino acids sequence of the protein, being either in untranslated regions, promoter sequences, intron sequences, or giving rise to a synonymous codon. Some studies evaluated the impact of such variations on PPARβ/δ expression levels, as mentioned in the table (Table 3), albeit no further mechanistic studies have been done so far. However, we believe that at this present time of major expansion of individual genome analyses, GWAS analyses will be significantly empowered.
PPARβ/δ single nucleotide polymorphisms associated with disease.
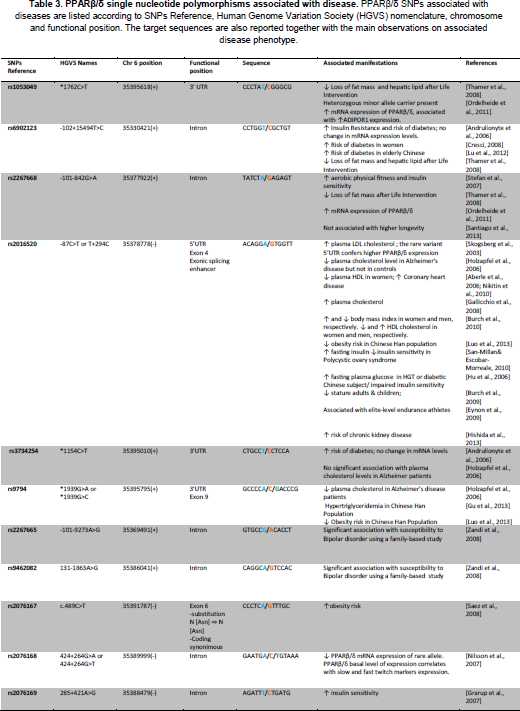
Skogsberg et al. [Skogsberg et al.,2003a] were the first to screen the proximal promoter and the 5'-UTR for functional polymorphisms in the human gene for use as tools in association studies. Four polymorphisms were detected: −409C/T in the promoter region, +73C/T in exon 1, +255A/G in exon 3, and T+294C (rs2016520) in exon 4. The authors found that only the rs2016520 showed significant association with a metabolic trait. Homozygotes for the rare C allele had a higher plasma LDL–cholesterol concentration than homozygotes for the common T allele. This association was verified in an independent cohort of 282 healthy men. Transfection studies showed that the rare C allele is more efficiently transcribed than the common T allele. Electrophoretic mobility shift assays demonstrated that the T+294C polymorphism influenced binding of Sp-1. Using the West of Scotland Coronary Prevention Study (WOSCOPS), the authors found that individuals carrying the rare PPARβ/δ T+294C allele had significantly lower high-density lipoprotein cholesterol (HDL-C) concentration than subjects homozygous for the common T-allele. Homozygous carriers of the C-allele also showed a tendency towards higher risk of Coronary Heart Disease (CHD) compared with homozygous carriers of the T-allele. In addition, a gene-gene interaction involving T+294C polymorphism and the PPARa L162V polymorphism may influence the plasma LDL-C concentration [Skogsberg et al.,2003b]. Finally, a recent study on another PPARβ/δ polymorphism, the rs9794, suggested its association with hypertriglyceridemia [Gu et al., 2013].
In 2004, in an effort to identify polymorphic markers in candidate genes for type 2 diabetes and associated phenotypes, Shin et al. [Shin et al., 2004] identified nine polymorphisms in PPARβ/δ: four in the intron, one in the 5’ untranslated region (UTR), and four in the 3’ UTR. Among identified polymorphisms, five were common sites, namely c.-13454G>T, c.-87T>C, c.2022-12G>A, c.2629T>C and c.2806C>G. While the authors did not find significant associations with the risk of type 2 diabetes, several positive associations of PPARβ/δ polymorphisms with fasting plasma glucose and BMI were detected in non-diabetic control subjects [Shin et al., 2004]. Intriguingly, in a study encompassing more than 11000 individuals, association of PPARβ/δ polymorphisms with BMI, HDL, leptin and TNFα was positive only in a gender-dependent manner, for example significant association of rs2016520 and increased HDL cholesterol in males, whereas HDL cholesterol tended to be decreased in females [Burch et al., 2010]. Increased risk of diabetes was also observed in female but not male carriers of the C allele of rs6902123 in a study including 769 subjects participating in the STOP-NIDDM trial [Andrulionyte et al., 2006]. Along this line, but independent of gender, the C allele of the T+294C polymorphism was associated with higher fasting plasma glucose concentrations in both normoglycemic and diabetic subjects in a study of 663 Shangaï men and women [Hu et al., 2006]. The same polymorphism (genotype CC) is associated with increased risk of coronary artery disease [Aberle et al.,2006; Nikitin et al., 2010]. In other studies, association of the T+294C allele with the metabolic syndrome or with Type 2 diabetes was at best suggested [Robitaille et al., 2007] or denied [Villegas et al., 2011].
A series of studies revealed the interaction between polymorphisms. One in particular, is the interaction of PPARβ/δ polymorphisms with the gly482-to-ser polymorphism in PGC1α, shown to increase the risk for diabetes [Andrulionyte et al., 2006], or be associated with polycystic ovary syndrome [San-Millan and Escobar-Morreale, 2010]. Searching for interaction between the three PPAR isotypes, Luo et al. [Luo et al., 2012] found that in the group of 820 Chinese subjects (non obese and obese) rs2016520 (T+294C) is associated with lower obesity risk, in contrast to the observations of Skogsberg et al. [Skogsberg et al.,2003a]. However, the authors pointed out that interactions among rs2016520, rs9794 on PPARβ/δ and rs10865170 (PPARγ) are associated with higher obesity risk.
Because of the link between cholesterol metabolism and Alzheimer's disease (AD) pathogenesis, Holzapfel et al. [Holzapfel et al., 2006] investigated three frequent polymorphisms located in exons 4 (rs2016520) and 9 (rs3734254 and rs9794) of PPARD for their putative influence on the risk of Alzheimer's disease and on cholesterol, 24S-hydroxycholesterol and 27-hydroxycholesterol plasma levels. The study population consisted of 167 AD patients and 194 controls. No single PPARβ/δ haplotypes influenced the risk of AD. However, a two-marker haplotype consisting of rs2016520 and rs9794 in AD patients showed a significant effect on the relative plasma levels of 24S-hydroxycholesterol and 27-hydroxycholesterol, but not in non-demented controls. Their results suggest that PPARβ/δ haplotypes might influence levels of cholesterol metabolites in AD patients, but not act as risk factors for AD. Indeed, a Finnish study showed no association between PPARβ/δ polymorphism and AD [Helisalmi et al., 2008].
As described above, PPARβ/δ plays an important role in energy metabolism, particularly in muscle. Studies were therefore designed to investigate whether SNPs in the PPARD gene can modulate the effect of exercise training on aerobic physical fitness and insulin sensitivity. Stefan et al., genotyped and described in detail SNPs rs1053049, rs6902123 and rs2267668 in PPARD [Stefan et al., 2007]. After 9 months of lifestyle intervention (Tuebingen Lifestyle Intervention Program), the minor G allele of rs2267668 in PPARβ/δ and the gly482-to-ser polymorphism in PGC1α (PPAR gamma co-activator 1alpha) were independently associated with less increase in aerobic physical fitness, with a possible additive effect of these two SNPs. The interpretation is that the minor alleles of the tested PPARβ/δ SNPs would allow a lesser benefit from exercise and weight loss than the major alleles, thereby providing a mechanistic explanation for the reduced aerobic physical fitness and insulin sensitivity of these substances [Stefan et al., 2007]. These results are in line with those of Thamer et al. [Thamer et al., 2008], showing that SNPs rs1053049, rs6902123 and rs2267668 in PPARβ/δ after lifestyle intervention induced changes in overall adiposity, hepatic fat storage, and relative muscle mass. In contrast, no association of PPARβ/δ polymorphism with performance of marathon athletes was found [Tsianos et al., 2010], while the interaction of rs2016520 and Gly482-to-Ser polymorphism in PGC1α is suggested in elite level endurance athletes [Eynon et al., 2009].
In conclusion, some but not all studies indicate a significant association with either propensity for diabetes or obesity. These difficulties in pointing to a specific phenotype underline the role of PPARβ/δ as a subtle regulator rather than a master key in metabolic regulation. It is likely that the galloping development of the new generation sequencing technology will provide a wealth of difficult to handle but extremely rich information, particularly in the context of gene-gene interaction studies.
Conclusion and future perspectives
In the present review we have summarized different issues surrounding PPARβ/δ trying to sum up the present knowledge in a systems biology-oriented view. The different mouse models plus the ligand treatment strategies, microarray, and ChIP-seq highlighted that PPARβ/δ plays its role in three main fields: i) regulation of energy metabolism, ii) cell proliferation and differentiation and iii) protection in stress conditions such as oxidative stress and inflammation. Whereas interest in developing new PPARβ/δ-based therapeutics for obesity remain, this is impaired by its possible use as a doping substance. In addition, a design for tissue-specific delivery of PPARβ/δ agonists would limit the systemic effects due to improper activation of PPARβ/δ in other compartments. A better understanding and characterization of coregulators (coactivators and/or corepressors) interacting with PPARβ/δ in different cell types will be fundamental in this regard. However, our knowledge about the PPARβ/δ interactome remains poor because of the difficulties inherent in generating suitable tools, such as high performance antibodies or modified (e.g. epitope-tagged) PPARβ/δ.
In conclusion, while significant strides have been made in understanding PPARβ/δ function, a number of questions still remain open. Further studies are needed in order to better characterize this receptor in a more systemic manner, to strengthen the possibility that PPARβ/δ might be used as a therapeutic target in metabolic disorders, muscle endurance improvement and inflammation control.
Footnotes
Acknowledgements
The authors are grateful to Yaakov Barak for his in-depth reading and suggestions. We would also like to thank Federica Gilardi and all members of the BD Laboratory for their critical reading. The laboratory of Béatrice Desvergne is supported by the Fond National de Recherche Suisse, the Etat de Vaud, and the European Initial Training Networks NR-NET.