
Abstract
High-altitude pulmonary edema (HAPE) is a life-threatening noncardiogenic form of pulmonary edema (PE) that afflicts susceptible persons after rapid ascent to high altitude above 2500 m. Its pathogenesis is related to increased sympathetic tone, exaggerated hypoxic pulmonary vasoconstriction, uneven hypoxic pulmonary vasoconstriction with overperfusion of some regions of the pulmonary vascular bed, increased pulmonary capillary pressure, stress failure of pulmonary capillaries, and alveolar fluid leak across capillary endothelium resulting in interstitial and alveolar edema. Prevention of HAPE is most effectively achieved by gradual ascent with time for acclimatization, although recent small studies have highlighted a number of pharmacologic options. Inhaled salmeterol prevents HAPE presumably by increasing alveolar fluid clearance, the phosphodiesterase-5 inhibitor tadalafil works by acting as a pulmonary vasodilator, and dexamethasone seems to prevent HAPE by stabilizing the capillary endothelium, along with other potential effects. These investigations have yet to be validated in widespread clinical practice. Nifedipine, which prevents HAPE via its effects as a pulmonary vasodilator, has a longer history of clinical use. The most effective and reliable treatment of established HAPE is immediate descent and/or adequate flow supplemental oxygen to maintain arterial saturation above 90%, accompanied by rest from strenuous physical activity. Use of a portable hyperbaric chamber is an effective temporizing measure, and nifedipine may be used for treatment of HAPE, although only as an adjunct to descent and/or supplemental oxygen if these methods of treatment are not immediately available to a person with HAPE.
Introduction and clinical description
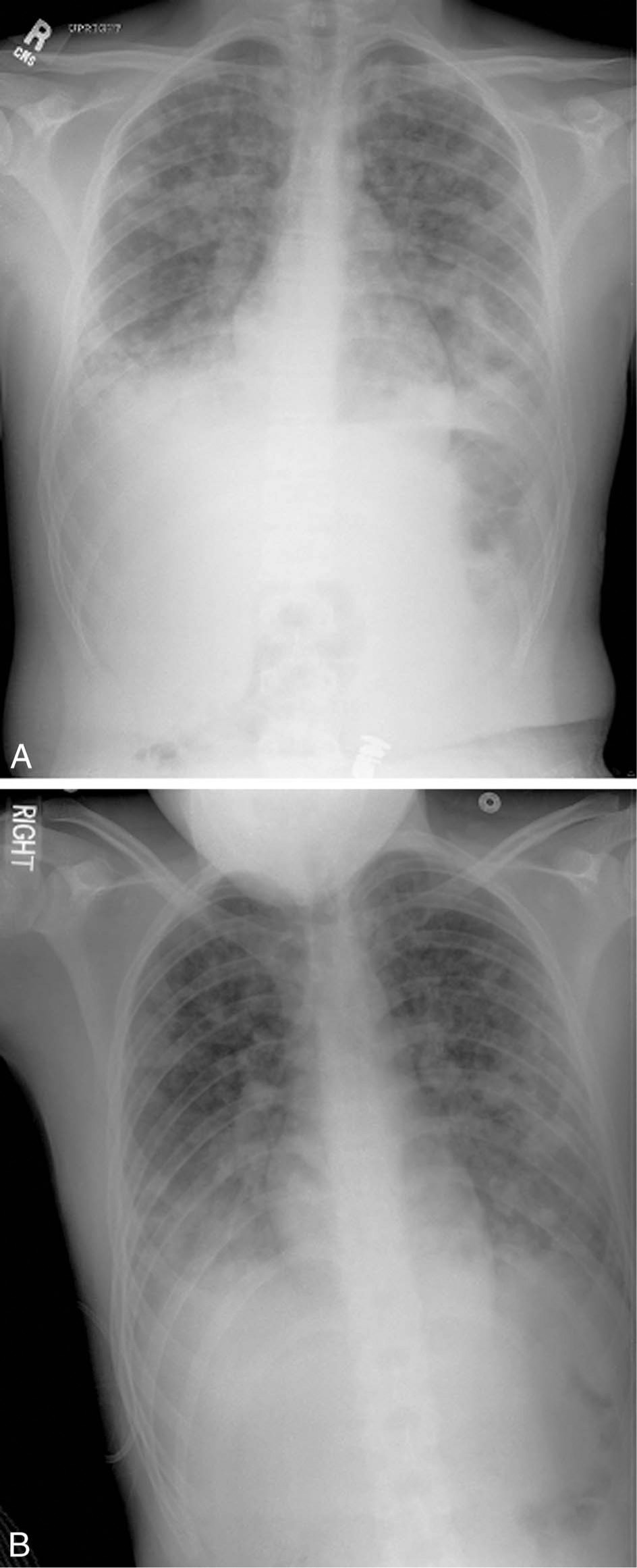
High-altitude pulmonary edema (HAPE) is a life-threatening noncardiogenic form of pulmonary edema (PE) that develops in nonacclimatized persons after rapid ascent to altitudes above 2000 to 3000 m. HAPE is primarily a pulmonary disorder, whereas acute mountain sickness (AMS) and the much less frequent high-altitude cerebral edema, are neurologic disorders. First described in the English language medical literature in 1960 by Houston 1 and Hultgren, 2 HAPE is characterized by cough, progressive dyspnea with exertion, and decreased exercise tolerance, generally developing within 2 to 4 days after arrival at high altitude. The cough typically begins as a dry cough that progresses to a cough productive of pink, frothy sputum, rarely producing frank blood. 3 Symptoms of AMS, including headache and nausea, may accompany HAPE, but 50% of persons with HAPE experience no AMS symptoms. 4 On physical examination, persons with HAPE have tachycardia, tachypnea, low-grade fever, and inspiratory crackles on lung auscultation, which are typically bilateral but may be unilateral in early HAPE, often heard in the right middle lobe. 4 The latter diagnostic finding is not particularly specific for early HAPE, because focal crackles may be heard in the absence of any high-altitude illness after ascent to high altitude.5,6 As HAPE progresses, crackles may become diffuse. On chest radiography, HAPE is characterized by patchy bilateral alveolar infiltrates that may be unilateral in the right hemithorax in early HAPE. 6 A retrospective review of 60 cases of severe HAPE (requiring hospitalization) found more homogeneous, confluent infiltrates in the late stage of the disease, suggesting progression from its initial patchy distribution with increasing severity (Figure 1). 7 HAPE is characterized by increased pulmonary artery (PA) pressure with a normal cardiac output and pulmonary capillary wedge pressure. HAPE-related hypoxemia is variable, with a mean arterial oxygen saturation of 74% observed in one study at a ski resort at 2928 m (normal average saturation at that altitude is approximately 92%) (Table 1).3,4
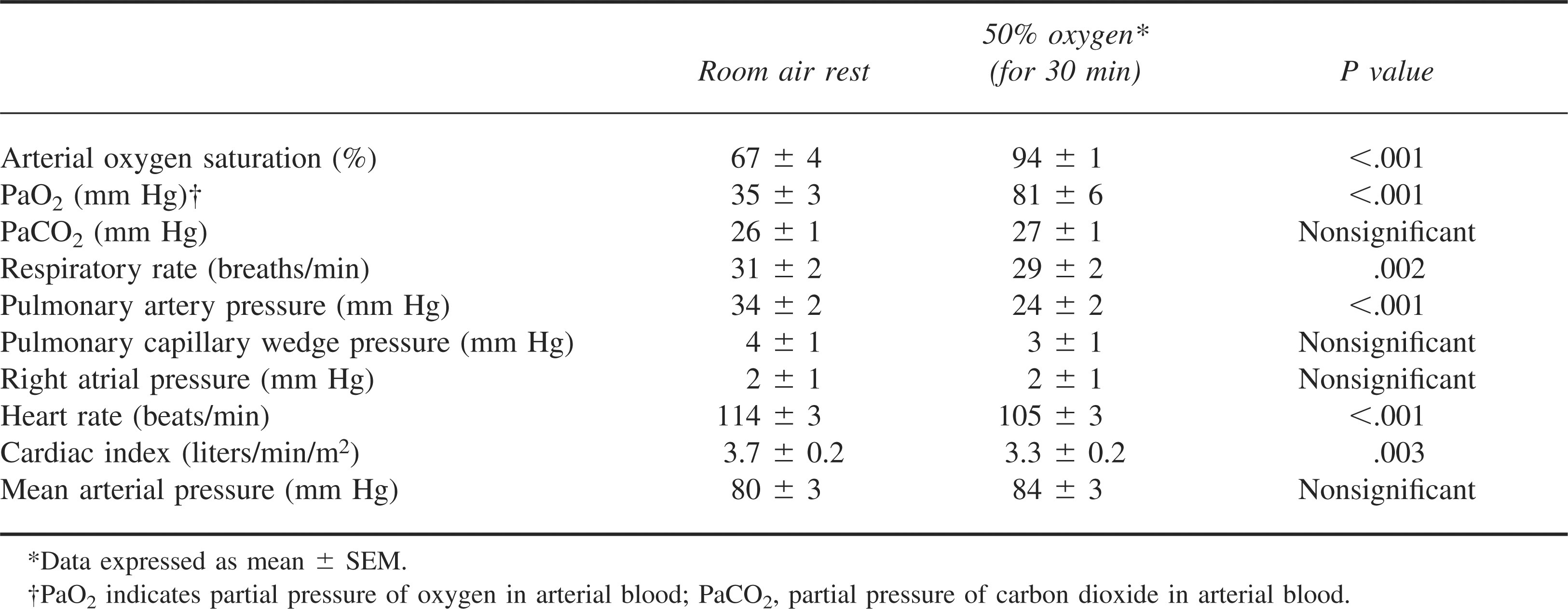
Hemodynamic and gas exchange parameters in 14 patients with high-altitude pulmonary edema at 3600 m before and after treatment with supplemental oxygen*95
Risk factors and susceptibility
Factors that increase the incidence of HAPE include a prior history of HAPE, 8 faster rates of ascent, higher altitudes, male sex,4,9 cold ambient temperatures,9,10 preexisting respiratory infection, 11 and intense exercise. 12 Most HAPE-susceptible persons (at least 1 prior episode of HAPE) can travel to high altitude on subsequent trips without developing HAPE if they ascend at a gradual rate to allow time for acclimatization once above 2000 m. 13 As demonstrated in children by Durmowicz and colleagues 11 and anecdotally observed in adults, preexisting respiratory tract infection is a risk factor for the development of HAPE, presumably due to the alteration of vascular permeability by inflammatory mediators. Priming with endotoxin and preexisting viral respiratory infection has been found to increase pulmonary vascular permeability and protein content in edema fluid of rats exposed to high altitude, further supporting this theory.14,15
Evidence also exists for an association between a predisposition to HAPE and the HLA-DR6 and HLA-DQ4 alleles, suggesting an immunogenetic susceptibility to HAPE. 16 Because these studies were conducted in the somewhat restricted gene pool of entirely Japanese individuals, more comprehensive investigation of this intriguing possibility is warranted. 17
Preexisting conditions or anatomic abnormalities that lead to increased pulmonary blood flow or intravascular pressure may predispose to HAPE, even at altitudes less than 2500 m. These include primary pulmonary hypertension, 18 congenital absence of one pulmonary artery (PA), 19 or intracardiac shunt. The latter includes left-to-right shunts, such as atrial septal defect, 20 ventricular septal defect, 21 and the more well-studied patent foramen ovale (PFO). 22 It has been theorized that, as a result of rising pulmonary vascular resistance (PVR) during hypoxic pulmonary vasoconstriction (HPV), a PFO may reverse direction and begin shunting blood from the right to left heart, further exacerbating hypoxemia. 23 This effect has been demonstrated in HAPE-prone persons at high altitude, and the presence of a PFO has been found to be 4 times more frequent in HAPE-susceptible individuals. The size of the PFO directly correlates to the degree of arterial hypoxemia, with a trend toward increased risk of developing HAPE in those with large PFOs. 22 Considering these findings, cases of HAPE that occur at low to moderate altitude certainly warrant investigation into a preexisting cardiopulmonary abnormality, and these patients should seek consultation with a physician knowledgeable in high-altitude illness before traveling to such altitudes.
Although particular genetic, anatomic, or pathologic characteristics may increase the risk of HAPE in certain individuals, complete resistance may not exist. Several authors have postulated that any person may develop HAPE if the ascent rate is fast enough to a high enough altitude. 13 This assertion is based on studies that show subclinical increased extravascular lung water in the majority of persons who ascend to high altitude.24,25
Long-term inhabitants of high-altitude regions constitute a special group at risk for HAPE because of “reentry HAPE” that may occur upon reascent after a sojourn at low altitude.2,26 Children appear to be more susceptible to this condition. 26
Pathophysiology
HAPE is characterized by exaggerated HPV27,28 and high-permeability noncardiogenic PE.29,30 HAPE is caused by stress failure of pulmonary capillaries31,32 in focal areas of the pulmonary circulation due to increased pulmonary capillary transmural pressures 33 and resultant permeability edema, leading to alveolar flooding and progressive hypoxemia (Figure 2). The exaggerated hypoxia-induced pulmonary hypertension in HAPE-susceptible individuals may be accompanied by blunted HPV, thereby worsening hypoxemia. 34 –36

Proposed pathogenesis of high-altitude pulmonary edema.
The etiology of the abnormal increase in PA pressure in HAPE-prone persons is likely multifactorial, involving augmented sympathetic activity, 37 increased levels of endothelin-1, 38 and decreased availability of nitric oxide (NO).30,39,40 It is readily apparent that vigorous exercise and cold ambient temperatures in mountainous areas can contribute to an increase in sympathetic activity. 41 In HAPE-susceptible persons, this increase in sympathetic activation is exaggerated in response to hypoxia at both low and high altitude and is directly correlated to the increase in PA pressure. 37 The concept of a sympathetic contribution to the pulmonary hypertension of HAPE is supported by the efficacy of alpha-blockade by phentolamine infusion in decreasing PA pressure in subjects with HAPE, more than other vasodilators without antiadrenergic properties. 42 Levels of endothelin-1, a potent endothelium-derived pulmonary vasoconstrictor, directly correlate with increased PA pressure in HAPE-susceptible persons at high altitude, with levels 33% higher than those in persons without a history of HAPE. 38 More recent data indicate that antagonism of endothelin-1 with bosentan decreases PA pressures in healthy volunteers at high altitude. 43
As a mediator of exaggerated HPV, decreased NO availability may play a major role in the susceptibility to and development of HAPE and is thus a potential target for prevention and/or treatment. At high altitude, HAPE-prone individuals demonstrate significantly reduced levels of exhaled NO with an associated increase in PA pressure.39,40 Additionally, bronchoalveolar lavage fluid in HAPE-susceptible persons at high altitude held reduced concentrations of nitrates and nitrites.
44
Berger and colleagues recently implicated impairment of the NO-mediated vasodilator pathway, demonstrating that HAPE-prone persons exposed to normobaric hypoxia had a decreased systemic endothelium-dependent vasodilator response to intra-arterial acetylcholine.
45
Several factors may contribute to the decreased bioavailability of NO, including downregulation or defect of endogenous NO synthase (eNOS), inhibition of eNOS by
Studies of bronchoalveolar lavage fluid demonstrate mild pulmonary hemorrhage and transvascular leak of proteins into the alveolar spaces in early HAPE, without elevation of cytokines and other inflammatory mediators.44,49 Maggiorini and coworkers have demonstrated that increased pulmonary capillary pressure is an initiating factor in the development of HAPE, presumably resulting in injury to the capillaries and a high-permeability edema. These investigators found an altitude-induced increase in mean pulmonary capillary pressure to 19 mm Hg in HAPE-susceptible persons, compared with a mean of 13 mm Hg in controls, after rapid ascent to 4559 m. Furthermore, a capillary pressure of 19 mm Hg appears to be a threshold for the development of HAPE, because only individuals with pressures exceeding this value went on to develop HAPE during the study. 33 The development of increased permeability edema in the setting of elevated pulmonary capillary pressures seems to result from structural alterations in the barrier formed by the alveolar epithelium and capillary endothelium, proposed by West and colleagues as “pulmonary capillary stress failure.” 31 This concept describes the traumatic breakdown of the basement membrane and pulmonary capillary endothelium resulting from excessive intravascular pressures.32,50 Indeed, upon ultrastructural examination of pulmonary capillaries in thoroughbred racehorses, stress failure has been demonstrated in the setting of overt pulmonary hemorrhage as a result of extreme exercise. 51 Increased levels of proteins and erythrocytes have also been observed in the bronchoalveolar lavage fluid of elite athletes after intense exercise. 52 However, the activation of platelets and coagulation factors that would be expected to stem from exposure of the basement membrane in this model of stress failure only occurs in very advanced cases of HAPE. 53
Another mechanism for the alveolar fluid leak in HAPE may be a transient pressure-induced increase in the transvascular transport of fluid, erythrocytes, and proteins, without overt injury to the underlying structure. This may be mediated by the intercellular passage of fluid and proteins by relaxation of tight junctions or perhaps an increase in transcellular fluid flux via pores, fenestrae, and/or vesicular channels. 44 Indeed, studies in isolated rabbit lungs verify that hydrostatic edema with capillary pressures of 14 to 29 mm Hg is associated with numerous disruptions of the alveolar epithelial layers, with a relatively intact endothelial barrier. 54 Notably, in frogs with intravascular pressures high enough to rupture capillaries, more than 80% of the pressure-induced wall openings passed through endothelial cells, with the basement membrane remaining intact. 55 In both frogs 55 and rabbits, 56 these transcellular channels readily close when intravascular pressure is reduced to normal. These findings bear a conceptual similarity to the rapid reversibility of HAPE, which can even allow some afflicted persons to reascend to altitude after only a few days of descent and rest. From this evidence, we may conclude that as intravascular pulmonary capillary pressure increases in HAPE, dynamic changes are triggered in the structural permeability of the blood-gas barrier, allowing alveolar flooding to occur.
One question that remains a point of contention in the pathogenesis of HAPE is the etiology of increased capillary pressures that lead to PE. A commonly accepted theory is that the process of HPV is unevenly distributed in the pulmonary arteriolar bed, resulting in overperfusion of some regions of microvasculature that are not protected from high intravascular pressures by upstream vasoconstriction. 27 The role of increased pulmonary blood flow as an etiologic factor is supported by case reports in which the absence of the right pulmonary artery leads to the development of HAPE limited to the left lung after ascent to moderate altitudes.19,57 In a HAPE-susceptible individual who experiences an abnormal increase in PA pressure upon rapid ascent to high altitude, this results in portions of the pulmonary capillary bed being subjected to increased hydrostatic pressures that may cause stress failure. First proposed by Hultgren, 58 the concept of “uneven HPV” has been demonstrated in microsphere studies by nonuniform spatial distribution of HPV in pigs and cephalization of pulmonary perfusion in HAPE-susceptible individuals.59,60 Recent investigations using magnetic resonance imaging perfusion scanning reveal increased heterogeneity of pulmonary blood flow after hypoxic challenge in persons prone to HAPE,61,62 who also demonstrate an exaggerated exercise-induced increase in ventilation-perfusion mismatch. 25 These findings support the idea that, in susceptible persons, the hypoxia of high altitude causes an augmented and more unevenly distributed pulmonary vasoconstrictor response, which in turn may lead to regional overperfusion of pulmonary capillaries, transient structural changes in the permeability barrier, and resultant interstitial and alveolar edema.
An area of further interest in the pathophysiology of HAPE is the possible role of impaired alveolar sodium and fluid clearance as a permissive factor in the development of alveolar flooding. Studies in rat lungs demonstrate that hypoxia downregulates the expression and activity of sodium channels and Na+, K+-ATPase, as well as decreases apical sodium influx via amiloride-sensitive channels. 63 –66 Furthermore, mice with a targeted lesion in the gene coding for the alpha-subunit of the amiloride-sensitive sodium channel have significantly decreased alveolar transepithelial fluid clearance and higher risk of developing PE. 67 There are also data to suggest that impaired alveolar fluid clearance may contribute to the pathogenesis of HAPE in humans. 68
Prevention
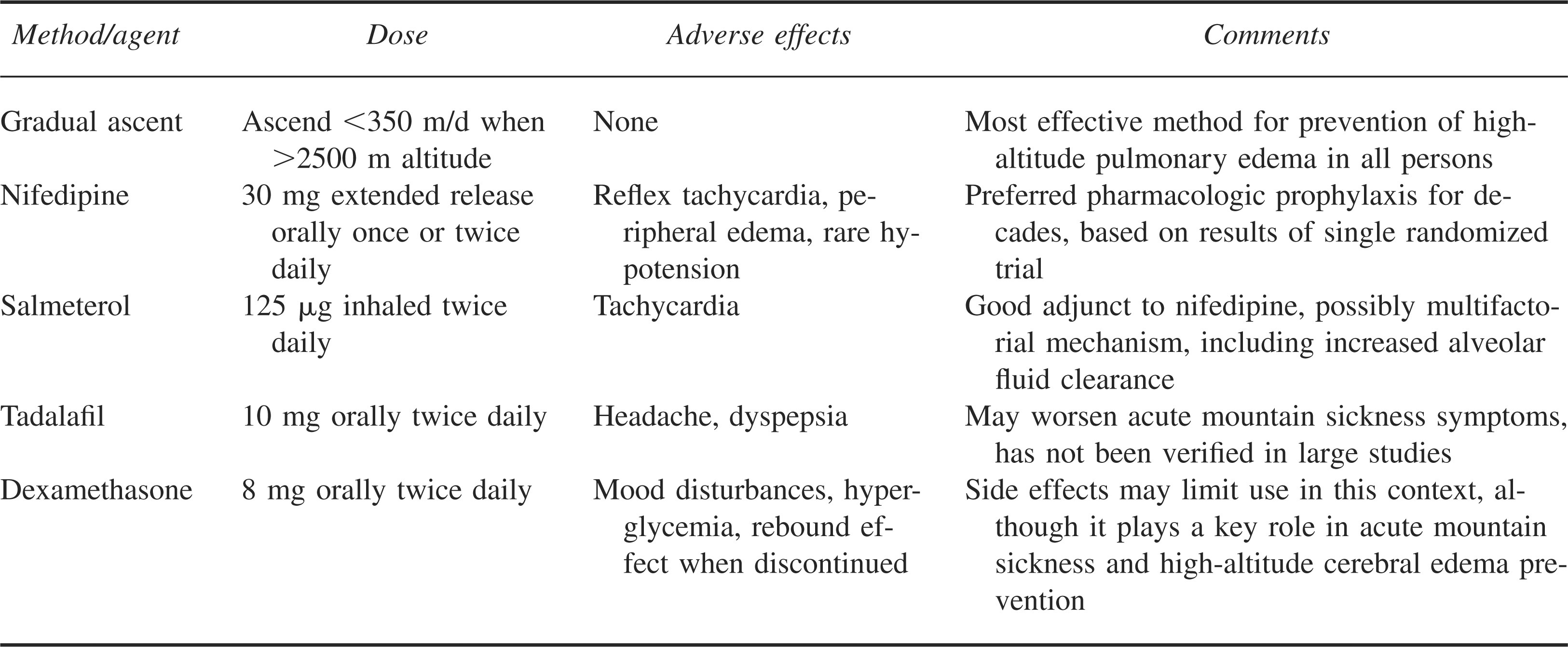
Gradual ascent remains the best method for prevention of HAPE and other high-altitude illnesses, with the recommended ascent rate of 300 to 350 m per day at altitudes above 2500 m. 13 These refer to sleeping altitudes, and an extra day of acclimatization should be added for every 600 to 1200 m above 2500 m. Although this strategy is optimal, pharmacologic prophylaxis is recommended as adjunctive therapy for persons with a prior history of HAPE and for those who must ascend more than 3000 m in a 24-hour period, as in some rescue situations. 69
With regard to prophylactic methods, inhibition of altitude-induced pulmonary hypertension has been the mainstay in pharmacologic prevention of HAPE. Nifedipine (30 mg extended release form orally once or twice daily), a calcium-channel blocker, has long been a recommended prophylactic agent, based on a single randomized double-blinded clinical trial. In HAPE-susceptible individuals, it mitigated the increase in PA pressure, reduced the incidence of HAPE, and improved gas exchange upon rapid ascent to 4559 m. 8
As potent pulmonary vasodilators via the NO/cGMP pathway, phosphodiesterase-5 inhibitors have been recently scrutinized as a prophylactic measure for HAPE. A 1-time dose of sildenafil (50 mg per oral [PO]) improves cardiac output and exercise capacity and mitigates the increase in PA pressure in healthy persons exposed to normobaric hypoxic challenge and ascent to 5400 m. 47 In a more recent study, sildenafil (40 mg PO 3 times daily) given to healthy subjects for several days after arrival to high altitude improved gas exchange and attenuated altitude-induced pulmonary hypertension without significant effects on systemic blood pressure. 48 Although neither of the sildenafil studies examined HAPE incidence, Maggiorini and colleagues demonstrated that prophylactic use of the phosphodiesterase-5 inhibitor tadalafil (10 mg PO twice daily, started 1 day prior to ascent) reduces the risk of developing HAPE by 65% in HAPE-prone individuals. 70 However, although tadalafil effectively relieved the altitude-induced increase in PA pressure, 2 of 10 individuals in the tadalafil group were incapacitated by severe headache after rapid ascent to 4559 m, which may reflect drug effect and/or worsening of AMS symptoms.
Maggiorini et al also compared dexamethasone (8 mg PO twice daily, started 1 day prior to ascent) with tadalafil in the prevention of HAPE in susceptible individuals. Dexamethasone, commonly used for both treatment and prevention of AMS, was predictably superior in preventing AMS (50% reduction) and decreasing AMS symptom severity. Surprisingly, dexamethasone, with a 78% reduction in HAPE incidence, was superior to tadalafil in the prevention of HAPE, reduction of PA pressure, and improvement of hypoxemia. 70 It appears that in order to protect against the development of HAPE, dexamethasone should be administered 1 day before ascent, because HAPE has occurred in persons who were given dexamethasone for treatment of AMS after ascent. 71 The mechanism by which dexamethasone prevents HAPE remains unclear. Maggiorini et al observed a significantly lower heart rate in the dexamethasone group of the above study, suggesting that it may contribute to protection from HAPE by attenuating sympathetic activation. 70 In animal studies, dexamethasone has been shown to increase expression and activity of eNOS, as well as cGMP concentration, which supports its possible role as a pulmonary vasodilator.72,73 Dexamethasone may also reduce transvascular fluid transport by “tightening” the pulmonary capillary endothelium. 74 Another suggested mechanism is a dexamethasone-mediated increase in alveolar fluid clearance by increased expression of epithelial sodium channels and augmented activity of Na+, K+-ATPase.75,76
Intriguing findings by Sartori and colleagues that prophylactic inhalation of the beta-adrenergic agonist salmeterol (125 μg twice daily) reduces the risk of HAPE by 50% in HAPE-prone persons support the role of impaired alveolar fluid resorption in the pathogenesis of HAPE. 68 Data show that beta agonists enhance transepithelial sodium and fluid transport during hypoxia and mechanical lung injury in animal studies64,77 and decrease extravascular lung water in humans with acute lung injury. 78 Although salmeterol may prevent HAPE by increasing fluid uptake through epithelial sodium channels and thus inhibiting alveolar flooding, it may also contribute via additional mechanisms, including tightening of endothelial intercellular junctions and the production of pulmonary NO.79,80
An area of budding interest in the prevention of HAPE is the effect of the carbonic anhydrase inhibitor acetazolamide, which, like dexamethasone, is already an accepted drug for prophylaxis and treatment of AMS. Berg and coworkers have shown its efficacy in preventing HAPE-like alveolar protein leak in rats exposed to hypobaric hypoxia, likely via mitigation of HPV. 81 In humans with no history of HAPE, Teppema et al demonstrated that acetazolamide (250 mg PO 3 times daily for 3 days prior to hypoxic challenge) decreases normoxic PVR and blunts the pulmonary vasoconstrictor response to hypoxia. 82 Other animal studies have verified acetazolamide's attenuation of HPV, which seems to be independent of carbonic anhydrase inhibition or effects on the NO vasodilation pathway but rather mediated by relaxation of the pulmonary vascular musculature. 83 –86 The applicability of these conclusions to the prevention of HAPE is yet unknown, but these recent findings warrant further study of acetazolamide as another drug that may provide dual prophylaxis against both AMS and HAPE.
For the prevention of HAPE in all individuals, we emphasize that gradual ascent is the optimal strategy (Table 2). However, we recommend oral nifedipine as adjunctive pharmacologic prophylaxis in those with a history of HAPE. With few significant adverse effects other than reflex tachycardia, it has withstood the test of time for protection from HAPE. Inhaled salmeterol is another optional adjunct for HAPE prevention, because it is logistically convenient and has only minor side effects due to increased beta-adrenergic tone. Cautious use of phosphodiesterase-5 inhibitors may be an acceptable alternative for selected individuals who have already been observed to tolerate these drugs without ill effects, because they may worsen symptoms of AMS, including headache. The finding of dexamethasone as an effective agent in the prevention of HAPE is intriguing, as are the preliminary studies with acetazolamide. Both drugs may prevent AMS and HAPE, 2 disorders that have traditionally required prophylaxis with separate agents (acetazolamide or dexamethasone for AMS and nifedipine for HAPE), because nifedipine does not confer protection from AMS. 87 The concept of one drug working to prevent both AMS and HAPE is of particular relevance to those persons with a history of HAPE who continue to pursue high-altitude endeavors on a frequent basis. However, because the use of dexamethasone is known to be associated with insomnia, euphoria, hyperglycemia, and adrenal suppression, it is not recommended as the initial choice for pharmacologic prevention of HAPE, except in rescue operations in which unacclimatized persons are deposited at high altitude, when it will also provide prophylaxis against AMS and high-altitude cerebral edema. Acetazolamide has not been studied as a preventive measure for HAPE in humans but warrants further study in this role, because it may prove useful for dual prophylaxis of AMS and HAPE.
Prevention of high-altitude pulmonary edema
Treatment
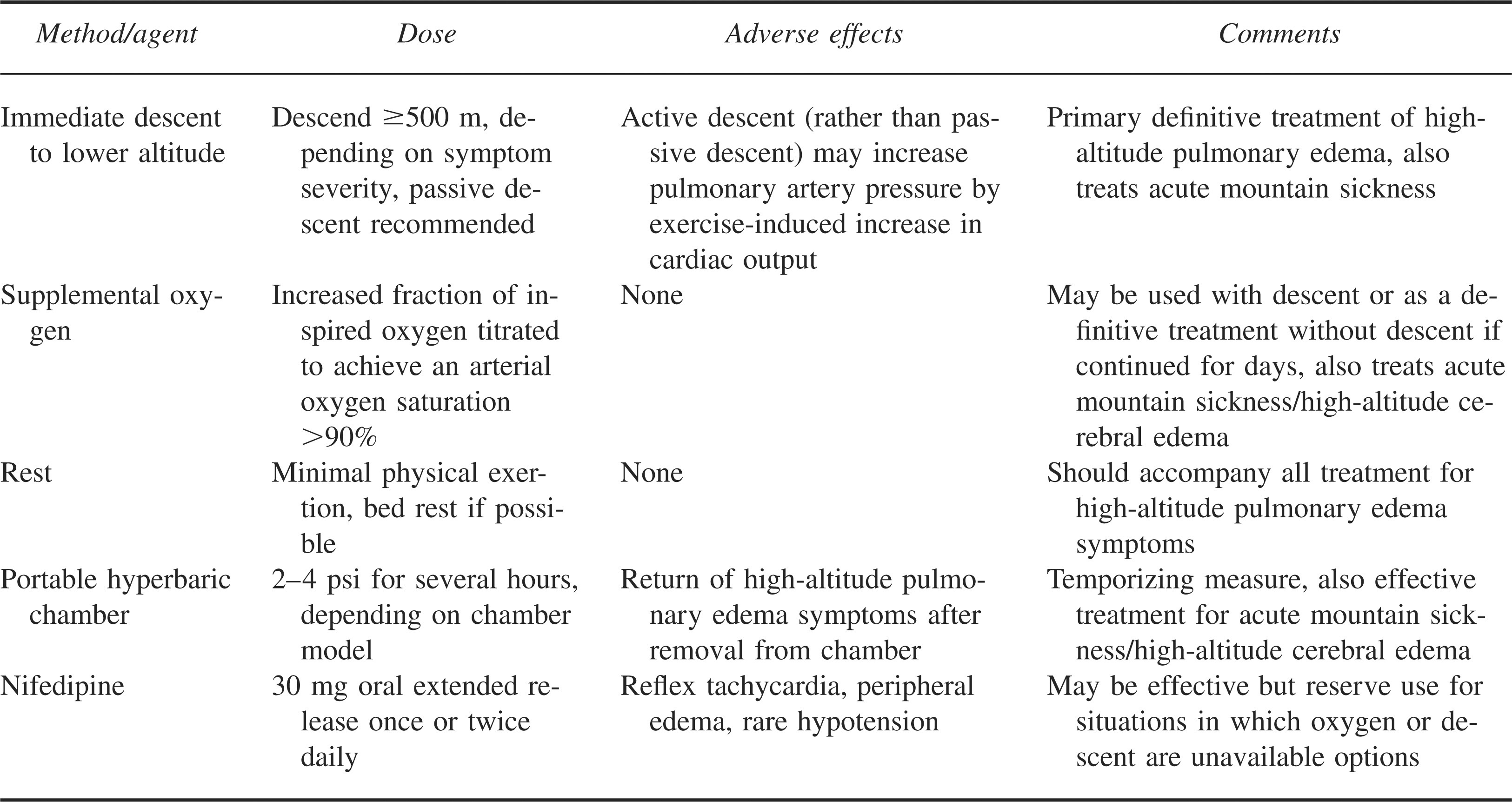
The most reliable treatment for HAPE is immediate descent (at least 500 to 1000 m), supplemental oxygen, or both. Supplemental oxygen and rest while remaining at high altitude are sufficient treatment for mild to moderate HAPE in persons without preexisting cardiopulmonary disease, and it is a common method of treatment for destination tourists who develop HAPE at high-altitude ski resorts.88,89 Oxygen should be provided at a high enough fraction of inspired oxygen to achieve an arterial oxygen saturation of greater than 90%. If oxygen is not available and descent is unsafe or impossible, a portable hyperbaric chamber can simulate a descent of 1500 m or more and is a good temporizing measure before definitive therapy is possible with descent to a lower altitude.90,91 Optimal therapy includes ensuring passive descent and keeping the patient warm, which will minimize any additional exercise- or cold-induced sympathetic contribution to the condition.
If none of the above methods is feasible for someone suffering from HAPE, adjunctive pharmacologic therapy may be considered, focusing primarily on reduction of PA pressure with vasodilators. Nifedipine (10 mg sublingually, then 20 to 30 mg PO extended release every 12 hours) can be considered but should not be regarded as a substitute for descent or oxygen. In a limited study of individuals with mild HAPE, Oelz and coworkers demonstrated that nifedipine decreased systolic PA pressure (by 50%), narrowed the alveolar-arterial oxygen gradient, and led to improving radiographic scores as PE cleared, although without a significant improvement in arterial oxygenation.92,93
Hackett and colleagues compared several vasodilators as treatment for HAPE, finding that nifedipine (10 mg sublingually) reduced mean PA pressure and PVR by about 30%, with improved oxygenation and slight decreases in mean arterial blood pressure. 42 Intravenous hydralazine (10 to 20 mg) behaved similarly, albeit with corresponding decreases in systemic blood pressure and heart rate. Interestingly, phentolamine infusion (1 mg/min for 10 minutes, then 0.5 mg/min for 20 minutes) induced significantly greater decreases in PA pressure and PVR, proving itself superior to nifedipine, hydralazine, or even oxygen. Phentolamine did, however, cause a 12.5 mm Hg drop in mean arterial pressure. When oxygen was added to phentolamine, the investigators observed a further reduction of 10 to 15% in PA pressure.
Nitric oxide, a pulmonary vasodilator already implicated in the pathogenesis of HAPE, has also been studied as a pharmacologic therapy for established HAPE. Scherrer and colleagues found that inhaled NO (40 ppm) markedly decreased systolic PA pressure and improved arterial oxygenation in individuals with HAPE and was also found to redistribute pulmonary blood flow to nonedematous regions of lung in these patients without significant effects on systemic blood pressure, minute ventilation, or end-tidal carbon dioxide. 94 More recently, Anand et al investigated the use of inhaled NO (15 ppm) with or without oxygen in patients with severe HAPE at a field hospital. The authors demonstrated similar effects of NO or oxygen alone on PA pressure and arterial oxygenation, with NO significantly more effective at decreasing PVR. When NO was combined with supplemental oxygen in participants with HAPE, the reduction in PA pressure and PVR, as well as the increase in oxygenation, was greater than with either gas alone. These additive effects of oxygen and inhaled NO on pulmonary hemodynamics and gas exchange are likely due to improvement in ventilation-perfusion matching in patients with HAPE. 95
Some researchers have investigated the use of expiratory positive airway pressure, which has been demonstrated to improve gas exchange in persons with HAPE, albeit transiently. 96 However, expiratory positive airway pressure, like inhaled NO, may present logistical difficulties in isolated mountain areas or hospitals at destination ski resorts where HAPE frequently occurs. Given the efficacy of inhaled beta agonists in the prevention of HAPE, as well as their convenience and minimal side effects, there may be a role for these agents in the treatment of HAPE, but this requires further study. For practical purposes, we recommend descent, supplemental oxygen, or both as the primary methods of HAPE treatment, using all other modalities of treatment, including pharmacotherapy, as adjunctive or temporizing measures (Table 3).
Treatment of high-altitude pulmonary edema
Although drugs may act as adjuncts to definitive therapy for HAPE, there is no abundant evidence for their efficacy and they may not always be readily available to a person who develops HAPE in the mountains. In our experience, treatment of HAPE with descent or supplemental oxygen to achieve an arterial oxygen saturation of 90% results in relief of symptoms and, given adequate time, resolution of PE and gas exchange abnormalities.