
Abstract
Connective tissue growth factor (CTGF) is a member of the CCN family of growth factors. CTGF is important in scarring, wound healing, and fibrosis. It has also been implicated to play a role in angiogenesis, in addition to vascular endothelial growth factor (VEGF). In the eye, angiogenesis and subsequent fibrosis are the main causes of blindness in conditions such as diabetic retinopathy. We have applied three different models of angiogenesis to homozygous CTGF−/− and heterozygous CTGF+/− mice to establish involvement of CTGF in neovascularization. CTGF−/− mice die around birth. Therefore, embryonic CTGF−/−, CTGF+/−, and CTGF+/+ bone explants were used to study in vitro angiogenesis, and neonatal and mature CTGF+/− and CTGF+/+ mice were used in models of oxygen-induced retinopathy and laser-induced choroidal neovascularization. Angiogenesis in vitro was independent of the CTGF genotype in both the presence and the absence of VEGF. Oxygen-induced vascular pathology in the retina, as determined semi-quantitatively, and laser-induced choroidal neovascularization, as determined quantitatively, were also not affected by the CTGF genotype. Our data show that downregulation of CTGF levels does not affect neovascularization, indicating distinct roles of VEGF and CTGF in angiogenesis and fibrosis in eye conditions.
C
However, combined exogenous administration of CTGF and VEGF in the back of a mouse or in a mouse model of hindlimb ischemia inhibits VEGF-induced angiogenesis because of the binding of VEGF by CTGF (Inoki et al. 2002; Jang et al. 2004). When CTGF is upregulated by VEGF (Suzuma et al. 2000; He et al. 2003), it can reduce the bioavailability of VEGF by VEGF—CTGF binding. Furthermore, we recently demonstrated a strong and highly significant correlation of human vitreous CTGF levels with degree of fibrosis, but not with neovascularization, in a large series of patients with various vitreo-retinal disorders, including PDR (Kuiper et al. 2006).
On the basis of the latter findings, we want to challenge the concept that CTGF is an intrinsic ocular angiogenesis factor. We hypothesize that in the eye, CTGF is primarily a pro-fibrotic factor and that CTGF is not essential for angiogenesis.
To test our hypothesis, we investigated the role of CTGF in three angiogenesis models in transgenic mice with absolute or relative CTGF deficiency (CTGF−/− or CTGF+/−, respectively). In these mice, we investigated whether CTGF is necessary for spontaneous and VEGF-induced angiogenesis in embryonal bone explants in vitro (Deckers et al. 2001; Van der Pluijm et al. 2003), in oxygen-induced retinopathy in neonatal mice (Agostini et al. 2005), and in laser-induced choroidal neovascularization in adult mice (Lambert et al. 2001, 2003).
Materials and Methods
Genetically Modified Mice
Male BalbC/129sv CTGF+/− mice (Ivkovic et al. 2003) were crossbred with CTGF+/− female C57Bl/6J mice (Harlan; Horst, The Netherlands). Seventeen-day-old embryos (CTGF+/−, CTGF+/+, and CTGF−/−) were removed from pregnant CTGF+/− mice, and metatarsals were dissected as described previously (Deckers et al. 2001). In addition, male BalbC/129sv CTGF+/− mice were crossbred with CTGF+/+ female C57Bl/6J mice (Harlan). The offspring (CTGF+/− and CTGF+/+ mice) were used for the ocular angiogenesis studies. Homozygous knock-outs (CTGF−/−) were not available for these experiments, because these mice are not viable after birth (Ivkovic et al. 2003).
All mice were genotyped using PCR. Animal experiments were performed in compliance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. The animals were maintained under a 12:12-hr light/dark cycle and had free access to food and water.
In Vitro Angiogenesis Assay of Embryonal Mouse Metatarsals
Metatarsals were dissected, cultured, and stained as previously described (Deckers et al. 2001; Van der Pluijm et al. 2003). Briefly, four left metatarsals and four right metatarsals of each of 17 CTGF+/−, 4 CTGF+/+, and 2 CTGF−/− embryos were isolated and separately cultured in 24-well plates for 14 days. The experiment was conducted in triplicate (metatarsals of three breeds). After 72 hr of culture, the 10% fetal calf serum—containing medium was replaced by serum-free medium with (right metatarsals) or without (left metatarsals) 50 ng/ml recombinant human VEGF-A (Oncogene; Sanbio, Uden, The Netherlands). At day 14, the cultures were fixed in zinc macrodex formalin (ZnMF) fixative containing 0.1 mmol/liter Tris-acetate, pH 4.5, 0.5% ZnCl2, 0.5% Zn acetate, 5% dextran, and 3.8% paraformaldehyde for 15 min and subsequently stained using a rat anti-mouse monoclonal antibody against an endothelial cell marker [CD31, PECAM-1, or ER-MP12; kindly provided by Pieter J. M. Leenen (Slieker et al. 1993)]. Following this, metatarsals were washed three times in PBS and then incubated with 40% methanol/1% H2O2 in PBS for 30 min at room temperature to abolish endogenous peroxidase activity. After three washes with PBS, nonspecific staining was blocked by incubation in a solution of 0.3% skimmed milk powder (Boehringer; Mannheim, Germany) in TTBS (50 mmol/liter Tris-HCl, pH 7.4, 150 mmol/liter NaCl, and 0.05% Tween 20) for 1 hr at 37C, followed by incubation with the ER-MP12 antibody (dilution, 1:33) overnight at 4C.
The following day, after three washes in 100 mmol/liter Tris-HCl, pH 7.4, 150 mmol/liter NaCl, and 0.05% Tween 20, cultures were incubated with biotinylated sheep anti-rat secondary antibody (dilution, 1:300; Amersham, Buckinghamshire, UK) for 45 min and streptavidin-horseradish peroxidase complex for 30 min at 37C. Peroxidase activity was visualized using its chromagenic substrate 3-amino-9-ethyl carbazole (Sigma; St. Louis, MO).
Mouse Model of Oxygen-induced Retinopathy
Retinopathy was induced as previously described (Agostini et al. 2005). Briefly, 7-day-old litters and the mother were kept for 5 days in a sealed container ventilated by a mixture of oxygen and compressed air with a final oxygen concentration of 76 ± 2% (Oxygen Monitor 180, Kontron Instruments, Bletchley, UK; sensor, Linde Heimox; Unterschleissheim, Germany). Afterwards, animals were kept under normal conditions for 6 days. On postnatal day 18, the litters were deeply anesthetized by inhalation of 2.5–3.5% isoflurane mixed with oxygen, with a flow of 2 liters/min in a vaporizer (Vapor 19.3; Dräger, Lubeck, Germany). Retinal vessels of the litters were visualized by perfusion with fluorescein isothiocyanate (FITC) coupled to high-molecular-weight dextran (FD 2000 S; Sigma-Aldrich, Steinheim, Germany) dissolved in 0.9% NaCl at a concentration of 50 mg/ml. Perfusion was performed by injecting 1 ml of the fluorescein solution into the right atrium with a 30-gauge cannula, after carefully opening the thorax. Three to 4 min after perfusion, the flow of isoflurane was increased and the mice were sacrificed. Eyes were enucleated, and pieces of the tail were collected for PCR genotyping.
After enucleation, the eyeballs were fixed for 4 hr in 4% buffered formaldehyde at room temperature. After removal of the anterior segment, retinas were mounted on siliconized microscope slides (SuperFrost Plus; Menzler-Gläzer, Braunschweig, Germany) using Vectashield mounting medium (Vector; Burlingame, CA), an anti-fading agent, and 15 mmol/liter NaN3 (Dako; Glostrup, Denmark). Retinal whole mounts were covered by a coverslip and kept in the dark at 4C until analysis.
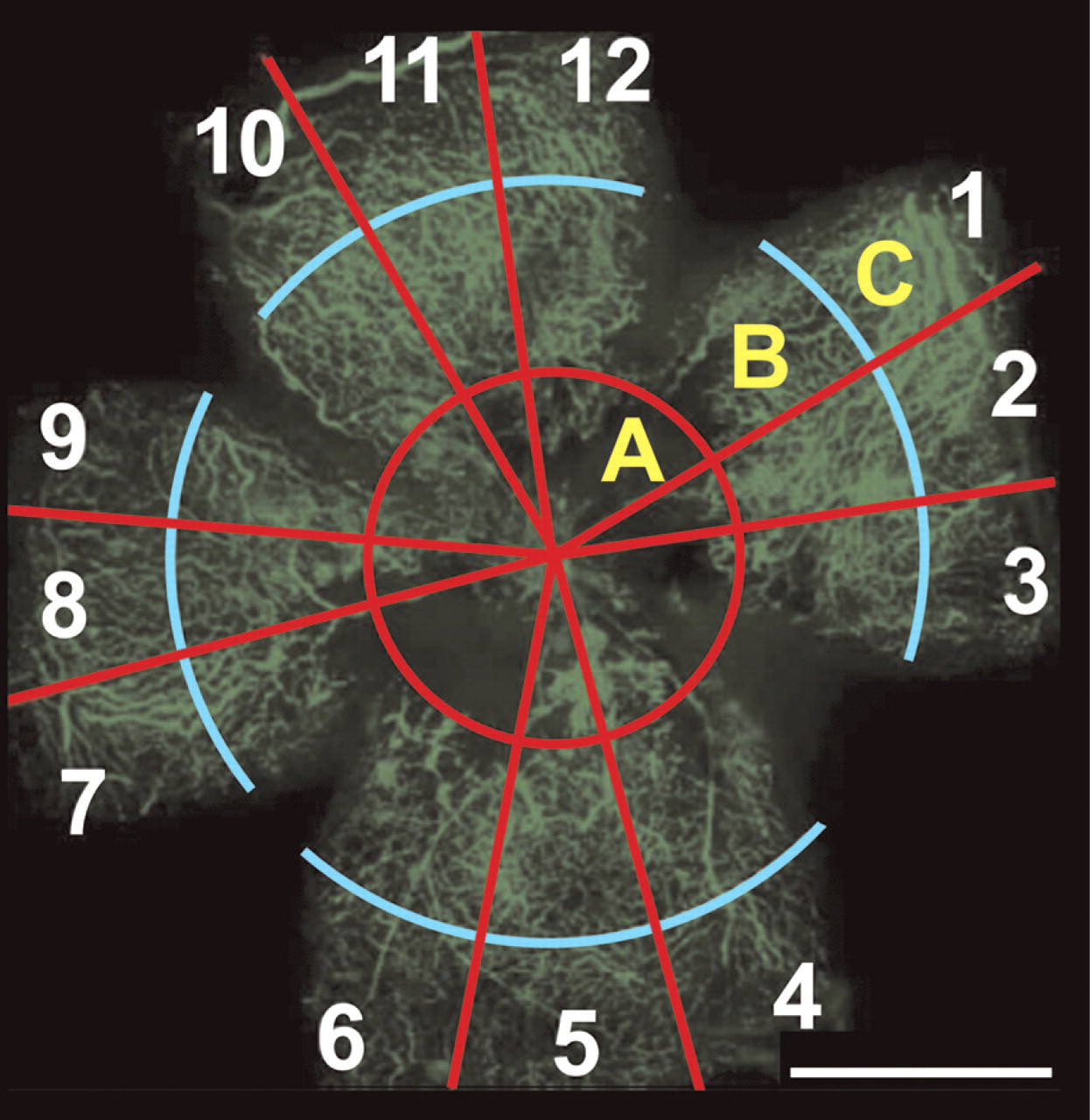
Retinal flat mount of mouse eye with oxygen-induced retinopathy showing blood vessels after perfusion with fluorescein isothiocyanate (FITC) coupled to high-molecular-weight dextran. The retina was divided into three concentric zones: the inner zone around the optic disc (
Criteria for retinal vascular pathology scores in retinal whole mounts in oxygen-induced retinopathy
Mouse Model of Laser-induced Choroidal Neovascularization
Choroidal neovascularization was induced in mice by laser burns as previously described (Lambert et al. 2001, 2003). Briefly, mice were anesthetized with an intraperitoneal injection of Avertin (Aldrich; Milwaukee WI). Both pupils were dilated with topical administration of 0.5% tropicamide (0.5% Tropicol; Bournonville Pharma, The Hague, The Netherlands) and four burns were delivered at the 3, 6, 9, and 12 o'clock positions around the optic disc, using a green argon laser (532 nm; 50-μm diameter of the spot size; 0.05-sec duration; 400 mW; Figure 2) and a coverslip as contact lens. Animals were sacrificed after 14 days.
To quantify choroidal neovascularization, mice were injected intravenously with 200 μl of 50 mg/ml FITC-conjugated dextran (Sigma) in PBS (pH 7.4). Immediately after, one eye per mouse was fixed in 1% paraformaldehyde, pH 7.4, for 60 min at room temperature. Retinas were removed, and the choroid was mounted flatly for confocal microscopy using Vectashield mounting medium. The other eye was fixed in buffered 3.5% paraformaldehyde and frozen in liquid nitrogen for cryostat sectioning.
In addition, choroidal neovascularization was also quantified by confocal visualization of flat-mount choroids. The retinas were removed, and the choroid was mounted flatly for confocal microscopy using anti-fading Vectashield mounting medium. The spatial distribution of FITC fluorescence was examined using a Leica TCS SP2 inverted confocal laser microscope (Leica Microsystems; Mannheim, Germany) equipped with an argon laser and an acousto-optical tunable filter for excitation intensity. Digitized images were acquired using a 10× [numerical aperture (NA) 0.4] objective or a 63× (NA 1.2) Plan-Apo water-immersion objective at 1024 × 1024 pixel resolution. FITC was visualized by using an excitation wavelength of 488 nm. Emission light was recorded at 500–555 nm. For each lesion, serial optical sections were recorded with a z-step of 1.67 μm. After successive scanning for each interval, three-dimensional images of fluorescence were reconstructed by using Leica confocal software. Images were acquired under identical conditions, and we ensured that the maximal fluorescence signal was not saturating the photo-multiplier tubes. Captured images were exported as TIFF files and processed further using Photoshop software (Adobe; San Jose, CA). Quantification of the confocal images was realized by measurement of the surface areas of fluorescence (Scion Image for Windows beta 4.0.2; Scion, Frederick, MD).
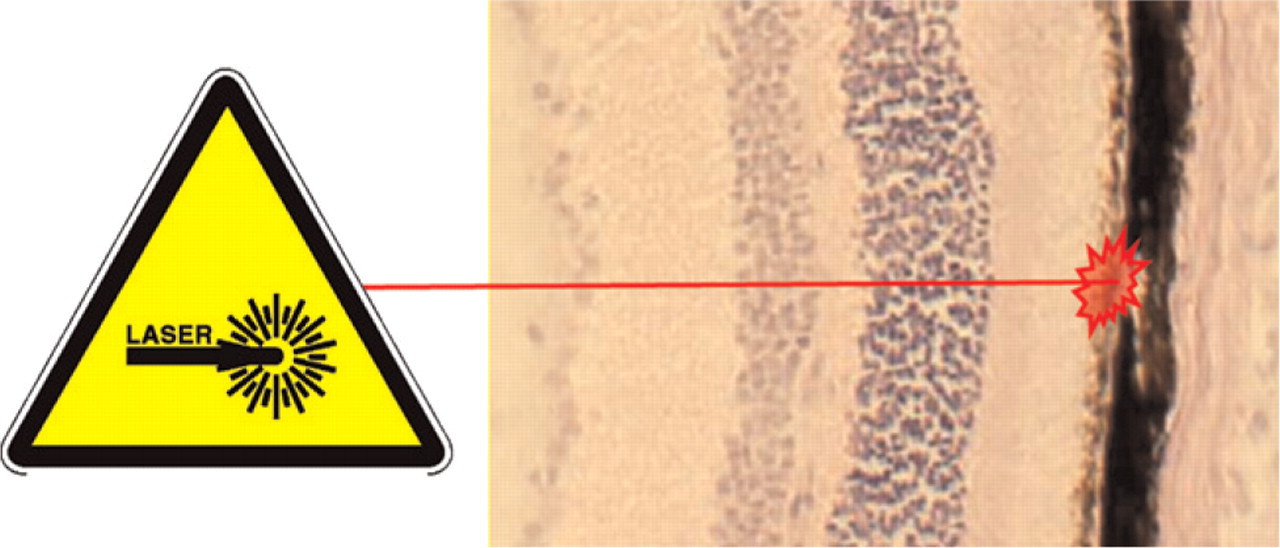
Induction of choroidal neovascularization in mice by laser photocoagulation.
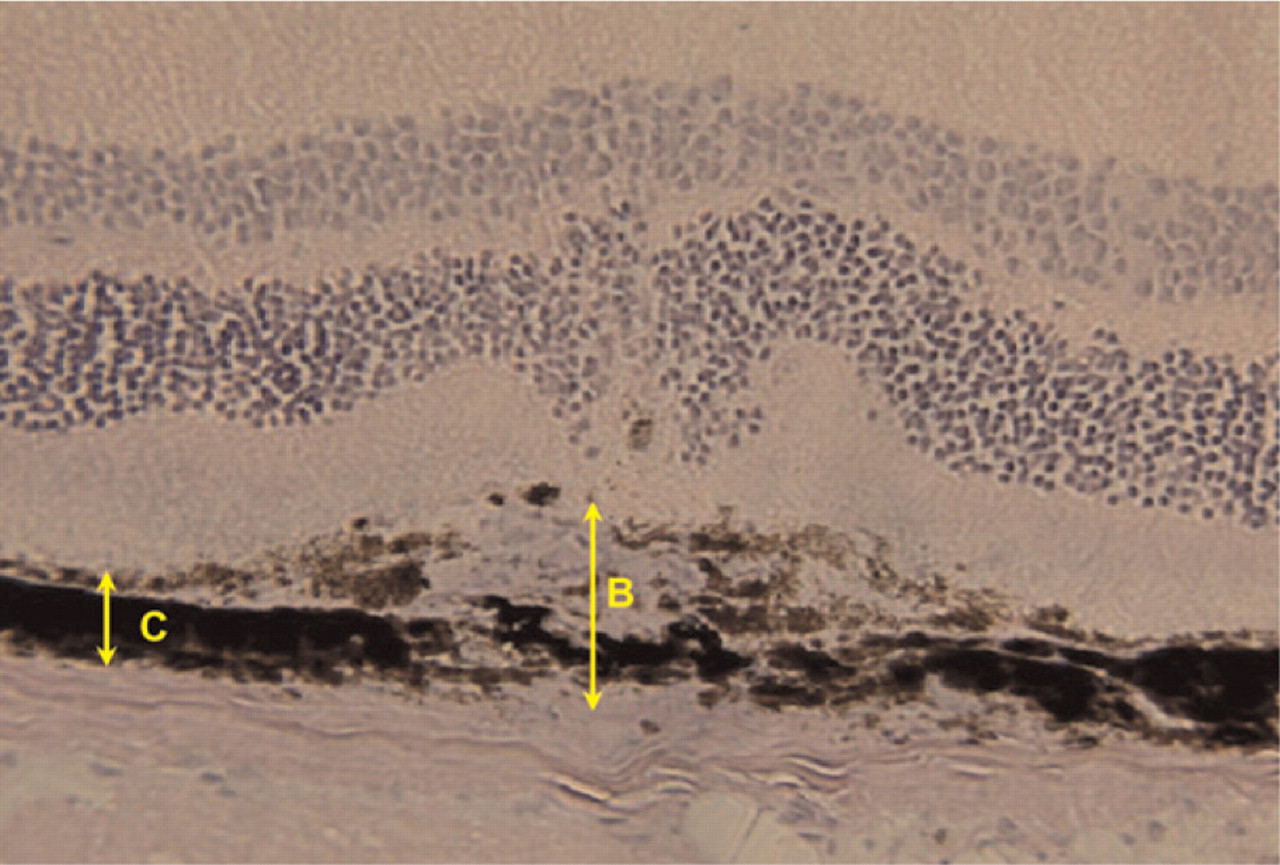
Estimation of choroidal neovascularization after induction by laser photocoagulation in hematoxylin-stained cryostat sections of the area containing the entire extent of the burn in mouse eyes. Neovascularization is determined by the ratio of the thickness of the lesion (B) to the adjacent intact pigmented choroidal layer (C).
Statistics
Differences between metatarsals incubated in the presence or absence of VEGF were calculated using the nonparametric paired Wilcoxon signed rank test. Data were corrected for age differences between breeds as determined by differences in degree of calcification of the bones and for the fact that part of the data were obtained from metatarsals of one mouse (repeated measurements). Thus, results for CTGF+/+, CTGF+/− and CTGF−/− mice (genotype effects and effects of VEGF) were calculated using a repeated measurement model with metatarsal as repeated subject with correction for degree of calcification. Results were considered significantly different at
Scores of retinal vascular changes in retinal whole mounts were not normally distributed and therefore were expressed as median and range. Differences between scores of retinal vascular changes and choroidal neovascularization in CTGF+/+ and CTGF+/− mice were statistically analyzed using the nonparametric Mann-Whitney test with
Results
In Vitro Angiogenesis Assay
In total, 92 metatarsals of 4 CTGF+/+, 17 CTGF+/−, and 2 CTGF−/− mouse embryos were cultured in the presence or absence of VEGF, and each of these metatarsals demonstrated outgrowth of vessels (Figure 4). Owing to failure of attachment of some of the metatarsals, 15 out of 16 CTGF+/+, 60 out of 68 CTGF+/−, and 7 out of 8 CTGF−/− metatarsals incubated in the absence of VEGF, and 15 out of 16 CTGF+/+, 50 out of 68 CTGF+/−, and 5 out of 8 CTGF−/− metatarsals incubated in the presence of VEGF were analyzed. Metatarsals of CTGF+/+, CTGF+/−, and CTGF−/− embryos showed similar outgrowth of vessels. A genotype effect could not be detected, whether treated with VEGF or not (treated group,
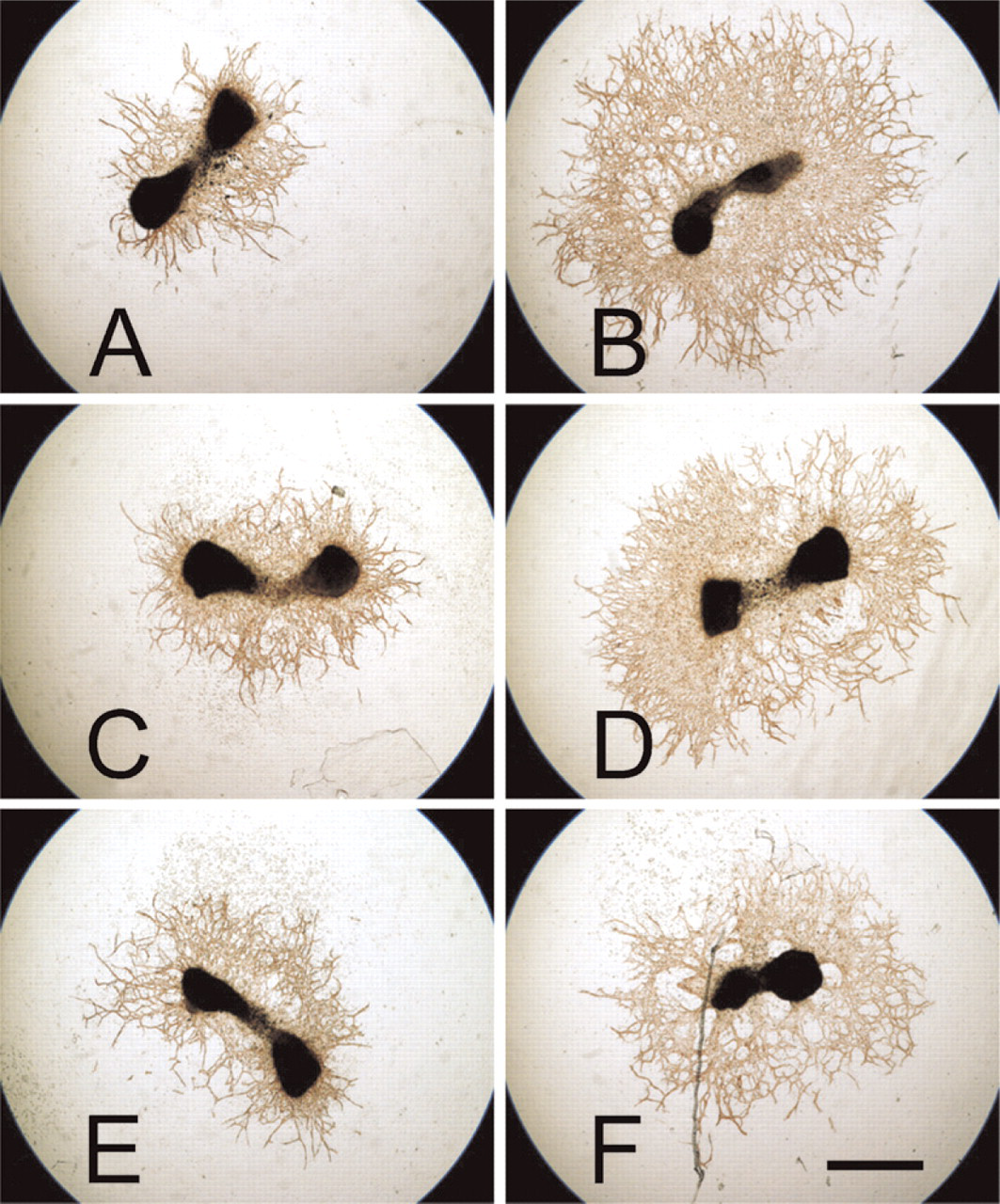
Vascular outgrowth of metatarsals as stained immunohistochemically with anti-CD31 antibodies after being cultured in the absence (
Oxygen-induced Retinopathy
In total, 13 CTGF+/+ mice (26 eyes) and 8 CTGF+/− mice (16 eyes) were analyzed by FITC—dextran perfusion to examine the retinal response after oxygen exposure (Figure 1). Seven eyes of CTGF+/+ mice and one eye of a CTGF+/− mouse were excluded from analysis because of incomplete perfusion. In total, 19 CTGF+/+ and 15 CTGF+/− retinal whole mounts were analyzed using the scoring system shown in Table 1. There was no difference in median score of the retinal vascular changes between the CTGF+/+ retinal whole mounts (5.0; range, 1–9) and the CTGF+/− whole mounts (6.0; range, 4–10; Mann Whitney,
Laser-induced Choroidal Neovascularization
In total, four CTGF+/+ mice and six CTGF+/− mice received four laser burns per eye. Twelve out of the 16 spots in the eyes of the CTGF+/+ mice and 20 of the 24 spots in the eyes of the CTGF+/− mice induced neovascularization and were included in the analysis of the B/C ratios in cryostat sections.
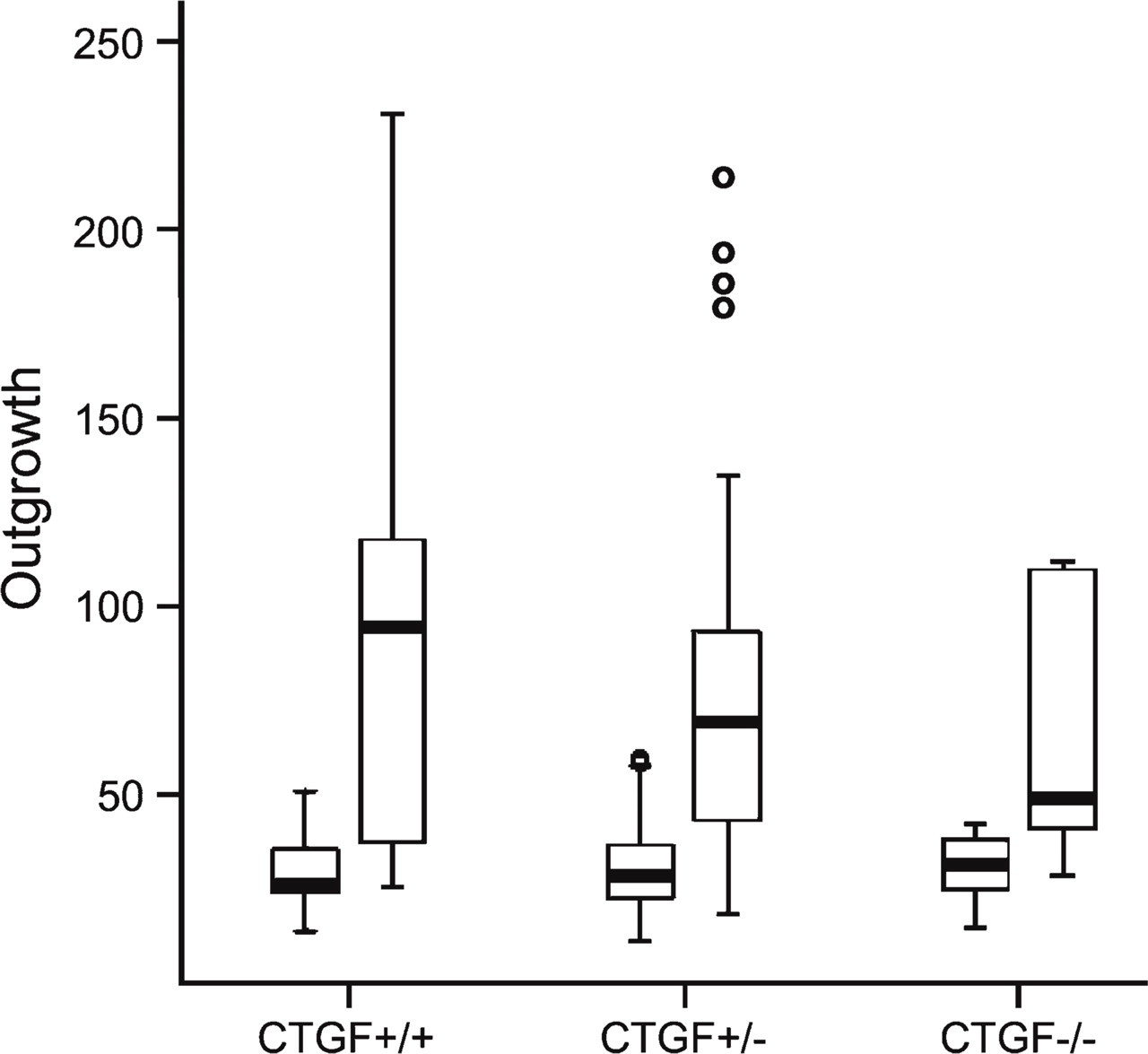
Graphic representation of the differences in median area of vascular outgrowth (expressed as numbers of pixels × 1000) between the metatarsals cultured with and without VEGF for CTGF+/+, CTGF+/−, and CTGF−/− mice. Data are expressed as median with range. The box indicates values from the 25th to the 75th quartile.
Figure 7 shows confocal images of FITC—dextran-perfused mouse retinas. Only 8 of 16 choroidal impact areas of CTGF+/+ mice and 11 of 24 choroidal impact areas of CTGF+/− mice could be included for quantitative analysis. Background fluorescence was too high in the areas of the other impacts. Neither quantification method showed statistical differences between experimental choroidal neovascularization in CTGF+/+ and CTGF+/− mice (Mann Whitney,
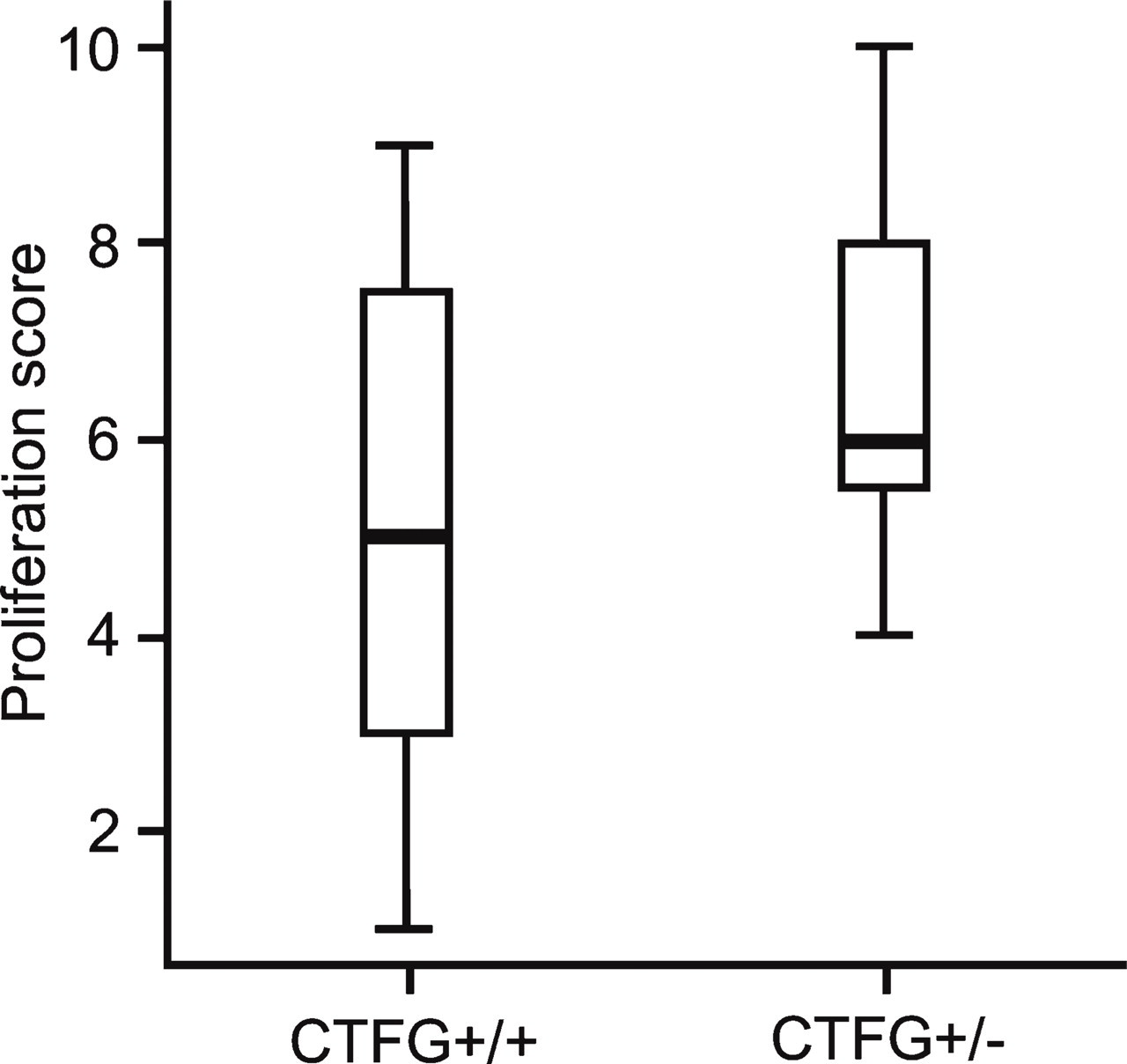
Median scores with range of vascular pathology in retinal whole mounts of CTGF+/+ and CTGF+/− mice in the oxygen-induced retinopathy model. There is no significant difference in neovascular response between CTGF+/+ and CTGF+/− mice.
Discussion
The observations in the mouse models in the present study are in line with our previous findings in patients that intravitreal CTGF levels do not correlate with degree of neovascularization (Kuiper et al. 2006). Vascular outgrowth from embryonic metatarsal explants in vitro of homozygous CTGF−/− knock-out mice did not differ significantly from vascular outgrowth from metatarsals of wild-type CTGF+/+ mice or heterozygous CTGF+/− mice, in both the presence and absence of VEGF. However, the large variation in all experimental groups that were incubated in the presence of VEGF may hide a different response in the three genotypes. Nevertheless, our data indicate that CTGF is not required for (VEGF-induced) angiogenesis in this model. Our findings do not exclude that CTGF on its own and/or excessive levels of (exogenous) CTGF may induce angiogenesis, but redundancy of CTGF in ocular angiogenesis in vivo is also suggested by the lack of significant differences between heterozygous mice and wild-type mice in the two ocular angiogenesis models. Heterozygous CTGF+/− mice have been identified as a valid model of decreased CTGF availability, because they express ∼50% lower protein levels of CTGF (Kuiper 2006; Roestenberg 2007). Furthermore, these mice fail to develop a thickened glomerular basement membrane during experimental diabetes mellitus (Roestenberg 2007).
The fact that angiogenesis is not impaired in the CTGF knock-out mouse is novel and surprising. Previous studies have shown that CTGF is sufficient to induce angiogenesis, directly or indirectly, under experimental conditions in vitro and in vivo (Aiello et al. 1998; Babic et al. 1999; Ferrara 1999a, b; Shimo et al. 1999; Campochiaro and Hackett 2003). In these studies, exogenous CTGF induced angiogenesis in rat cornea (Babic et al. 1999), in chicken chorio-allantoic membrane, and in rCTGF-containing implants in the backs of mice (Shimo et al. 1999). Our findings in human PDR (Kuiper et al. 2006) and in three distinct angiogenic mouse models as described here question the applicability of this concept to ocular disease. The ocular models used in our study mimic pathological conditions occurring in the human eye. In the oxygen-induced retinopathy model, preretinal angiogenesis is caused by retinal hypoxia and VEGF overexpression, a situation similar to retinopathy of prematurity and PDR (Witmer et al. 2003). In the laser-induced model of choroidal neovascularization, the angiogenic response is part of a wound-healing response after disruption of Bruch's membrane, a situation similar to myopic and age-related macular degeneration. Our data suggest that as far as the final angiogenic response is concerned, CTGF is a dispensable factor in the complex interplay of hypoxic signaling and VEGF- and wound-healing—driven angiogenesis. Our previous studies indicated that CTGF plays a role in the later stages of PDR, after neovascularization, and induces subsequent fibrosis (Kuiper et al. 2006). CTGF can be induced by increased VEGF (Suzuma et al. 2000; He et al. 2003) and/or advanced glycation end products (Hughes et al. 2007) that are the result of diabetic conditions.
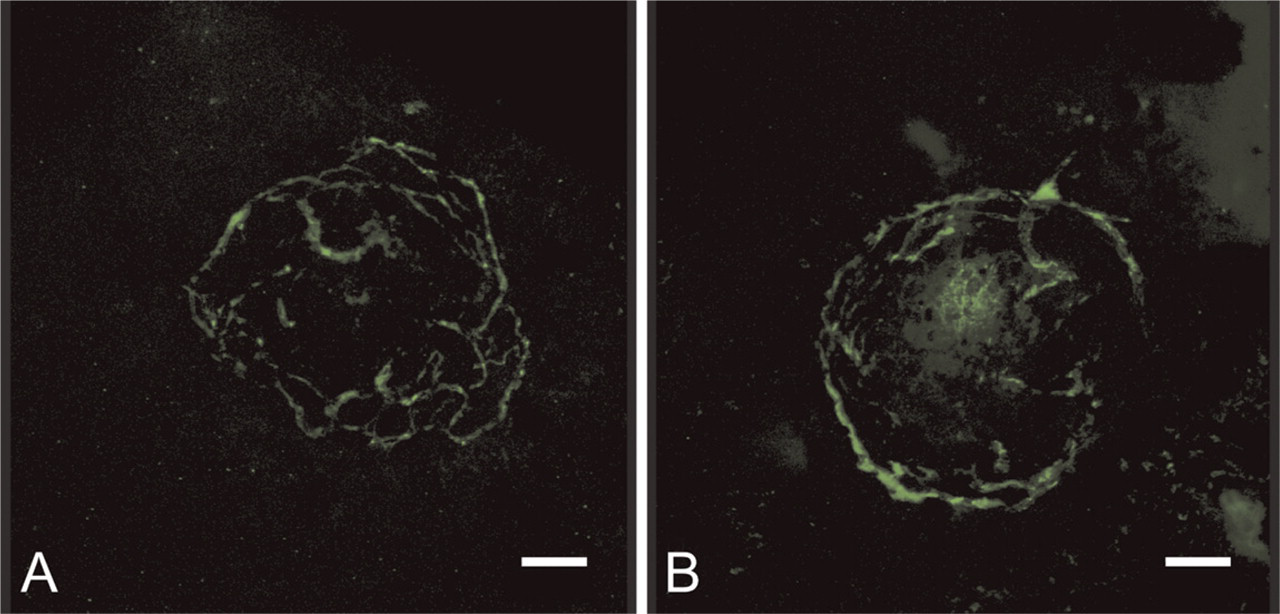
Confocal images of FITC—dextran-perfused retinas showing the vasculature in a CTGF+/+ (
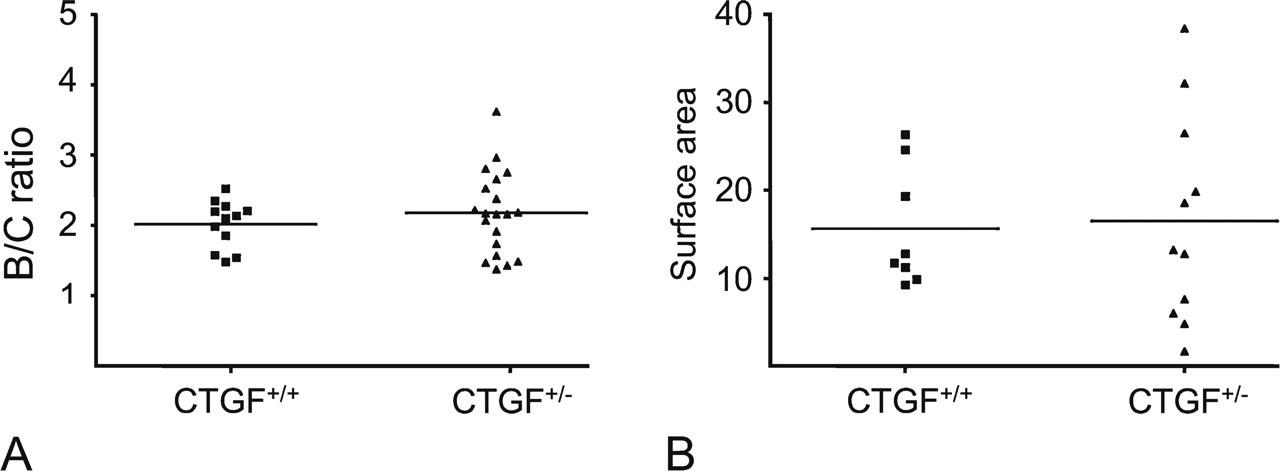
Induction of choroidal neovascularization by laser in retina of CTGF+/+ and CTGF+/− mice. There is no difference between the two groups as determined by B/C ratio (
The literature supports this concept. In other organs, the role of CTGF as a pro-fibrotic factor is well-established (Franklin 1997; Grotendorst 1997; Duncan et al. 1999; Mori et al. 1999; Goldschmeding et al. 2000; Yokoi et al. 2001, 2004; Perbal 2004; Van Nieuwenhoven et al. 2005). Furthermore, it has been shown recently that hypoxic induction of CTGF is directly mediated by the transcription factor hypoxia-inducible-factor-1 (HIF-1; Higgins et al. 2004), whereas CTGF inhibits VEGF-dependent angiogenesis by promoting proteasomal HIF-1α degradation in tumors (Chang et al. 2006; Smith et al. 2006; Tosetti et al. 2006). This shows that the interplay between VEGF and CTGF in angiogenesis and fibrosis is complex, but evidence is accumulating that CTGF does not induce angiogenesis.
In the eye, CTGF (mRNA and protein) is upregulated by VEGF in retinal endothelial cells (Suzuma et al. 2000; He et al. 2003) and colocalizes with VEGF in neovascular subretinal membranes of age-related macular degeneration patients (Watanabe et al. 2005). In these membranes, CTGF is also found in retinal pigment epithelial cells (Watanabe et al. 2005), which can transdifferentiate into myofibroblasts, the main cell type driving fibrosis. CTGF expression has also been found in myofibroblasts in fibrovascular membranes of patients with PDR (Watanabe et al. 2005) and in pericytes in the retina of PDR patients (Kuiper et al. 2004). Pericytes and myofibroblasts are closely related cell types.
In summary, our data, obtained under various experimental conditions in vivo and in vitro, show that CTGF gene expression, or the level of expression, is not a critical determinant for angiogenesis induced by VEGF, hypoxia, or tissue damage. However, our results do not exclude that CTGF by itself can induce angiogenesis under certain conditions. These data and those from our previous studies suggest that CTGF drives fibrosis rather than angiogenesis in pathological eye conditions.
Footnotes
Acknowledgements
This study was supported by a grant from the Diabetes Fonds Nederland (Grant 2001.042), the Edmond and Marianne Blaauwfonds, and by Les Amis des Aveugles (Ghlin).
The authors thank Lotte Wieten (Department of Pathology, University Medical Center, Utrecht, The Netherlands) for technical assistance, Patrick Motte (Unité de Biologie Cellulaire Végétale, Department of Life Sciences, University of Liege, Belgium) for the confocal imaging, and Dr. Pieter J.M. Leenen (Department of Immunology, Erasmus University, Rotterdam, The Netherlands) for the anti-mouse endothelium antibody.