
Abstract
The neuronal apoptosis inhibitory protein (NAIP) gene, also known as the baculovirus inhibitor of apoptosis repeat-containing protein 1 (BIRC1) gene, is a member of the inhibitors of apoptosis (IAP) family and was first characterized as a candidate gene for spinal muscular atrophy (SMA). The expression of NAIP has been thoroughly studied in the central nervous system and overlaps the pattern of neurodegeneration in SMA. Recent studies have pointed to a role for NAIP in non-neuronal cells. We report here the production of a specific anti-NAIP antibody and the profile of NAIP expression in human adult tissues by Western blot and immunohistochemical detection methods. NAIP was detected in a number of tissues by Western blot analysis, but immunohistochemistry revealed that NAIP's presence in certain tissues, such as liver, lung, and spleen, is most likely due to macrophage infiltration. In the small intestine, the expression of NAIP coincides with the expression of p21WAF1. This observation, coupled with findings from other groups, suggests a role for NAIP in increasing the survival of cells undergoing terminal differentiation as well as the possibility that the protein serves as an intestinal pathogen recognition protein. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
Keywords
P
The distribution of NAIP-like immunoreactivity was examined in the rat CNS by using an affinity-purified polyclonal antibody (E1.0) against human NAIP (Xu et al. 1997b). This investigation demonstrated a wide distribution of NAIP-like species in the rat CNS. Although low-to-moderate levels of NAIP-like immunoreactivity were observed in the cortex and hippocampus, intense staining was detected within a wide variety of subcortical brain regions (Xu et al. 1997b). These included the thalamus, cranial nerve nuclei, brainstem relay nuclei, cerebellum, and Clarke's column of the spinal cord. With the exception of the striatum, all of these regions display pathologic hallmarks in severe cases of SMA (Xu et al. 1997b). Further studies in human neuronal tissues utilizing the same antibody were performed (Pari et al. 2000), demonstrating high immunoreactivity only in spinal cord motor neurons and in pyramidal cells of the motor cortex (Pari et al. 2000). The highest expression of NAIP was found in the cells of the choroid plexus. Interestingly, choroid plexus is a CNS site where peripheral macrophages can proliferate and gain entry to the brain. A study by Gotz et al. (2000) looked at the developmental expression and tissue distribution of six isoforms of NAIP mRNA in adult mouse tissues. Full-length NAIP mRNA was widely expressed in all tissues analyzed.
Prenatal studies showed expression of NAIP mRNA in the brain and spinal cord of E9.5-E14.5 mouse embryos (Ingram-Crooks et al. 2002). NAIP transcripts were found in the marginal zone of the lateral ventricle, the follicles of the vibrissae, the retina, and intestinal villi at a later stage of development (E16.5) (Ingram-Crooks et al. 2002). Northern blot analysis on adult mice showed a predominant expression in the liver, lung, spleen (Matsumoto et al. 1999; Diez et al. 2000), intestine, and macrophage (Diez et al. 2000). In human adult tissues, NAIP transcripts were detected using Northern blot analysis in placenta, liver (Roy et al. 1995; Yamamoto et al. 1999), spleen, lung, and peripheral blood leukocytes (Yamamoto et al. 1999), with minimal expression in a number of other tissues, including fetal tissues (Yamamoto et al. 1999).
Although the studies on the protein's expression in neurons are consistent with an SMA-modulating role for NAIP specifically and a CNS-protective effect generally, the presence of transcripts in other tissues suggests that NAIP might have a broader function. The deletion of NAIP was found to influence cell survival (Liston et al. 1996; Xu et al. 1997a), and its neuroprotective role was revealed by the increased apoptotic vulnerability of CA1 hippocampal neurons to kainate-induced seizure in mice null for NAIP1 (Holcik et al. 2000). It has been shown that NAIP is capable of inhibiting the effector caspases-3 and −7 (Hutchison et al. 2001; Maier al. 2002) and can associate with caspase-9 (Davoodi et al. 2004).
In an analysis of adipocyte differentiation, a peak of NAIP expression was observed at day 4 of an 8-day-long differentiation process (Magun et al. 1998). This peak correlated with an enhanced resistance against apoptosis induced by growth factor deprivation (Magun et al. 1998). NAIP has been shown to be a determining factor in macrophage infection with Legionella pneumophila (Diez et al. 2000, 2003), the causative agent of Legionnaires' disease (Fraser et al. 1977; McDade et al. 1977). Additionally, NAIP has been found to be expressed in the granulosa cells of developing ovarian follicles (Matsumoto et al. 1999). The suppression of NAIP expression using antisense oligonucleotides reduced the number of morphologically normal oocytes (Matsumoto et al. 1999). These latter studies (Magun et al. 1998; Matsumoto et al. 1999; Diez et al. 2000) clearly indicate NAIP expression in a cellular context that is disconnected from the CNS and not yet fully understood.
We report here the generation and characterization of a novel and highly specific polyclonal antibody against human NAIP and the detailed tissue profile of NAIP protein expression in humans. NAIP, the presence of which is found mostly in cells undergoing or having gone through differentiation, overlaps with the cell cycle inhibitor p21 in the intestinal villi and localizes to a region consistent with it being a pathogen response protein (Meylan et al. 2006). This work provides a basis for further investigation leading to the elucidation NAIP's function(s).
Materials and Methods
Generation and Purification of NAIP Antigen
The NAIP antigen construct of the BIR domains [encoded by amino acids 1 to 490 (Genbank accession number #U19251)] was generated by PCR amplification of cDNA using the following primer pair: 5′ forward primer AAA GGATCC ATG GCC ACC CAG CAG AAA GCC and 3′ reverse primer AAA CTCGAG CCA GAT GCC CAC AGA AAA GCT AT (Invitrogen Corp.; Carlsbad, CA). The PCR reaction was performed for 30 cycles (1 min at 96C, 1 min at 55C, 1 min at 72C) using a 9600 Perkin Elmer thermocycler (Perkin Elmer; Wellesley, MA). The PCR product was subcloned in TOPO-vector using the TOPO TA cloning kit (Invitrogen Corp.). After sequencing, the construct was subcloned in pGEX-4T3 cloning vector (Pfizer, Inc.; New York, NY) and transformed in the bacterial Escherichia coli strain BL21 (EMB Biosciences, Inc.; Madison, WI). Recombinant protein was generated using isopropyl β-D-1-thiogalactopyranoside (0.1 mM) induction for 7 hr. The cells were harvested by centrifugation at 5000 × g, and pellets were resuspended in 1 × STE buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 5 mM DTT) and broken with lysozyme (1 mg/ml in STE buffer). Recombinant glutathione S-transferase (GST)-NAIP protein was solubilized with N-lauroylsarcosine (Sigma-Aldrich, Inc.; St. Louis, MO) at a final concentration of 1.5% (w/v) followed by sonication (Sonics and Materials, Inc.; Newtown, CT) (30-sec pulses for 4 min). Soluble proteins were separated by centrifugation at 27,000 × g. Triton X-100 (Sigma-Aldrich, Inc.) was added to the soluble fraction at a final concentration of 4% (v/v) before being mixed with glutathione-Sepharose-4B beads (Pfizer, Inc.). The GST fusion protein and bead mixture was incubated overnight then washed with 1 × PBS, pH 8.0, 5 mM DTT, 0.25% NP-40 (Sigma-Aldrich, Inc.) and eluted with several 300-μl rounds of 50 mM Tris, pH 9.5, 15 mM reduced glutathione (Sigma-Aldrich, Inc.), and 0.25% (v/v) NP-40. Following dialysis in buffer A [25 mM Tris, pH 9.5, 100 mM NaCl, 5 mM DTT, 0.25% (v/v) NP-40], further purification was achieved by anion exchange chromatography using HiTrap-Q anion exchange columns (Pfizer, Inc.) equilibrated in buffer A. The proteins were eluted with a salt gradient up to 2-M NaCl in buffer A. Protein concentration was determined using a BioRad Protein Assay (BioRad, Inc.; Hercules, CA) with BSA as a protein standard. The purity was assessed by SDS-PAGE analysis (precast 4-15% Tris-HCl gradient gels; BioRad, Inc.) and generally found to be >90%.
Immunization Procedure
Procedures involving animals were performed in accordance with the University of Ottawa Animal Care and Veterinary Services guidelines. Polyclonal anti-NAIP antibodies were generated in an adult male rabbit (New Zealand white rabbit) by subcutaneous injection into three sites [0.1 μg of antigen in complete Freund's adjuvant (Sigma-Aldrich, Inc.) per site] once a week for 1 month. The anti-NAIP antibody content in the serum of the stimulated animal compared with preimmune serum was evaluated by Western blot analysis of samples extracted from NAIP-expressing cells or tissues.
Purification of Anti-NAIP Antibody
Recombinant GST-NAIP and GST protein were bound to CnBr-activated Sepharose-4B beads (Pfizer, Inc.) according to the manufacturer's protocol. The affinity columns were equilibrated and kept in buffer A. Whole-blood samples from immunized animals were centrifuged at 6000 × g, and serum was collected and kept at 4C. Rabbit immune globulins were separated by purification on recA-HiTrap column (Pfizer, Inc.). The serum was loaded on the recA affinity column and washed with buffer A, followed by elution of the immune globulins in buffer C (25 mM Tris, pH 2.0, 150 mM NaCl, 1 mM DTT). The eluate was neutralized in buffer D (25 mM Tris, pH 12.5, 150 mM NaCl, 1 mM DTT) to a final pH of 8.0. GST-NAIP-bound beads were added and incubated for 2 hr at 4C and then washed in buffer A. Nonspecifically bound protein was eluted with buffer B (25 mM Tris, pH 8.0, 1 M NaCl, 1 mM DTT). The anti-GST-NAIP immunoglobulins were eluted with buffer C, and the eluate was neutralized in buffer D until a final pH of 7.5-8.0 was obtained. GST-bound Sepharose-4B beads were added to the eluted fraction and incubated for 1 hr at 4C, and the unbound fraction was then collected. Anti-GST antibody was eluted in buffer C with subsequent equilibration of the GST affinity column in buffer A. This procedure was repeated until an elution peak was undetectable (BioRad ECONO-system; BioRad, Inc.). Final purification of the anti-NAIP antibody (removal of trace amounts of anti-GST and other nonspecific antibodies) was achieved by addition of 1 mg of GST-XIAP protein bound to glutathione beads and subsequent incubation for 20 min. The unbound fraction containing the anti-NAIP antibody was collected and concentrated with MicroFilters (Centricon-30 with a cut-off size of 10 kDa; Milipore, Inc., Billerica, MA) at 3500 × g to a final concentration of 5 mg/ml with sodium azide [0.05% (w/v)]. This purified fraction was termed J2.
Western Blot Analysis
Standardized amounts (25 μg to 50 μg) of total cell lysates from mouse tissue or human tissue protein extracts (Protein Medley; Clonetech, Inc., Mountain View, CA) in BioRad protein sample buffer (BioRad, Inc.) were denatured at 95C for at least 5 min before separation on SDS-PAGE (precast 4-15% Tris-HCl gradient gels; BioRad, Inc.). Proteins were transferred to Immobilon-P membrane (0.45-μm pore size; Milipore, Inc.) with the BioRad semi-dry protein transfer system (BioRad, Inc.). Membranes were blocked with 5% skim milk in buffer PBST (1 × PBS, pH 7.0, 0.1% Tween-20) overnight at 4C. The blots were then washed five times in PBST (15 min each) before incubation with J2 (1/5000 and 1/1000 from a 1-mg/ml stock) for 1 hr at room temperature. The membrane was washed five times in PBST (15 min each) and incubated with horseradish peroxidase conjugated to antibodies raised against rabbit immunoglobulins (1/2000; GE Bio-Sciences AB, Uppsala, Sweden) in PBST for 1 hr at room temperature. After ten washes in PBST (each 10 min), antibodies were detected using an enhanced chemiluminescence detection kit (GE Bio-Sciences AB).
Materials for Immunohistochemistry
The samples of human tissue were kindly provided by Dr. S. Robertson from the Department of Pathology at the General Hospital, Ottawa, Ontario, Canada. The source of the human samples was autopsy material from adults, with full informed consent of the patients' families under Institutional Review Board-approved protocols for research purposes. For all samples, the cause of death was unrelated to the tissue being studied. The tissues were immediately fixed in 5% (w/v) paraformaldehyde, dehydrated through a graded series of ethanol solutions, and embedded in paraffin.
Immunohistochemistry on Human Tissues
Sections (5 μm) of paraffin-embedded tissues were dried overnight at room temperature before being dewaxed in xylene (Sigma-Aldrich, Inc.) and rehydrated in a series of diminishing ethanol concentrations in aqueous solution. Endogenous peroxidase activity was blocked by treating the sections with 3% (v/v) hydrogen peroxide in TBS (50 mM Tris-buffered 150 mM NaCl, pH 7.6) for 20 min. The sections were then incubated in blocking buffer containing 2% (v/v) normal rabbit serum in TBS and then with polyclonal rabbit anti-NAIP antibody (1/100 of 1 mg/ml stock solution) in blocking buffer in a humidified chamber for 30 min at room temperature. After rinsing extensively with TBS, the sections were incubated for a further 30 min with a biotinylated secondary goat anti-rabbit antibody (Vector Laboratories Ltd.; Burlingame, CA) diluted 1/100 in the blocking solution. The sections were then rinsed thoroughly in TBS and subsequently incubated in streptavidin-horseradish peroxidase complex (Vectastain Elite ABC reagent; Vector Laboratories Ltd.) for 30 min. Antibody binding sites were visualized by incubation in 1 mg/ml DAB (3′3-diaminobenzindine; Sigma-Aldrich, Inc.) plus 0.03% (v/v) H2O2 (Sigma-Aldrich, Inc.).
Immunohistochemistry on Mouse Tissues
Tissue sections (5 μm) were fixed in freshly prepared 3% (w/v) paraformaldehyde (Sigma-Aldrich, Inc.) for 60 min, paraffin-embedded, and blocked in 5% (v/v) serum (goat serum for anti-p21, rabbit serum for J2 antibody) in PBS before immunostaining. Cultured cells were washed three times in 10% FCS-PBS for cytospin preparations, which were subsequently fixed with 3% paraformaldehyde, permeabilized for 20 min with 0.2% (v/v) Triton X-100 in PBS, and washed four times (4 min each) with PBS at room temperature. The cells were blocked in PBS containing 5% (v/v) serum for 1 hr and washed three times with PBS. The tissues or cells were then incubated with primary antibody J2 (1/50) and/or anti-p21 (1/25), in PBS for 1 hr. After washing in PBS, they were incubated with secondary antibody (anti-goat FITC or anti-rabbit CY3) for 1 hr, counterstained with Hoechst 33258 (Invitrogen Corp.) or 4′-6-diamidino-2-phenylindole (DAPI) (Invitrogen Corp.) according to the manufacturer's instructions, and thoroughly washed five times (2 min each) in PBS. The cells were mounted with Vectashield (Vector Laboratories Ltd.), and all samples were examined with a Zeiss Axiovert 10 microscope (Zeiss; Jena, Germany). Images were captured as digital files using the NORTHERN ECLIPSE (Empix Imaging, Inc.; Mississauga, Canada) software package.
Generation of Peripheral Blood Mononuclear Cells
Blood samples were mixed 1:1 with PBS (30 ml total) and gently layered onto 15 ml of Ficoll (Sigma-Aldrich, Inc.). Cell separation was achieved by gentle centrifugation at 1000 × g for 25 min. The serum was removed, and the remainder was transferred into a new Falcon tube (Invitrogen Corp.) and complemented with 40 ml of PBS. Following a second centrifugation step (1000 × g for 5 min), the peripheral blood mononuclear cells (PBMCs) were allowed to adhere in RPMI 1640 complete media (Invitrogen Corp.) supplemented with 10% (v/v) human AB serum, penicillin, streptomycin, and glutamine. After a 2-hr incubation, the non-adherent peripheral blood lymphocytes were removed and the adherent cells were kept in culture (T75 flasks) for 12 days before being used.
Results
Generation and Purification of Recombinant NAIP Antigen
To generate highly purified protein for immunization and affinity purification, an expression vector encoding an NAIP-GST fusion was constructed. The recombinant fusion protein was eluted from glutathione beads using a Tris buffer at pH 9.5, resulting in a soluble protein at the anticipated molecular mass of 82 kDa (data not shown). Following further purification by anion exchange chromatography, a portion of the purified GST-NAIP protein was used for rabbit immunization. The majority of the eluted GST-NAIP protein was then covalently bound to Sepharose-4B beads and later used as an affinity column for antibody purification.
Determination of J2 Specificity
Following affinity purification of the novel anti-NAIP antibody designated J2, Western blot analysis was performed with a variety of tissue samples to determine the antibody quality. Mouse primary cortical neurons were infected with adenovirus (Ad) containing genes for lacZ (control of infection), XIAP, or NAIP. A Western blot of the neuronal extracts probed with J2 revealed a band of the expected size for the NAIP construct (∼140 kDa) exclusively in the Ad-NAIP sample (Figure 1). The murine NAIP protein was not detected under these conditions. Tissues were harvested from mice that express the human NAIP gene under the regulation of the chicken β-actin-cytomegalovirus promoter (which drives constitutive expression of human NAIP in these mouse tissues). NAIP expression was assessed in cellular extracts from the heart, muscle, and brain of transgenic (Tg) and wild-type animals by Western blot analysis. The results demonstrate specific NAIP signals only in Tg samples (Figure 1). Supplemental Figure SF1 shows a Western analysis of total protein from human cells with J2. Because neither of these cell lines expresses detectable levels of NAIP endogenously (data not shown), they were transfected with plasmid expressing full-length NAIP. Antibody specificity was also tested using protein extracts from stable transfectants of human HeLa cells promoter. The stable HeLa cell line used for this work contains the NAIP cDNA under the control of a tetracycline-inducible TET-OFF construct. Expression of NAIP from the pTRE-NAIP vector is constitutive until turned off by the addition of TET. For the mock system, pTRE-GFP was present instead of pTRE-NAIP. No doxycycline or tetracycline was added to the cultures. A band of the expected size for this construct (∼160 kDa) was found in the NAIP stable transfectants, but not in the mock-transfected cells (Figure 1). The endogenous NAIP was not detected, suggesting the absence or undetectable levels of NAIP in the HeLa cells.
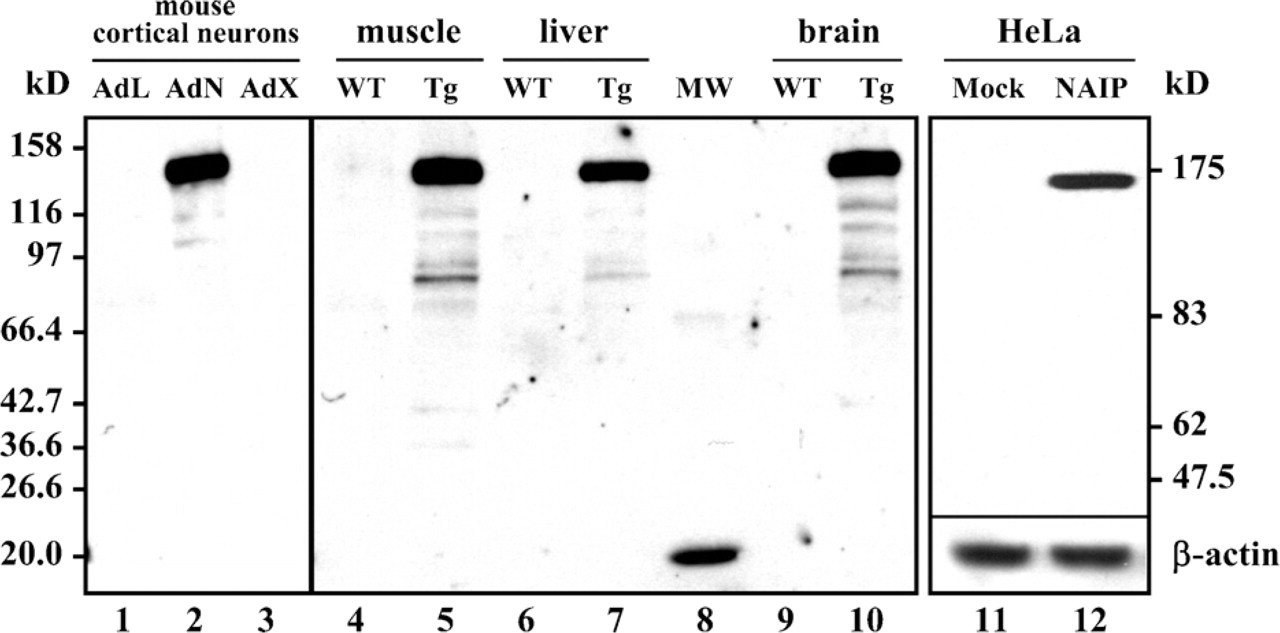
The polyclonal anti-neuronal apoptosis inhibitory protein (anti-NAIP) antibody detects NAIP in transgenic cells and mouse tissues. All panels were probed with the polyclonal anti-NAIP antibody (J2) at a 1/1000-1/2000 dilution. Primary mouse cortical neurons infected with adenovirus expressing LacZ (1), NAIP (2), or XIAP (3). (Anticipated molecular mass for NAIP is 138 kDa.) Samples from wild type (WT) and NAIP-transgenic (Tg) mouse tissues. WT muscle (4), Tg muscle (5), WT liver (6), Tg liver (7), broad range molecular mass markers (8), WT brain (9), and Tg brain (10). (Anticipated molecular mass of NAIP in Tg mouse is 138 kDa.) Detection of NAIP TET-OFF construct in stable HeLa transfectants: mock-transfected, in which pTRE-GFP was used instead of pTRE-NAIP (11) and stable full-length NAIP from the pTRE-NAIP vector (12). (Anticipated molecular mass of NAIP is 158 kDa.) β-actin loading control using a monoclonal anti-β-actin at a 1/1500 dilution.
Tissue Distribution of NAIP Protein
NAIP levels were determined in human tissues using Western blot analysis probed with the J2 antibody. Endogenous NAIP (∼150 kDa) was detected in brain, kidney, lung, ovary, testis, spleen, and placenta. NAIP expression in brain correlated with the results of previous studies (Xu et al. 1997b; Pari et al. 2000). Transcript levels in placenta, kidney, spleen, and lung detected by Northern blot analyses (Roy et al. 1995; Matsumoto et al. 1999; Yamamoto et al. 1999), correlated with protein levels as determined by Western blot analysis (Figure 2). In contrast, the presence of NAIP transcripts in the liver did not translate into protein expression.
Western blot analysis revealed high NAIP levels in the placenta, minimal expression in the heart, and none in the liver and skeletal muscle (Figure 2). The ∼45-kDa band seen in all tissues is probably a truncated form of NAIP (Notarbartolo et al. 2002; Shin et al. 2003), as is the case in the mouse (Hutchison et al. 2001). Experiments done in our laboratory with different antibodies have indicated that human NAIP is processed to yield smaller stable fragments (Supplemental Figures SF2 and SF3). The presence of NAIP fragments seems cell-type specific, as we see in Figure 2. Notarbartolo et al. (2002) detected a 38-kDa NAIP isoform, the product of a splice variant missing exons 10-11 in HL60 cells. Ka and Hunt (2003) detected a 35-kDa polypeptide in the placenta, similar in size to one of the fragments shown in Figure 2, Lane 10. Our results clearly showed expression of NAIP in tissues that are not directly affected in the pathology of severe SMA cases, suggesting a function that is not restricted to a neuronal context.
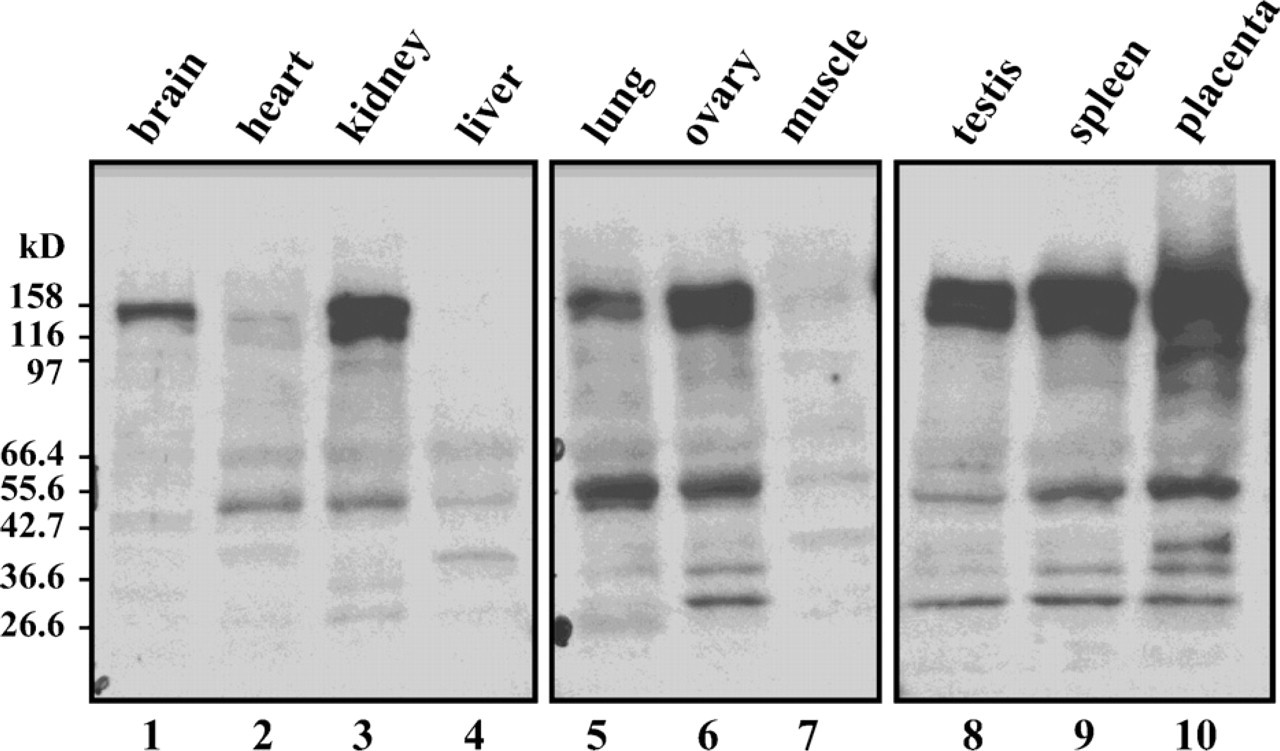
Distribution of NAIP in human tissues as determined by Western blot analysis using Protein Medley; Clontech, Inc. Membranes were probed with J2 (1/1000). An unstained broad-range molecular mass marker was used to determine sizes. Anticipated molecular mass for NAIP: 158 kDa. The lanes are: brain (1), heart (2), kidney (3), liver (4), lung (5), ovary (6), skeletal muscle (7), testis (8), spleen (9), and placenta (10).
Immunohistochemical Detection of NAIP Expression
Immunohistochemistry was performed on NAIP-Western blot-positive tissues (Figure 2) to evaluate the expression pattern of the protein within these tissues. Although expression of NAIP in the lung was positive on Western blots (Figure 2), it could not be detected in actual lung tissues such as the alveolar wall (Figures 3A and 3B). However, NAIP staining was observed around the capillaries, a region surrounded with white blood cells, especially lung macrophages (Figure 3B). Some regions within this tissue harbor carbonate deposits (arrows, Figure 3B), probably due to patient exposure to environmental pollutants such as tobacco smoke. To determine true NAIP-positive staining, we used lung tissue from mice raised in a pathogen-free environment (Supplemental Figure SF4). In these specimens, we do not observe artifactual staining caused by carbon-containing deposits. Although there is no staining in the lung epithelia, punctate staining is observed within distinct tissues such as veins and cartilage (Supplemental Figure SF4A), and cells such as adipocytes (Supplemental Figure SF4B) and macrophages (Supplemental Figure SF4C). Interestingly, there is a trend of decreasing staining with increasing branching of the trachea (Supplemental Figures SF4C and SF4D). We believe that this pattern may be due to increased air exchange, hence a greater exposure to pathogens higher up in the lung, leading to a greater presence of immunoreactive cells.
Attempts to use immunofluorescence for the detection of NAIP in the pathology samples were unsuccessful because of autofluorescence. Nevertheless, Figure 3B clearly demonstrates the absence of NAIP expression in inter-alveolar septae, indicating that protein expression (as demonstrated with Western blotting, Figure 2) may be attributable largely to macrophages residing in this tissue. A similar phenomenon might underlie the heterogeneity of staining within the spleen (Figures 3C and 3D), a location that contains numerous macrophages, possibly indicating a categorization of macrophages among the pericentric and sub-capsular regions. The absence of NAIP in the spleen cortex germinal centers corresponds to the previously demonstrated absence of macrophage staining (Witmer and Steinman 1984). To determine that the peripheral NAIP-positive staining is not an artifact caused by fixation and the embedding procedure, we performed immunohistochemical staining on the spleen, which was subjected to the antigen retrieval system (ARS) technique (Supplemental Figure SF5). The same pattern was observed with the NAIP-positive staining in the red pulp (periphery) (Supplemental Figures SF5A and SF5B). In addition, the ARS treatment uncovered staining within the germinal centers (Supplemental Figures SF5C-SF5F). We believe the specific granular staining is consistent with rough endoplasmic reticulum localization of selective lymphocytes (Supplemental Figures SF5E and SF5F).
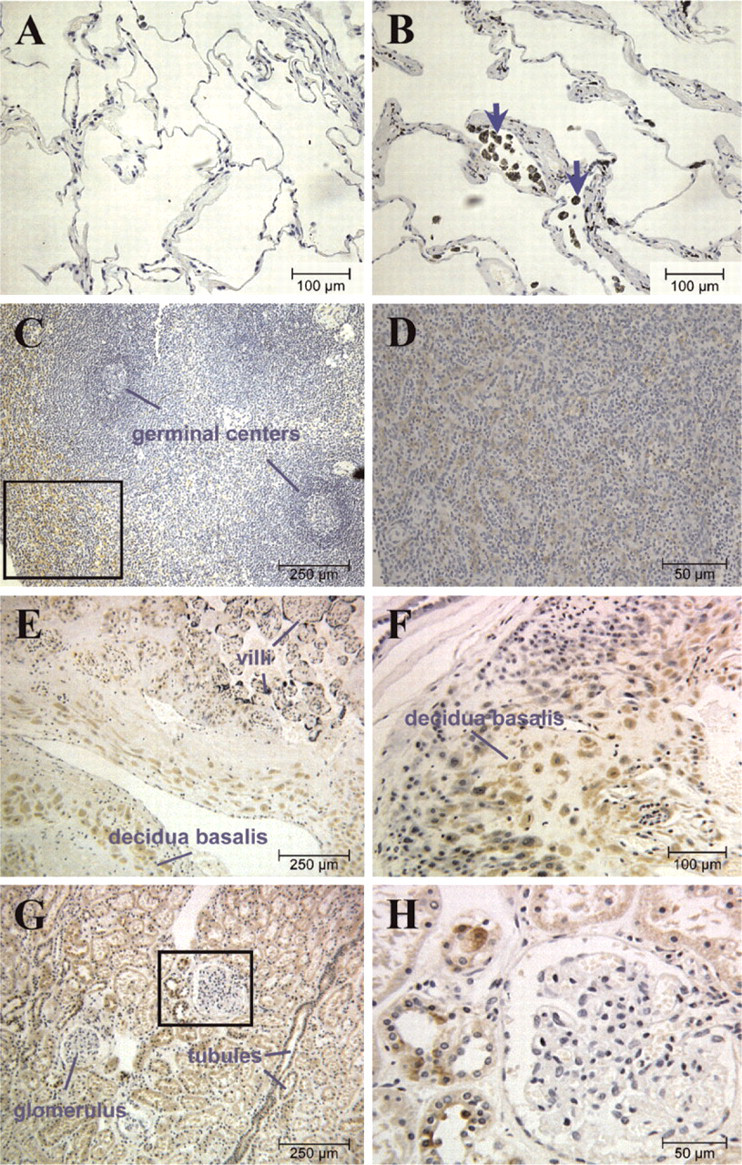
NAIP expression in human tissues assessed by immunohistochemistry. The tissues were obtained postmortem and were paraffin embedded. Sections were incubated with J2 (1/100), followed by a biotinylated secondary antibody and consecutive incubation with streptavidin-horseradish peroxidase complex and 3′3-diaminobenzindine (DAB), which results in a brown stain. Tissues were counter-stained with hematoxylin (blue stain). NAIP immunoreactivity in the lung (
A strong immunohistochemical expression of NAIP was found in placenta, consistent with the results obtained from Northern blot (Roy et al. 1995; Yamamoto et al. 1999) and Western blot analyses (Figure 2). NAIP expression was stronger in the decidual cells of the decidua basalis, the maternal portion of the placenta (Figures 3E and 3F). Specific NAIP expression was also found in the kidney. In the cortex, NAIP was seen exclusively in cells of proximal and distal convoluted tubules and not in supportive tissues, interstitium, or the glomerulus (Figures 3G and 3H). The same structures exhibited NAIP expression in the medulla (Figures 3I and 3J), with stronger staining observed in the proximal convoluted tubules than in the distal tubules.
Immunohistochemical analysis of all the tissues examined so far correlated with NAIP-positive tissues on Western blot analysis (Figure 2). The sparse but highly selective nature of NAIP staining observed within tissues such as kidney and spleen exemplified possible difficulties in obtaining positive results when studying whole tissues for the expression of NAIP at both the RNA and protein levels. Because the proportion of NAIP-expressing cells may have been diluted by using cellular extracts from whole tissues, those samples that tested negative by Western blotting (Figure 2) were also examined by immunohistochemistry. As expected, very low NAIP immunohistochemical staining was detected in the heart tissue (Figures 3K and 3L), with the exception of the strong positive staining observed in the smooth muscle epithelium around a subset of heart arterioles, as shown in Figure 3L. Although previous studies have detected NAIP transcripts in liver (Roy et al. 1995; Yamamoto et al. 1999), NAIP staining was not detected by immunohistochemistry (Figure 3M), in accordance with our Western blot results (Figure 2). In contrast to the Western blot analysis, NAIP staining was not observed in the testis by immunohistochemistry (Figure 3O), but immunohistofluorescence revealed a highly selective expression of NAIP (Figure 4B). It is unclear whether the expressing cells are spermatogonia or Sertoli cells. High levels of NAIP were observed in the intestinal epithelium (Figures 3N and 4D). No NAIP expression was observed in skin (Figure 3P). The positive result obtained by Western blot analysis for ovary (Figure 2) could not be confirmed by immunohistochemistry due to nonspecific staining obtained in the absence of the primary antibody (data not shown). This result precluded further investigation for NAIP expression in ovary using the J2 anti-NAIP antibody.
NAIP Expression in Macrophages
Previous studies reported NAIP expression in murine macrophages (Diez et al. 2000). PBMCs were thus cultured, differentiated, and examined by indirect immunofluorescence for the presence of NAIP (Figure 5). As expected, NAIP was detected in freshly prepared PBMCs (Figure 5B) and continued to be expressed during the differentiation process. The specificity of the anti-NAIP antibody in this technique was determined by probing the PBMCs with the J2 antibody preincubated with antigen, resulting in no detectable immunofluorescence (Figure 5D). The cytoplasm of these cells is very thin, and so NAIP staining shows as a halo around the nucleus of the PBMC. Although it is quite clear that the staining is largely cytoplasmic, there seems to be a merging of nuclear and NAIP staining in certain cells, but because this is not an image acquired using confocal microscopy, we think that this is caused by NAIP staining in the cytoplasm that is above the nucleus of these cells (Figure 5E). We have performed confocal microscopy imaging on the subcellular localization of NAIP in neuronal cells using the J2 antibody, demonstrating nuclear staining (data not shown).
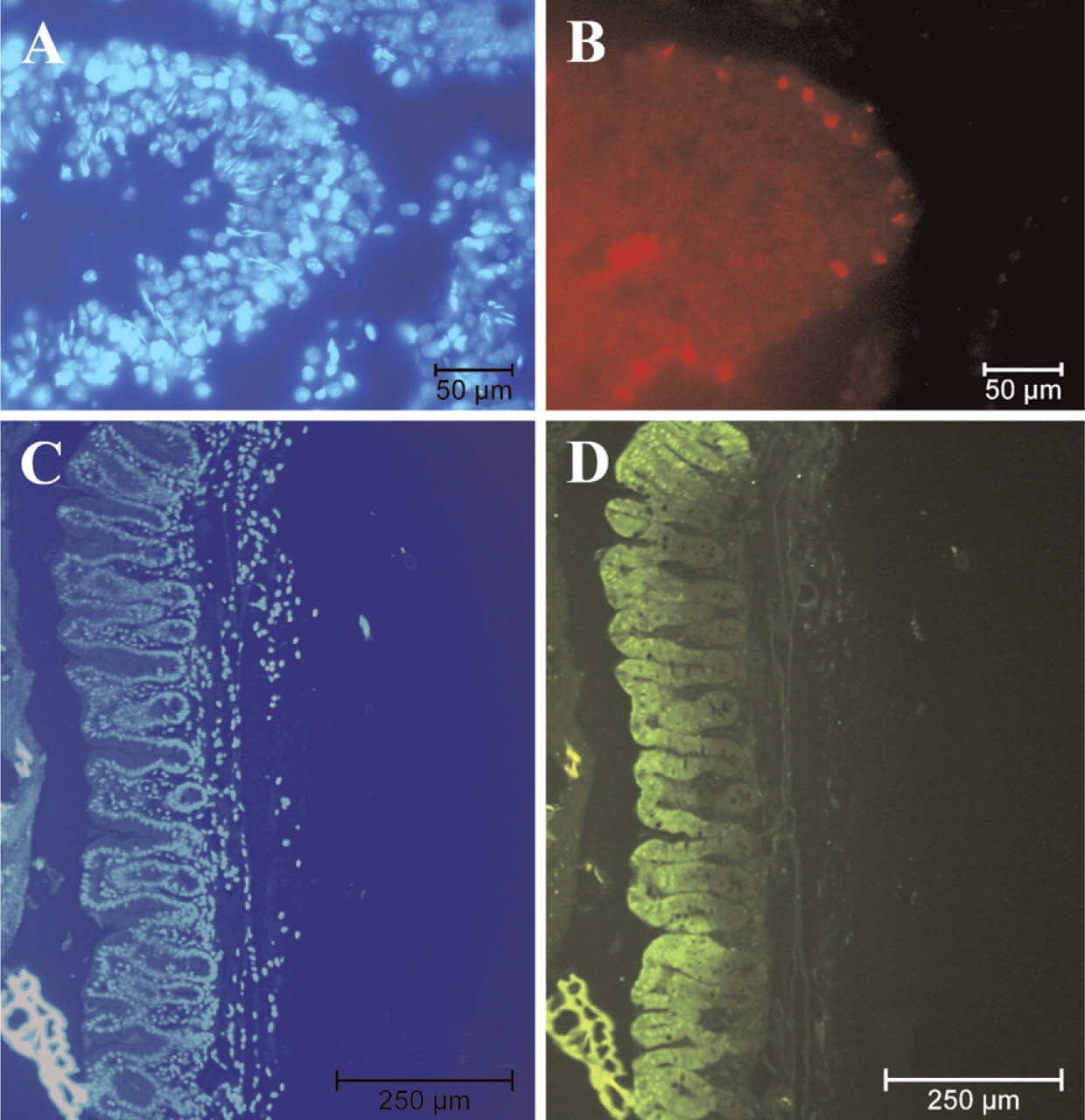
Expression pattern of NAIP in testis and small intestine by indirect immunofluorescence. Testis (

The presence of NAIP in freshly prepared peripheral blood mononuclear cells (PBMCs) as determined by indirect immunofluorescence. Phase contrast (
NAIP Is Expressed in Cells Undergoing Differentiation
Because NAIP transcripts have been reported in the small intestine (Yamamoto et al. 1999; Diez et al. 2000), NAIP expression was investigated further in this tissue. Freshly dissected mouse intestine was chosen for additional study, because the autopsy material might not reflect a representative physiological context for NAIP expression in this tissue. The epithelium of the gastrointestinal tract has a highly conserved organization characterized by a continuous and rapid cell proliferation, resulting in a complete turnover every 3 to 5 days. Highly selective expression of NAIP was observed in the upper crypt region as well as in the surface layers of epithelial cells connecting the crypts (Figure 4D).
NAIP Expression in Small Intestine Is Correlated With Expression of p21WAF1
The pattern of NAIP expression in the small intestine (Figure 4D) appeared similar to that reported for the cell-cycle regulator protein p21WAF1, as determined by in situ hybridization (Gartel et al. 1996). We thus used indirect immunofluorescence to examine the protein expression of p21WAF1 in this tissue. As expected, the protein expression of p21WAF1 matched what was previously determined by in situ hybridization. The patterns of p21WAF1 (Figure 6A) and NAIP (Figure 6B) expression were found to coincide, as revealed by the overlay of both immunofluorescent images (Figure 6D).
Discussion
The discovery of NAIP as a candidate gene for SMA, an inherited neurodegenerative disorder, led to the assumption that the protein is expressed and acts chiefly, and possibly exclusively, within the CNS. This hypothesis derives support from an NAIP expression study in rat showing expression in the CNS and other structures affected in the pathology of severe SMA cases (Xu et al. 1997b). The absence or extremely low expression of NAIP transcripts in non-neuronal tissues was further indication for NAIP function being of exclusive relevance within neurons (Roy et al. 1995). Additional studies in both human and rodent revealed a pattern of NAIP expression that is not exclusively neuronal (Magun et al. 1998; Matsumoto et al. 1999; Yamamoto et al. 1999; Diez et al. 2000; Ingram-Crooks et al. 2002; Shin et al. 2003).

Expression patterns of p21WAF1 and NAIP in small intestine are identical. Expression of p21WAF1 using the polyclonal anti-p21WAF1 and a CY3-conjugated secondary antibody (
To obtain a detailed account of NAIP expression in adult human tissues (neuronal and non-neuronal) as well as the cellular and functional context of this protein, a polyclonal NAIP antibody was generated that specifically recognizes NAIP's N-terminus, including the three characteristic BIR domains (Roy et al. 1995). The polyclonal NAIP antibody (termed J2) was affinity purified and evaluated on transfected samples by Western blot analysis (Figure 1) prior to application in adult human tissues. Cell extracts originating from a variety of human tissues were then tested by Western blot analysis. The expected 150 kDa band representing NAIP was found in brain, spleen, testis, ovary, lung, kidney, and placenta, with the latter showing the highest expression (Figure 2). A strong ∼45-kDa band was detected in most tissues (Figure 2), and was postulated to be a truncated form of NAIP, similar to that found in the mouse (Hutchison et al. 2001). In the work presented in this paper, an antisense construct to the first exons of NAIP was transfected in mouse neuroblastoma cells, and caused the reduction in intensity of all fragments recognized by the antibody. A number of experiments done in our laboratory with different antibodies have indicated that human NAIP is processed to yield smaller stable fragments. The presence of NAIP fragments seems cell type specific, as we see in Figure 2. Notarbartolo et al. (2002) detected a 38-kDa truncated NAIP, the product of a splice variant in HL60 cells. Ka and Hunt (2003) detected a 35-kDa polypeptide in the placenta, similar in size to one of the fragments shown in Figure 2, Lane 10.
The Western blot analyses clearly show expression of NAIP in human tissues that are not affected in the pathology of severe SMA, such as the kidney (Figures 3G and 3H), testis (Figures 3N and 4D), and intestinal epithelium (Figures 3K and 5D).
Immunohistochemistry was undertaken with J2 anti-NAIP antibody, revealing a highly selective NAIP expression pattern restricted to a certain subset of cells within a number of tissues. The selective expression was particularly noteworthy in decidual cells of the placenta (Figures 3C and 3D), the proximal and distal convoluted tubules of the kidney (Figures 3G and 3H), testis (Figures 3N and 4D), and intestinal epithelium (Figures 3K and 5D). This selectivity of expression may explain the low rates of transcripts found in previous studies using whole-cell extracts from tissues (Roy et al. 1995; Yamamoto et al. 1999).
The immunohistochemical analysis also revealed that a positive result on Western Blot does not necessarily reflect expression of protein within the tissue itself but may, in fact, be due to NAIP expression in macrophages that are associated with the tissue (Diez et al. 2000). The absence of NAIP in the liver (Figure 3L) is consistent with this interpretation.
The observation of highly selective expression of NAIP within tissues that have no apparent functional or physiological link prompts the question of a common cellular context that ties these ostensibly disparate tissues together for one such anti-apoptotic function. In this regard, NAIP's expression pattern in the small intestine is of particular note (Figure 4D). Pluripotent stem cells anchored near the base of the crypts are characterized by rapid proliferation; the daughter cells ascend along the crypt-villus axis, losing the ability to proliferate and differentiating into one of four distinct cell types (absorptive enterocytes, goblet cells, Paneth cells, and enteroendocrine cells). The tissue thus is comprised of three spatially defined cellular stages (proliferation, differentiation, apoptosis), allowing for analysis of protein expression pattern within specific cellular stages. NAIP expression is restricted to the non-proliferative compartment of the intestinal villi (Figure 4D).
Structurally, NAIP is a member of the pattern recognition receptor (PRR) family. More specifically, it belongs to the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR, also known as NOD-LRR, NACHT-LRR, and CATERPILLER) family, which is characterized by three distinct domains: a CARD or pyrin effector domain, a NOD or “NACHT” domain, and a variable number of carboxy-terminal leucine-rich repeats (LRRs) (Meylan et al. 2006). The finding that NAIP underlies murine L. pneumophila susceptibility is consistent with it playing a role in the innate immunity of an organism. Recent evidence suggests that the LRR of NAIP in macrophages binds bacterial flagellin (legionella, salmonella), resulting in an interaction with the cytosolic mammalian protein IPAF (also called CARD12 and CLAN) and the subsequent recruitment of caspase-1 [interleukin-1 (IL-1)-converting enzyme or ICE] to the NAIP CARD domain and caspase-1 activation. This leads to the generation of the chief proinflammatory cytokine IL-1β and IL-18 (Molofsky et al. 2006; Ren et al. 2006; Zamboni et al. 2006).
Recent work from our laboratory has shown that the binding of ATP to the NAIP NOD domain results in the oligomerization of NAIP, thereby activating caspase-9 inhibition. This suggests that the activation of caspase-1 may be associated with the inhibition of caspase-9, inhibiting cell death and directing NAIP into a more immunomodulatory role (Davoodi et al. 2004).
The high levels of NAIP found in the villi in the present study are in keeping with a role as an intestinal pathogen recognition receptor (Meylan et al. 2006). The class of toll-like receptors (TLRs) represents a second important component of innate immunity. These transmembrane proteins contain extracellular domains comprising LRRs that recognize various pathogen-associated molecular patterns, transducing signals into the interior of host cells that initiate defensive responses. Interestingly TLR5, which binds flagellin, has recently been shown to exist solely in the base of intestinal crypts, with negligible amounts expressed in the villi. It may be that NAIP and TLR5 comprise the flagellin recognition system of the intestinal tract, functionally redundant but spatially distinct.
Interestingly, the NAIP expression pattern expression in the intestinal villi matched perfectly with that of the cell cycle regulator protein, p21WAF1 (Figure 6B) (Gartel et al. 1996). The expression of p21WAF1 in intestine is associated with growth inhibition, cell cycle withdrawal, or terminal differentiation and is believed to be independent of p53 expression (Beauchamp et al. 1996; Doglioni et al. 1996; Gartel et al. 1996). Similar to this finding is the expression of NAIP during adipocyte differentiation (Magun et al. 1998) that parallels the upregulation of the cdk inhibitor p21WAF1 during differentiation processes and is associated with both postmitotic as well as apoptosis-resistant states (el-Deiry, 1998). Magun et al. (1998) reported higher apoptotic susceptibility during adipocyte differentiation due to the increased metabolic activity associated with this process, and linked NAIP expression to a compensatory cytoprotection. Caspase activation has also been implicated in the maturation and differentiation of multiple cell types (reviewed in Abraham and Shaham 2004), and it is hypothesized that NAIP is induced to serve as a regulatory mechanism in this process.
The expression of p21WAF1 during monocyte differentiation has been previously reported (Asada et al. 1999) and correlates with NAIP expression in PBMCs treated to differentiate into macrophages (Figure 5C). In a similar manner, p21WAF1 expression is upregulated in the granulosa cells of the ovarian follicle during maturation (Kiyokawa et al. 1996; Robker and Richards 1998). It could be the case that (among other factors) the expression of p21WAF1, and the subsequent withdrawal from the cell cycle, triggers NAIP expression. A connection between IAPs, p21WAF1, and apoptosis regulation has been previously reported (Suzuki et al. 1998; Suzuki et al. 2000a; Steinman and Johnson 2000). The regulator protein p21WAF1 is believed to bind to the prodomain of procaspase-3 (Zhang et al. 1999). Interaction of this procaspase-3-p21WAF1 complex with XIAP was demonstrated to regulate the susceptibility to FAS-mediated apoptosis (Suzuki et al. 1998; Suzuki et al. 1999). Survivin interaction with cdk4 mediates release of p21WAF1 from the p21WAF1-cdk4 complex, and the subsequent complex formation with procaspase-3 causes inhibition of apoptosis (Suzuki et al. 2000b). The proteasome-dependent degradation of HIAP-2 was also inhibited by p21WAF1, resulting in cytoprotection of etoposide-treated K562 leukemic cells (Steinman and Johnson 2000). The direct or indirect upregulation of NAIP by p21WAF1 could be an additional way to promote survival of cells during the differentiation process.
Our observation of NAIP expression patterns provides support for a model in which NAIP is highly expressed in, and protects, cells (within a subtype of tissue) that are undergoing terminal differentiation.
Footnotes
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research and the Muscular Dystrophy Association of Canada. A.E.M. is a Burroughs-Wellcome clinical translation awardee.