
Abstract
Recent studies have shown that, in mast cells, membrane microdomains rich in cholesterol and glycosphingolipids called lipid rafts play an important role in Fc∊RI signaling. The present study demonstrates that, in RBL-2H3 cells following stimulation, the mast cell-specific gangliosides associated with Fc∊RI are internalized from lipid rafts along with the receptor. When the cells are labeled with iodinated antibodies against the gangliosides or against Fc∊RI and the cell components are then fractionated on Percoll density gradients, in stimulated cells the gangliosides are internalized with the same kinetics as Fc∊RI and at 3 hr are present in the dense lysosome fraction. Using transmission electron microscopy, with antibody against the gangliosides conjugated to horseradish peroxidase and antibody against Fc∊RI conjugated to colloidal gold, it was possible to demonstrate that the gangliosides and Fc∊RI are internalized in the same coated vesicles. At 5 min, the gangliosides and Fc∊RI can be identified in early endosomes and at 3 hr are found together in acid phosphatase-positive lysosomes. This study demonstrates that the mast cell-specific gangliosides are internalized from lipid rafts in the same vesicles and traffic intracellularly with the same kinetics as Fc∊RI. This study contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
M
Lipid rafts are specialized microdomains of the plasma membrane and are enriched in cholesterol and glycosphingolipids that play an important role in Fc∊RI signaling (Holowka et al. 2000; Wilson et al. 2000, 2001,2004; Holowka and Baird 2001; Rivera et al. 2001). The concept of lipid rafts originated from studies in the early 1970s on the mobility of proteins in the plasma membrane that showed that plasma membrane-associated proteins can undergo a selective redistribution and internalization (Taylor et al. 1971; Tsan and Berlin 1971; Edelman et al. 1973; Oliver et al. 1974; Oliver and Berlin 1979). Later studies suggested that certain membrane proteins are restricted to lipid rafts. Brown and London (1992) demonstrated lateral segregation of newly delivered glycosylphosphatidylinositol-anchored proteins into plasma membrane microdomains that were enriched in sphingomyelin, cholesterol, and glycolipids. These microdomains (lipid rafts) are preferentially resistant to solubilization with non-ionic detergents such as Triton X-100 and compose ∼30%-40% of the plasma membrane (Yu et al. 1973; Apgar and Mescher 1986; Harder et al. 1998; Holowka et al. 2000). In unstimulated cells, Fc∊RI is loosely dispersed throughout the plasma membrane but upon activation rapidly aggregates (Stump et al. 1988,1989; Wilson et al. 2000,2001) and can then be found on the cell surface in lipid rafts in association with GM1, a ganglioside typically associated with lipid rafts (Wilson et al. 2004). Following aggregation, Fc∊RI is then internalized in coated vesicles (Stump et al. 1988,1989; Mao et al. 1993).
Previous studies have shown that two rodent mast cell-specific gangliosides, derivatives of GD1b (Guo et al. 1989) that are the binding sites for MAb AA4, are closely associated with Fc∊RI. When the gangliosides are crosslinked by MAb AA4, the mast cells are partially activated but do not degranulate (Guo et al. 1989; Oliver et al. 1992; Swaim et al. 1994). The gangliosides recognized by MAb AA4 coimmunoprecipitate with the receptor (Stephan et al. 1991). Furthermore, the protein tyrosine kinase p53/56Lyn, the first kinase to be activated in the signal transduction pathway, is also closely associated with the gangliosides (Minoguchi et al. 1994). In stimulated RBL-2H3 cells, the gangliosides are localized in lipid rafts along with Fc∊RI and Lyn (Field et al. 1999; Sheets et al. 1999; Holowka and Baird 2001). The present study was undertaken to determine if these mast cell-specific gangliosides are internalized in the same vesicles and traffic with the same kinetics as Fc∊RI.
Materials and Methods
Cells
The rat mast cell line, RBL-2H3, was grown as monolayers as previously described (Barsumian et al. 1981).
Antibodies
The following primary antibodies were used: mouse antiganglioside MAb AA4 (0.125 μg/ml), mouse anti-α subunit of Fc∊RI MAb ER 14CA4IIC3 (2.5 μg/ml), mouse anti-α subunit of Fc∊RI MAb BC4 (10 μg/ml), and goat anti-LAT (0.125 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA). The following secondary antibodies were used: goat anti-mouse IgG-horsedradish peroxidase (HRP), donkey anti-mouse IgG-HRP, donkey anti-goat IgG-HRP, rabbit anti-mouse IgG-HRP, or rabbit anti-goat IgG-HRP (all secondary antibodies were from Jackson ImmunoResearch; Port Washington, PA).
For some experiments, F(ab) fragments of MAb AA4 were prepared by digestion of the antibody with papain (Pierce Biotechnology; Rockford, IL).
Mediator Release
MAb AA4 interferes with MAb BC4 binding to Fc∊RI, thus preventing cell activation and mediator release if RBL-2H3 cells are first incubated with MAb AA4. However, MAb BC4 activates the cells as soon as it binds to the receptor. Therefore, in experiments where the two antibodies were used jointly, they were mixed just prior to addition to the cells. To confirm that the cells were being stimulated normally by 125I-BC4 and BC4-gold, supernatants were collected from all samples in each experiment, and histamine (Basciano et al. 1986) or β-hexosaminidase release (Jamur et al. 2005) was measured. Cells stimulated with unconjugated MAb BC4 served as controls. No significant differences were observed in the histamine release between the control and experimental cells in any experiment. Furthermore, MAb AA4, 125I-AA4, and AA4-HRP did not induce histamine release.
Sucrose Density Fractionation of Cellular Membranes
Detergent-insoluble glycolipid-enriched microdomains, also called lipid rafts, were prepared essentially as described previously (Sada et al. 2001). Cells were cultured overnight with or without mouse IgE anti-TNP (1:5000 dilution from ascites fluid). Sensitized cells were stimulated with 50 ng/ml DNP48-HSA (Sigma-Aldrich; St Louis, MO), and mediator release was monitored by β-hexosaminidase activity. Ten min after the addition of DNP48-HSA, cells (2-5 × 107) were washed with cold PBS and harvested in 2.8 ml lysis buffer [150 mM NaCl, 25 mM MES (2-N-morpholinoethanesulfonic acid), 5 mM EDTA, 1 mM Na3VO4, 2 mM phenylmethanesulfonyl fluoride (PMSF), protease inhibitor cocktail (1:50; Sigma-Aldrich), and 0.05% Triton X-100 (v/v), pH 6.5] on ice. The cell suspension was then homogenized with 30 strokes in a Dounce homogenizer with a tight-fitting piston, and the homogenate was layered over 2.6 ml 80% sucrose (w/v) in MNEV solution (150 mM NaCl, 25 mM MES, 5 mM EDTA, and 1 mM Na3VO4, pH 6.5) in a 14-ml Beckman centrifuge tube and gently vortexed to give a final concentration of 40% sucrose. Then, 5.6 ml of 35% sucrose (w/v) in MNEV solution with protease inhibitor cocktail 1:100 and 1 mM PMSF was overlaid, and 2.8 ml of 5% sucrose (w/v) in MNEV solution with protease inhibitor cocktail 1:100 and 1 mM PMSF was then overlaid. Samples were centrifuged using a Beckman Ti 40 rotor (Beckman Coulter; Fullerton, CA) at 38,000 × g at 4C for 20 hr. A series of 1.4-ml aliquots taken from the top of the tube were collected. Two μl from each fraction was put onto Hybond membranes (GE Healthcare Life Sciences; Piscataway, NJ) and the membranes analyzed by immuno-blotting. Membranes were incubated for 1 hr at room temperature in Tris-buffered saline (TBS: 25 mM Tris/Tris-HCl, 0.15 M NaCl, pH 7.5) containing 5% non-fat dry milk, washed in TBS containing 0.05% Tween-20 (TBS-T; Sigma-Aldrich), and incubated for 1 hr at room temperature with the various antibodies diluted in TBS. After washing five times with TBS-T, membranes were incubated with secondary antibody, goat anti-mouse IgG-HRP, donkey anti-mouse IgG-HRP, donkey anti-goat IgG-HRP, rabbit anti-mouse IgG-HRP, or rabbit anti-goat IgG-HRP (1:20,000 in PBS; Jackson ImmunoResearch) for 1 hr at room temperature. Membranes were then washed in TBS-T five times and developed using chemiluminescence (ECL; GE Healthcare Life Sciences).
Iodination of Antibodies
Antibodies were iodinated using IODO-beads (Pierce) according to the manufacturer's instructions using 1 mCi 125I/100 μg of antibody and 200 μg of antibody/IODO-bead.
Cell Labeling
RBL-2H3 cells were plated in 60-mm dishes (5 × 105 cells/dish) in DMEM (Invitrogen; Carlsbad, CA) supplemented with 15% heat-inactivated fetal calf serum (Invitrogen), 2 mM glutamine, 100 U/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) and cultured for 24 hr at 37C in a humidified incubator with 5% CO2 in air. Cells were rinsed twice in DMEM containing 1% BSA, and the culture medium was then replaced with fresh medium at 37C containing 10 μg/ml of 125I-BC4, 125I-AA4, or 125I-AA4 plus unlabeled MAb BC4 to stimulate the cells. The dishes were returned to the incubator for a 10-min pulse and then rinsed in cold (4C) Hank's Balanced Salt Solution (HBSS; Invitrogen) and processed immediately (0 min) or rinsed in complete medium at 37C and returned to the incubator for 10 min, 30 min, 1 hr, 2 hr, or 3 hr of chase. After the chase, the cells were rinsed in cold HBSS to stop the endocytic process and fractionated on Percoll density gradients.
Percoll Gradients
Percoll gradients were prepared as previously described (Oliver et al. 1989). Briefly, after labeling, cells were rinsed in cold HBSS, cold HBSS without Ca2+ and Mg2+, and in HBSS with 0.76 mg/ml EDTA and removed from the dishes with pronase-EDTA at 4C, rinsed once with HBSS containing 2.5% BSA, rinsed once in triethanolamine buffer, pH 6.8, 10 mM triethanolamine, 4 mM EDTA, 1 mM EGTA, 1% aprotinin (Sigma-Aldrich), resuspended in the same buffer, and disrupted using a nitrogen cavitation bomb (Kimble/Kontes; Vineland, NJ) followed by homogenization using a Potter-Elvehjem homogenizer with a loose-fitting pestle. The postnuclear supernantant (3000 × g for 10 min) was layered on Percoll (GE Healthcare Lifesciences) prepared by layering a 35% Percoll stock (90% Percoll in 2.5 M sucrose) over a 2.5-M sucrose cushion. Samples were centrifuged using a Beckman VTi90 vertical rotor at 34,800 × g for 1 hr. One-ml fractions were collected and assayed for acid phosphatase (AcPase) activity or counted in a gamma counter for 125I.
AcPase activity, the classic marker for lysosomes (De Duve 2005), was assayed using the method of Rome et al. (1979) with 4-methylumbelliferyl phosphate (Sigma-Aldrich) as substrate. Samples were incubated for 30 min at 37C and the reaction stopped by the addition of glycine-carbonate, pH 10.0.
Preparation of Labeled Proteins for Electron Microscopy
Transferrin was saturated with iron according to the method of Rensoude et al. (1982) and conjugated to HRP using an EZ-Link Plus Activated Peroxidase Kit (Pierce) according to the manufacturer's directions. All preparations of transferrin-HRP were tested before use in experiments with RBL-2H3 cells to insure that the transferrin-HRP was transcytosed and not delivered to lysosomes. MAb AA4 was conjugated to HRP using the EZ-Link Plus Activated Peroxidase Kit (Pierce). The resulting preparation, AA4-HRP, was tested using RBL-2H3 cells to ensure that its activity was identical to the unconjugated antibody.
Fifteen-nm colloidal gold was made using a modification of Fren's method with sodium citrate as the reducing agent (Oliver 1999). Six-nm gold particles were prepared using tannic acid-sodium citrate as the reducing agent (Oliver 1999). Antibodies were conjugated to the gold according to the method of Birrell et al. (1987).
Scanning Electron Microscopy
Cells were plated at a density of 1 × 104 on 12-mm-round coverslips. At 24 hr, cells were stimulated with MAb BC4 conjugated to 15-nm colloidal gold (BC4-gold) for various times and then rinsed briefly in PBS at 37C and fixed for 1 hr at room temperature in 2% glutaraldehyde (Electron Microscopy Sciences; Hatfield, PA) in PBS (pH 7.2). Cells were then rinsed twice in distilled deionized water and silver enhanced using IntenSE M (GE Healthcare Life Sciences) and rinsed again in distilled deionized water. Samples were then dehydrated with a graded series of ethanol and critically point dried with CO2. After critical point drying, samples were mounted on aluminum stubs with colloidal graphite (Electron Microscopy Sciences) and coated with carbon. Both secondary and backscatter electron images of the cells (deHarvan and Soligo 1986) were observed using a JEOL JSM-35C scanning electron microscope (Jeol, Ltd.; Tokyo, Japan).
Electron Microscopy and Cytochemistry
For transmission electron microscopy, MAb BC4 conjugated to 6-nm colloidal gold (BC4-gold), MAb AA4 conjugated to HRP (AA4-HRP), and transferrin-HRP were used as markers. Cells were incubated continuously with the markers or pulsed for 30 min at 4C, rinsed in warm (37C) medium, and returned to the incubator for various chase times. There was no difference in the trafficking of the markers between protocols. After incubation, cell monolayers were washed twice with cold HBSS (Invitrogen) and the cells fixed for 1 hr with 2% glutaraldehyde-2% formaldehyde (both from Electron Microscopy Sciences) in 0.1 M cacodylate buffer (pH 7.4) and rinsed in cacodylate buffer. Some samples were further reacted for AcPase or HRP activities. AcPase activity was demonstrated by the method of Novikoff (1963) using cytidine 5′C-monophosphate as substrate. Samples were incubated for 1 hr at 37C, rinsed, and the reaction product visualized with 1% sodium sulfide. Samples incubated without substrate served as controls. All controls were negative for reaction product. HRP was localized using the method of Graham and Karnovsky (1966). Controls consisted of samples incubated without chromagen (DAB) or without substrate (H2O2). All controls were negative for reaction product. Samples were incubated for 1 hr at room temperature, rinsed, and postfixed in 1% osmium tetroxide containing 1.5% potassium ferrocyanide for 1 hr at room temperature (Karnovsky 1971). All other samples were postfixed for 1 hr in 2% osmium tetroxide at room temperature, rinsed in cacodylate buffer, and stained en bloc with 0.5% uranyl acetate. All samples were dehydrated through a graded series of ethanol, removed from the culture dishes with propylene oxide, and embedded in Spurr's resin (Electron Microscopy Sciences). Thin sections were cut with a diamond knife, mounted on bare copper grids, stained with Reynolds' lead citrate (Reynolds 1963), and examined in a JEOL 100CX electron microscope.
Results
Gangliosides Recognized by MAb AA4 Are Internalized With the Same Kinetics as Fc∊RI
Initial experiments characterized the distribution of the gangliosides recognized by MAb AA4 and Fc∊RI in detergent-resistant membrane fractions (lipid rafts) separated by sucrose density gradients (Figure 1). Aliquots of the resulting fractions were blotted onto nitrocellulose paper and probed with antibodies against the gangliosides (MAb AA4), the α subunit of Fc∊RI, and LAT, a marker of lipid rafts. In both unstimulated and stimulated cells, the gangliosides are localized along with LAT in the lipid rafts. However, in unstimulated cells, Fc∊RI is in fractions 5 to 9, outside the lipid rafts. Stimulation of the cells with MAb BC4, which aggregates Fc∊RI, results in a shift of the receptor toward the lipid raft fractions, essentially as described previously (Field et al. 1999; Sheets et al. 1999). In stimulated cells, Fc∊RI and the gangliosides recognized by MAb AA4 are both localized in the lipid raft fractions.
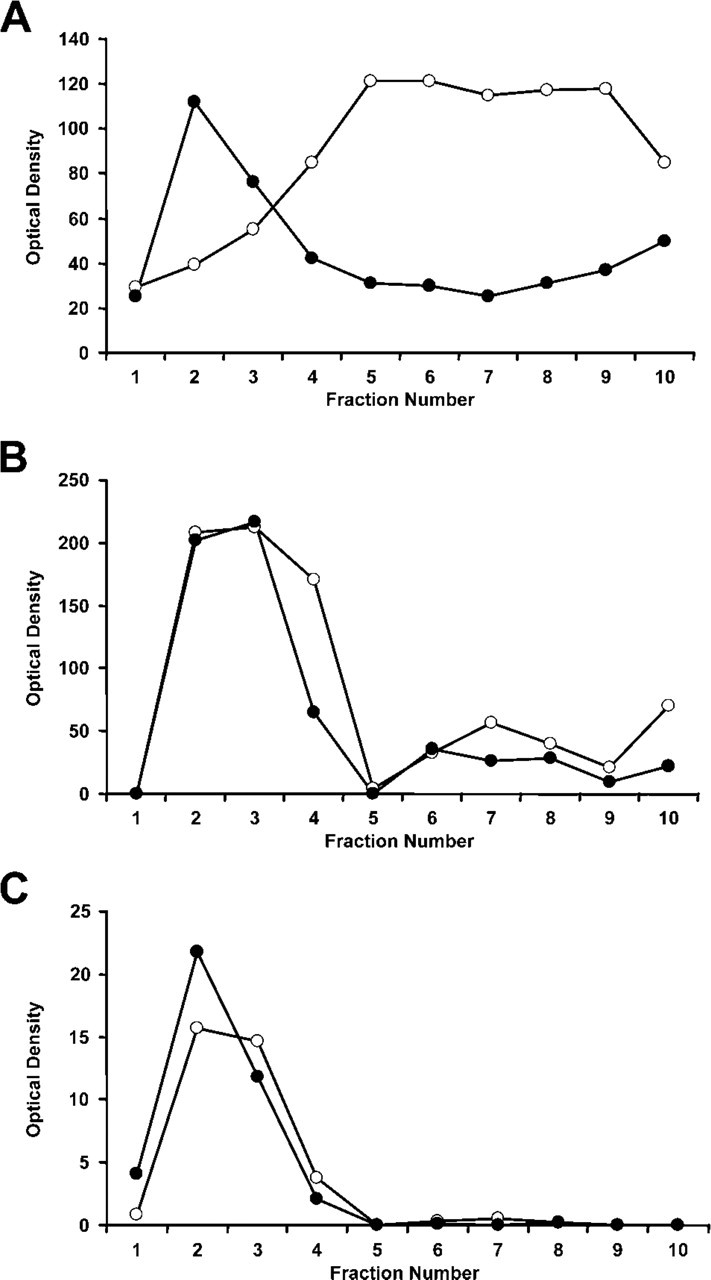
Association of Fc∊RI and gangliosides in lipid rafts after receptor aggregation. The detergent-insoluble fraction of Triton X-100 solubilized cells was separated on sucrose density gradients; aliquots from each fraction applied to nitrocellulose membranes; and the presence of Fc∊RI, the gangliosides (marked by MAb AA4), and LAT (used as a marker for the lipid rafts) was detected by immunoblotting. Optical density of each fraction was then graphed to illustrate the shift in position of the various components after stimulation via Fc∊RI. In unstimulated cells, Fc∊RI
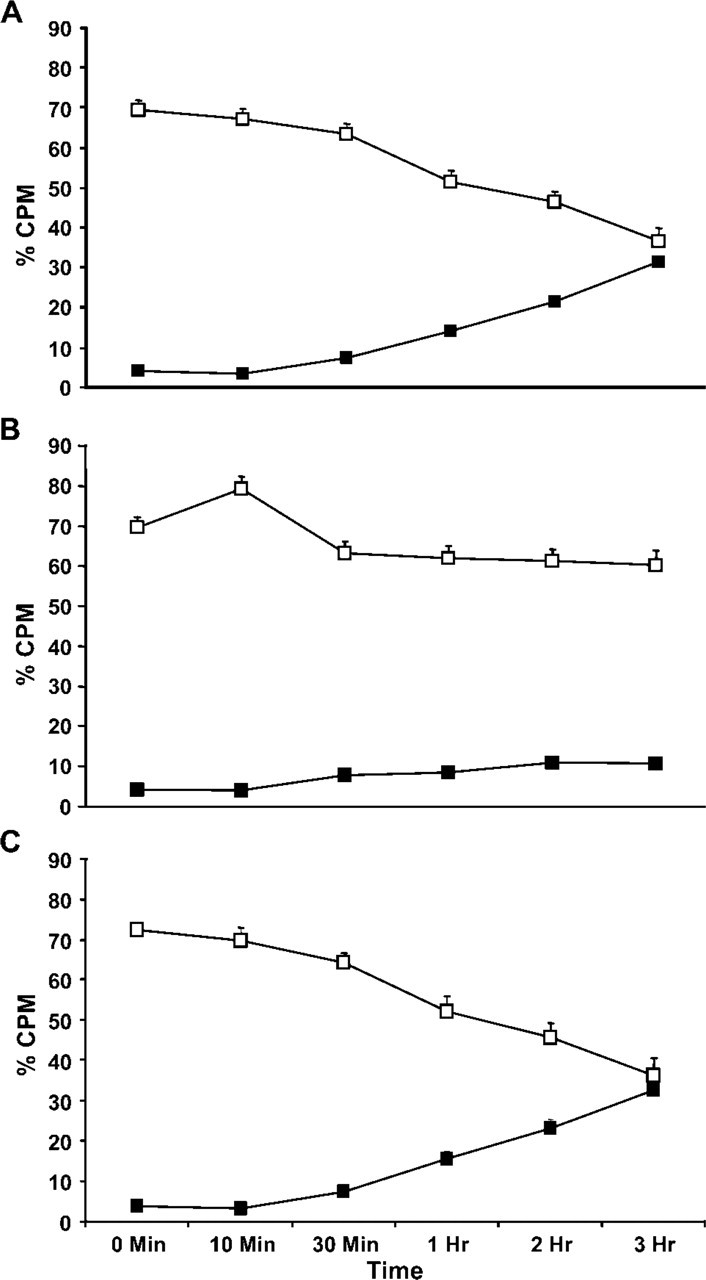
Fc∊RI and gangliosides derived from GD 1b have the same kinetics of internalization. RBL-2H3 cells were pulsed with the iodinated antibodies for 10 min, rinsed, and chased at 37C for varying times, and Percoll density gradient fractions were prepared. In cells incubated with 125I-BC4, with time the label decreases in the light membrane fraction and increases in the dense lysosome fraction
The kinetics of internalization and intracellular trafficking of the gangliosides and Fc∊RI were investigated using 125I-labeled antibodies and cell fractionation on Percoll density gradients. RBL-2H3 cells were pulsed for 10 min with 125I-BC4 and 125I-AA4 and then chased from 0 min to 3 hr before the Percoll density gradient fractions (as shown in online Supplemental Figures SF1, SF2, and SF3) were prepared. With time, 125I-BC4 decreases in the light membrane fraction and increases in the dense lysosome fraction (Figure 2A). In contrast, when unstimulated cells are labeled with 125I-AA4, there is little movement of the labeled antibody into the dense lysosome fraction (Figure 2B). However, when cells labeled with 125I-AA4 are stimulated by Fc∊RI, the kinetics are similar to that seen with 125I-BC4. With time, there is a decrease in 125I-AA4 in the light membrane fraction and an increase in the dense lysosome fraction (Figure 2C). These results show that the gangliosides were internalized and traffic with the same kinetics as Fc∊RI.
Fc∊RI Clusters on the Surface of RBL-2H3 Cells Incubated With BC4-Gold
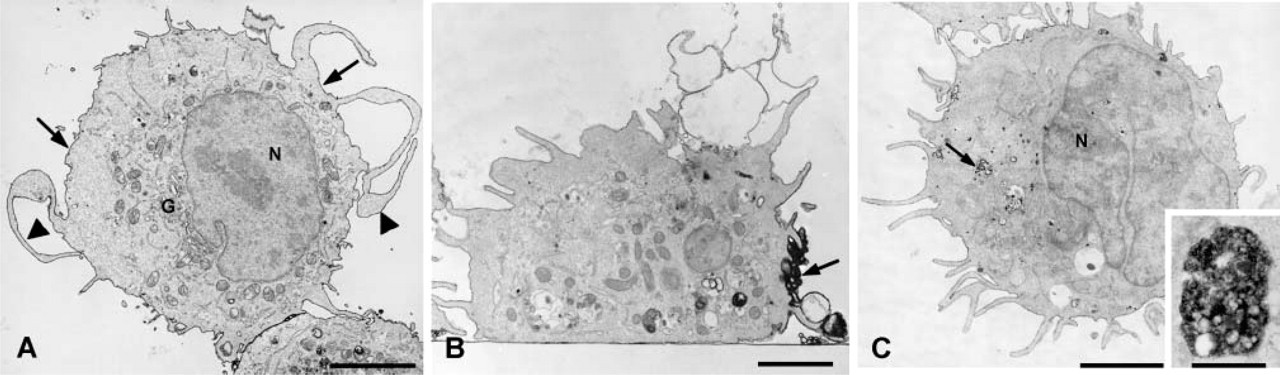
To confirm that conjugation of MAb BC4 to colloidal gold did not alter its function, RBL-2H3 cells were incubated with MAb BC4-gold, fixed at varying time intervals, and the surface distribution of the BC4-gold examined by scanning electron microscopy. Unstimulated RBL-2H3 cells have a fusiform shape and their surface is covered with fine microvilli (Figure 3A). When the cells are incubated with BC4-gold, the cells spread over the substratum and their surface becomes covered with membrane ruffles. Initially, the gold complexes are randomly distributed on the cell surface (Figure 3B), but with time they become aggregated (Figure 3C), demonstrating receptor clustering after stimulation. Histamine release was also examined after incubation with BC4-gold. When the RBL-2H3 cells are stimulated with unconjugated MAb BC4, at 30 min of incubation they release 41.6 ± 4% of their histamine. For cells incubated with BC4-gold, the histamine release was 45 ± 7%. Therefore, these results demonstrate that the ability of the BC4-gold to stimulate histamine release is unaltered.
Fc∊RI Is Internalized in Coated Pits and With Time Is Delivered to Lysosomes
To investigate the internalization of Fc∊RI, RBL-2H3 cells were incubated with MAb BC4 conjugated to colloidal gold and then examined at various time intervals by transmission electron microscopy. Transferrin-HRP was used as a marker of early endosomes, and AcPase activity was used to identify lysosomes. At the earliest times of incubation (<5 min), BC4-gold is found distributed on the cell surface (Figure 4A). At 5 min, the gold complexes can be found in coated vesicles budding from the cell surface (Figure 4B). At 10 min of incubation the BC4-gold is seen in smooth membrane vesicles near the plasma membrane (Figure 4C) that can be identified as early endosomes by the presence of HRP-transferrin within the vesicles (Figure 4D). At 30 min after stimulation, the majority of BC4-gold has been internalized and can now be found in compartments morphologically similar to CURL (compartment for uncoupling receptor and ligand) (Breitfeld et al. 1985), a type of early endosome (Figure 4E). These same compartments label with transferrin-HRP, confirming their identity as early endosomes (Figure 4F). At 1 hr of incubation with BC4-gold, the majority of the gold complex is now localized in multivesicular bodies (Figure 5A), and at 3 hr of incubation most of the BC4-gold is found in structures that morphologically resemble lysosomes (Figure 5B). Identity of these compartments as lysosomes can be confirmed by their lack of transferrin-HRP (Figure 5C) and by the presence of AcPase (Figure 5D).
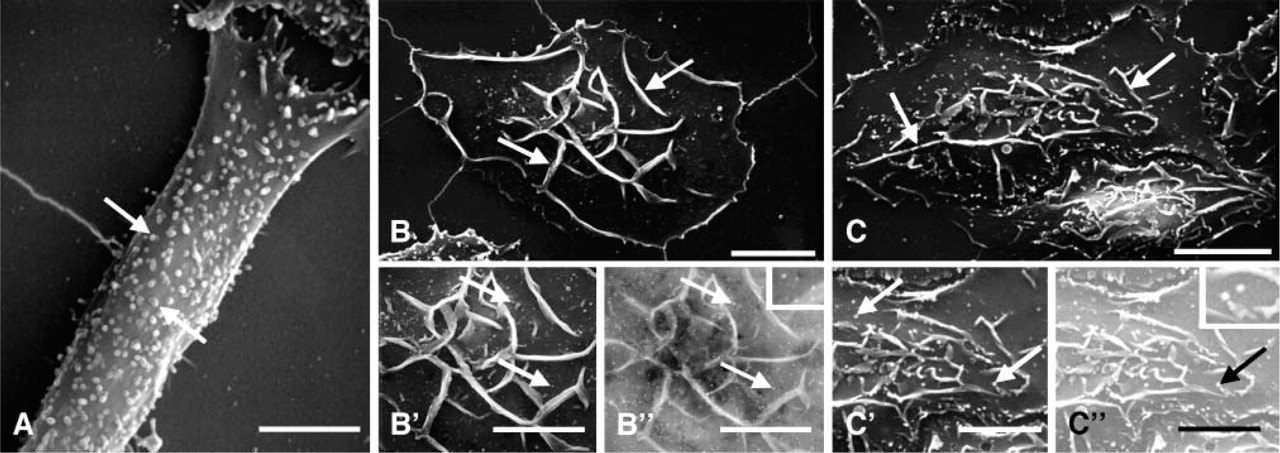
By scanning electron microscopy, stimulation of RBL-2H3 cells by BC4-gold results in clustering of the gold on the cell surface.
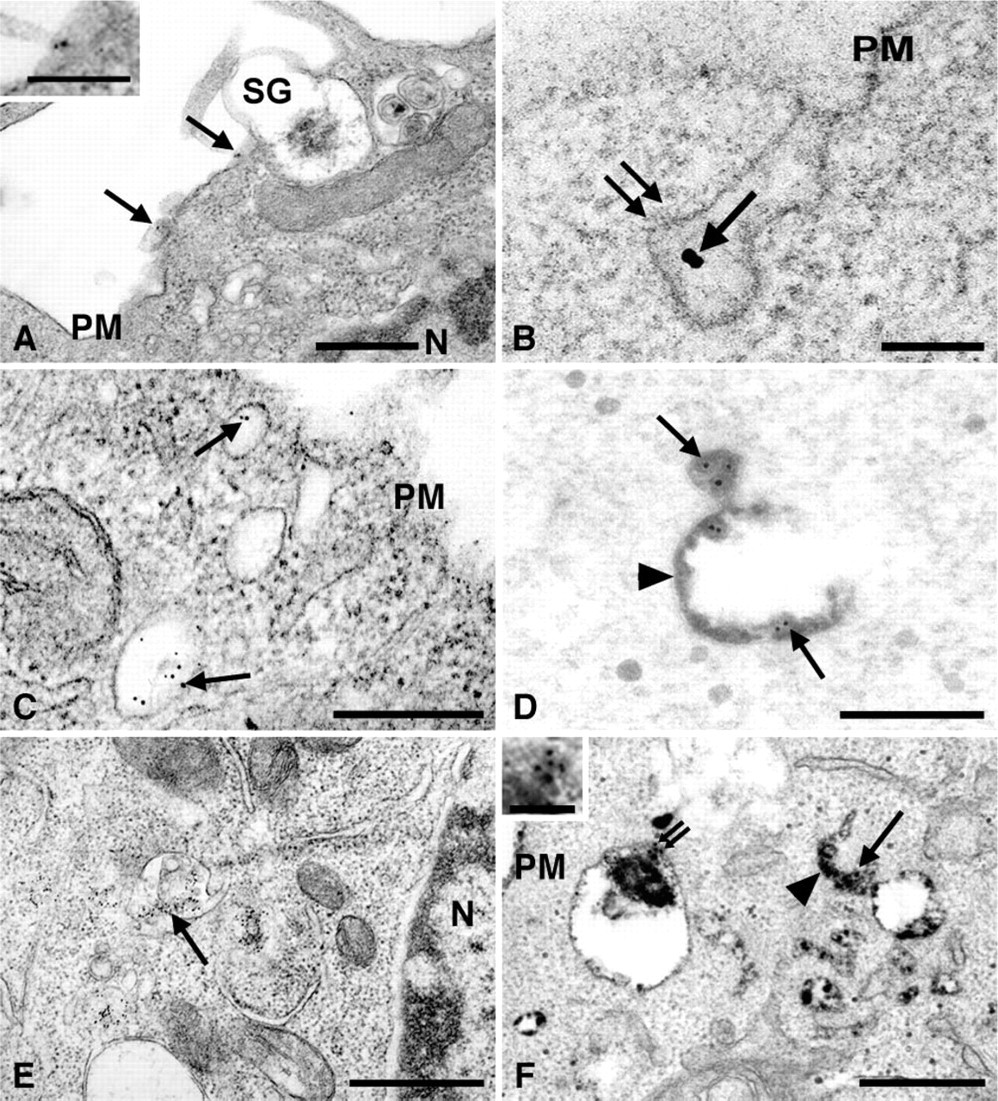
Until 30 min of incubation, the majority of the BC4-gold is found in early endosomes. RBL-2H3 cells were incubated with MAb BC4 conjugated to colloidal gold for varying times and observed by transmission electron microscopy.
Gangliosides Derived From GD1b Follow the Same Endocytic Pathway as Fc∊RI in Stimulated Cells
To evaluate the relationship between the endocytic pathway followed by Fc∊RI and the gangliosides derived from GD1b, BC4-gold was used in combination with AA4-HRP to trace the endocytic pathways. When AA4-HRP is used alone, at 30 min, although the cells appear activated, the majority of the AA4-HRP is on the cell surface and little HRP reaction product can be seen inside the cells (Figure 6A). If the AA4-HRP is crosslinked with rabbit anti-mouse IgG, although the AA4-HRP caps on the cell surface (Figure 6B), there is little internalization. However, when Fc∊RI is cross-linked with MAb BC4 (unlabeled) thereby simulating the cells, the AA4-HRP is rapidly internalized and, at 30 min, AA4-HRP reaction product can be seen in endocytic compartments inside the cell (Figure 6C). Therefore, significant internalization of the gangliosides occurs only when the cells are stimulated via Fc∊RI.
At later times the BC4-gold moves into lysosomes. RBL-2H3 cells were incubated with MAb BC4 conjugated to colloidal gold for varying times and observed by transmission electron microscopy.
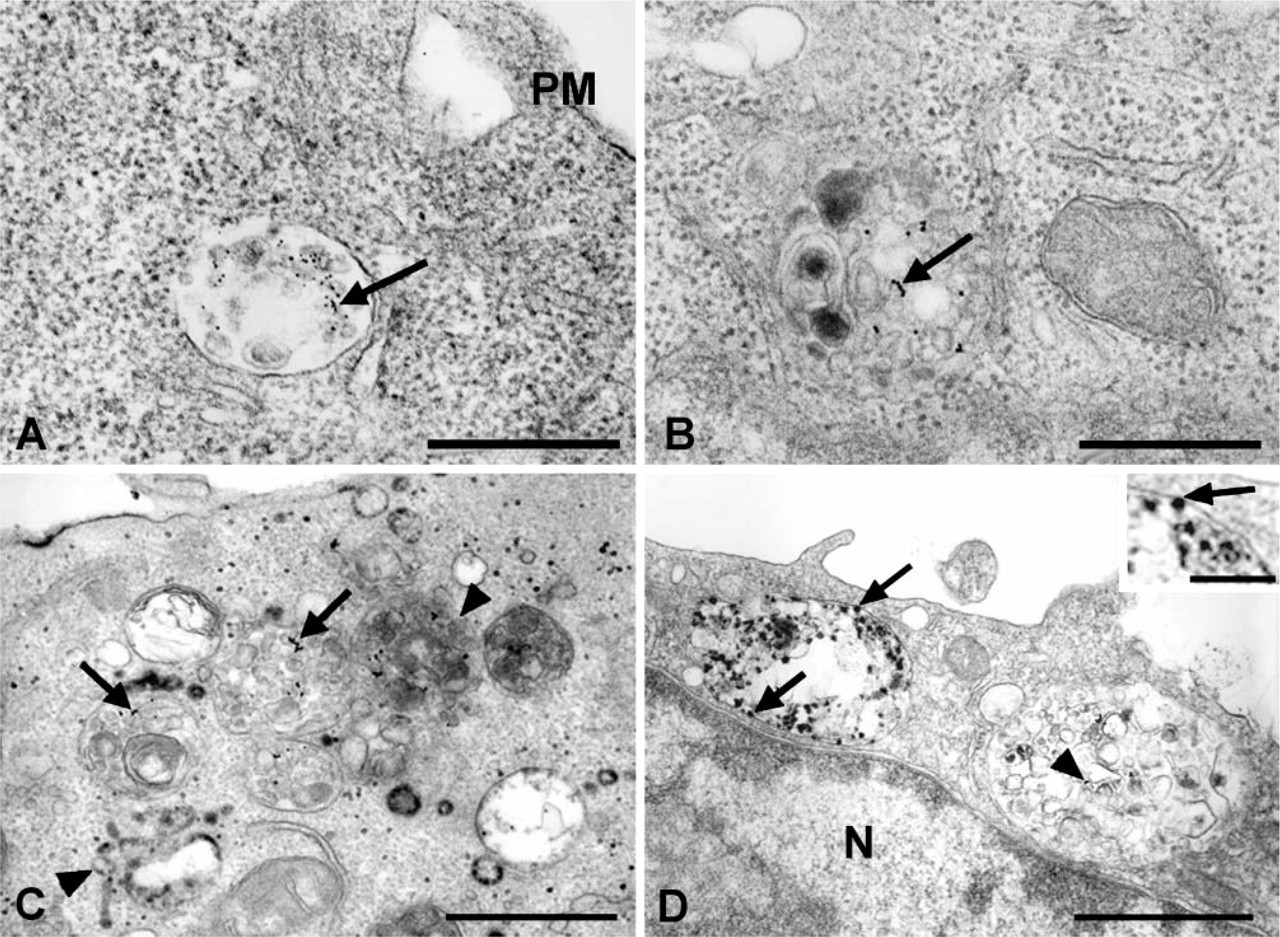
Internalization of the gangliosides was then examined using AA4-HRP in cells stimulated via Fc∊RI by BC4-gold. At 5 min of incubation, both markers could be seen in coated vesicles budding from the plasma membrane (Figure 7A). At 15 min, the BC4-gold and AA4-HRP were colocalized in early endocytic vesicles adjacent to the plasma membrane (Figures 7B and 7C). At 30 min, the tracers had moved into structures similar to late endosomes (Figure 7D) and at 3 hr could be found in vesicles resembling lysosomes (Figure 7E). Identity of these structures as lysosomes was confirmed by the presence of AcPase activity (Figure 7F). Monomeric F(ab) fragments of MAb AA4 gave identical results as the whole antibody (data not shown), indicating that the valiancy of the antibody did not affect its intracellular trafficking. These results demonstrate that the endocytic pathways for both Fc∊RI and the gangliosides are virtually identical, and that at all time points examined the markers are colocalized.
Discussion
Results of this investigation demonstrate that in RBL-2H3 cells stimulated via Fc∊RI, internalization of the mast cell-specific gangliosides recognized by MAb AA4 is augmented, and Fc∊RI and the gangliosides are internalized together and follow the same intracellular endocytic pathway. In unstimulated cells, although MAb AA4 binding partially activates RBL-2H3 cells (Oliver et al. 1992), little internalization of the gangliosides occurs.
Gangliosides recognized by MAb AA4 appear to be intimately involved with signaling through Fc∊RI. This MAb is one of a panel that was produced by immunizing mice with RBL-2H3 cells and selected due to its ability to inhibit sensitization with IgE. It was then found to inhibit, in a dose- and time-dependent manner, histamine release from RBL-2H3 cells (Basciano et al. 1986). MAb AA4 recognizes two gangliosides that are α-galactosyl derivatives of GD1b (Guo et al. 1989). Binding of MAb AA4 or F(ab)'2 fragments to the gangliosides on the surface of RBL-2H3 cells produces morphological and biochemical changes similar to those seen with activation of Fc∊RI but without histamine release (Oliver et al. 1992; Swaim et al. 1994; Stephan et al. 1997). Gangliosides coprecipitate with Fc∊RI (Stephan et al. 1991) as well as with the tyrosine kinase p53/56Lyn (Minoguchi et al. 1994) and previously have been shown to be associated with Fc∊RI and inner leaflet raft components in RBL-2H3 cells (Sheets et al. 1999; Pyenta et al. 2001). Results from this study together with the previously published data suggest that, in stimulated cells, gangliosides recognized by MAb AA4 and Fc∊RI are associated in the same lipid raft.
Analysis of the subcellular distribution of the gangliosides recognized by MAb AA4 and Fc∊RI on sucrose gradients showed that after activation of Fc∊RI there was a shift in the distribution of the receptor to the lighter (lipid raft) fractions where it was colocalized with the gangliosides. It is well established that in many cell types, including mast cells, important events in signal transduction occur in lipid rafts (Holowka et al. 2000,2005; Wilson et al. 2000,2004; Holowka and Baird 2001; Rivera et al. 2001; Pike 2003). The movement of Fc∊RI into the lipid rafts where it is in proximity to the gangliosides recognized by MAb AA4 may be another mechanism that regulates signal transduction in mast cells. Lipid rafts play an important role in signaling from many receptors that signal through tyrosine phosphorylation including the T-cell receptor (Harder 2004; Horejsi 2004), B-cell receptor (Petrie and Deans 2002), endothelial growth factor receptor (EGFR) (Mineo et al. 1999), platelet-derived growth factor (PDGF) receptor (Liu et al. 1997), and the insulin receptor (Gustavsson et al. 1999). Several of these receptor tyrosine kinases have a differential response to the addition of ligand (Pike 2003). Whereas EGFR moves out of the lipid rafts upon addition of ligand, the insulin receptor moves into the rafts, and the PDGF receptor is largely unaffected by the addition of ligand. In contrast, immune receptors are dispersed throughout the plasma membrane but upon activation move into lipid rafts.
Stimulation of RBL-2H3 cells is required for internalization of the gangliosides.
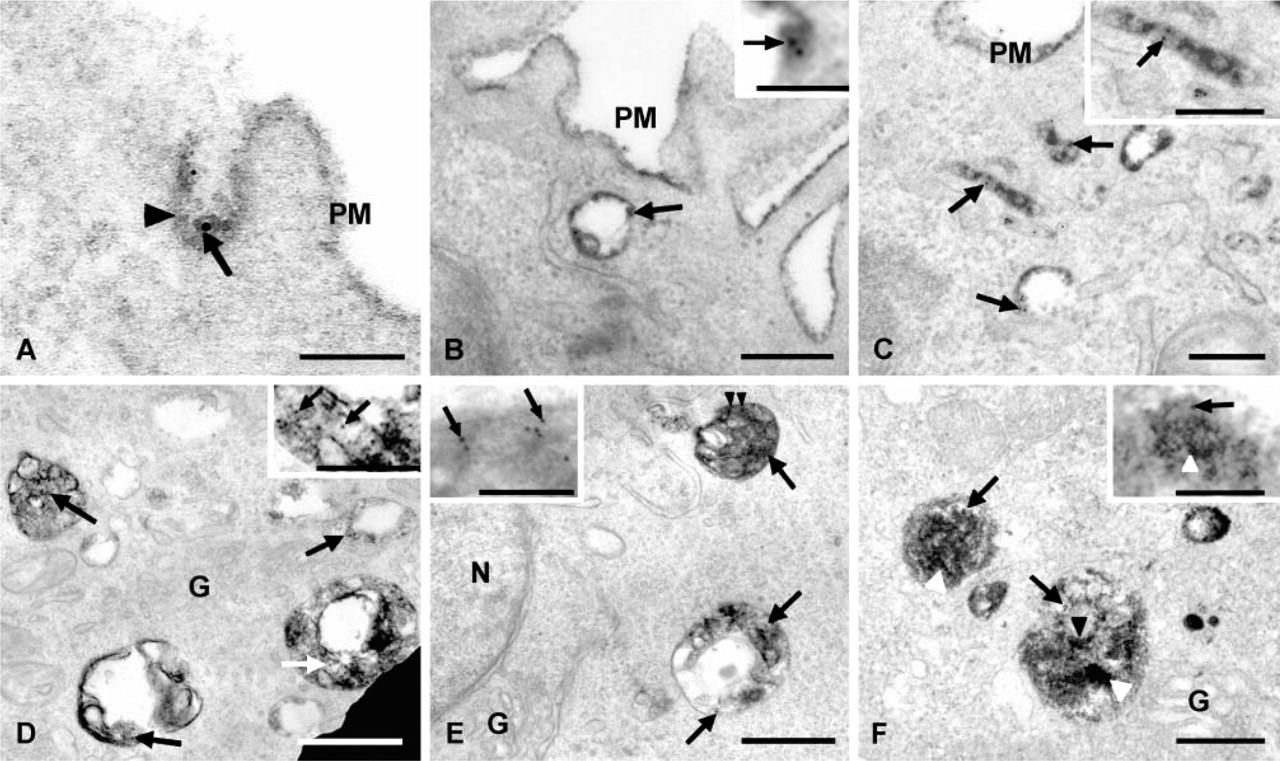
Gangliosides follow the same endocytic pathway as Fc∊RI.
The process of endocytosis itself may play an important role in signal transduction (McPherson et al. 2001). It has generally been accepted that endocytosis of receptor-ligand complexes leads to downregulation of the receptor (Baass et al. 1995; Burke et al. 2001; Wiley and Burke 2001). However, there is growing evidence that suggests that receptor-ligand complexes may still be active in signaling even if they are internalized into endosomes. Studies with the neurotrophin receptor, tropomyosin receptor kinase A, suggest that nerve growth factor binds to this receptor at axonal tips and is transported to the cell body in actively signaling endosomes (Grimes et al. 1996). Signaling from receptors internalized in endosomes may also be different from receptors localized on the cell surface. For example, endosomally localized EGFR can activate Ras but not phospholipase C-γ (Haugh et al. 1999a,b). Furthermore, internalized receptors may associate with different proteins. With EGFR, Shc is associated with the receptor at the cell surface and in endosomes, but other molecules such as Grb2 are associated primarily with the EGFR at the cell surface and still others such as Eps8 are found associated only with the intracellular receptors (Burke et al. 2001). The fact that the gangliosides are internalized along with Fc∊RI may facilitate the structural preservation of signaling complexes and the prolongation of the signal.
It is well established that intracellular degradation of gangliosides occurs in lysosomes (Allende and Proia 2002). Additionally, Möbius et al. (1999), using externally administered GM1, showed that GM1 can be found in all compartments of the endocytic pathway including lysosomes. The present study extends this observation by demonstrating that internalization of the gangliosides is stimulated by activation of RBL-2H3 cells through Fc∊RI, and that the receptor and the gangliosides are most likely internalized together and follow the same pathway to lysosomes. Thus, this cointernalization may be important in the downregulation of Fc∊RI by removing not only the receptor from the cell surface, but also the gangliosides associated with the lipid raft signaling complex.
Footnotes
Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo for financial support.
We also thank Mrs. Judy Waters, Ms. Lynda Weedon, and Mr. Anderson Roberto de Souza for technical help.