
Abstract
Tissue microarray (TMA) technology provides a possibility to explore protein expression patterns in a multitude of normal and disease tissues in a high-throughput setting. Although TMAs have been used for analysis of tissue samples, robust methods for studying in vitro cultured cell lines and cell aspirates in a TMA format have been lacking. We have adopted a technique to homogeneously distribute cells in an agarose gel matrix, creating an artificial tissue. This enables simultaneous profiling of protein expression in suspension- and adherent-grown cell samples assembled in a microarray. In addition, the present study provides an optimized strategy for the basic laboratory steps to efficiently produce TMAs. Presented modifications resulted in an improved quality of specimens and a higher section yield compared with standard TMA production protocols. Sections from the generated cell TMAs were tested for immunohistochemical staining properties using 20 well-characterized antibodies. Comparison of immunoreactivity in cultured dispersed cells and corresponding cells in tissue samples showed congruent results for all tested antibodies. We conclude that a modified TMA technique, including cell samples, provides a valuable tool for high-throughput analysis of protein expression, and that this technique can be used for global approaches to explore the human proteome.
I
In vitro cultured cell lines have been used extensively in basic biomedical research, and much knowledge today concerning malignant transformation, differentiation, and other cellular processes originates from experiments performed in cell lines. Although cell lines in several aspects are poor representatives of corresponding malignant tumors, in vitro cultured cells provide well-defined model systems for analyzing molecular events in a cellular context. The possibility to culture indefinite amounts of clonal cells in addition to an option to perform controlled manipulations is also advantageous. In this study we used IHC to analyze protein expression in 46 unique cell lines and 12 patient cell samples, using 20 different antibodies. Frequently used cell lines were selected to represent the major forms of solid tumors and hematological malignancies. As the cell TMA was designed as a complement to the TMAs containing human normal and cancer tissues used in an antibody-based proteomics project (Uhlen et al. 2005), cell types not present in the these TMAs (Kampf et al. 2004) were also included. One ambition with the cell TMA was to represent leukemias and lymphomas, both as cell lines and as fresh patient cells. With this design, the cell TMA enables studies not only of tumor progression but also steps of differentiation and normal hematopoiesis (Drexler and MacLeod 2003). Various methods for embedding of cultured cells in paraffin to facilitate subsequent sectioning have been described (Hoos and Cordon-Cardo 2001; Morgan 2001; Moskaluk and Stoler 2002), and the use of in vitro cultured cells as controls for quality assurance and analysis of variations in histoprocessing as well as immunostaining has previously been demonstrated (Riera et al. 1999; Wester et al. 2000). Recently, two separate studies described the use of a limited number of adherent cell lines embedded in paraffin for production of cell microarrays as a high-throughput method for protein profiling in cell lines (Ferrer et al. 2005; Waterworth et al. 2005).
Cells and cell lines included in the cell TMA


S, suspension; A, adherent.
F, female; M, male; U, unclear; §, cells are not of clonal origin.
DSMZ, German Resource Centre for Biological Material (http://www.dsmz.de/); ATCC, American Type Culture Collection (http://www.lgcpromochem-atcc.com/); KN, Prof. Kenneth Nilsson; BW, Prof. Bengt Westermark; GN, Prof. Gunnar Nilsson; NEF, Prof. Norbert E. Fusenig; NEH, Assoc. Prof. Nils-Erik Heldin; LCW, Prof. Lena Claesson-Welsh.
PBMC, peripheral blood mononuclear cells; CML, chronic myelogenous leukemia; APL, acute promyelocyte leukemia; AML, acute monocytic leukemia; ALL, acute lymphoblastic leukemia; CLL, chronic lymphoblastic leukemia.
The gold standard for IHC evaluation is manual examination of immunoreactivity using light microscopy. To reduce subjectivity and increase reproducibility, computerized image-analysis systems have been developed, both for enzyme-based (Ranefall et al. 1998) and fluorescence-based detection of IHC-stained cells (Camp et al. 2002). Although image analysis provides a valuable complementary method, instruments have not yet reached sufficient standards to replace manual evaluation and scoring of immunoreactivity performed by a certified pathologist. Such manual scoring is based on experience and the comparison with immunostaining of reference tissues. When applicable, manual scoring is performed as an evaluation of intensity of immunoreactivity (negative, weak, moderate, or strong), fraction of positive cells (estimated percentage), and subcellular localization (nuclear, cytoplasmic, membranous, and/or extracellular).
Antibody-based proteomics has been developed to enable mapping of protein expression in human tissues on a proteome-wide scale (Uhlen and Ponten 2005). Recently, a first version of a Protein Atlas (www.proteinatlas.org) was made publicly available (Uhlen et al. 2005). The ultimate goal for this effort is to create an atlas of protein expression patterns by generating antibodies toward all non-redundant human proteins using information from the human genome sequence (www.ensembl.org). Antibodies are subsequently applied to human tissue samples in a TMA format (Agaton et al. 2003; Nilsson et al. 2005).
Here we present a technique for the assembly of cells in a TMA format, including a review on how to create an artificial tissue by suspension of cultured cells in agarose. Optimal cell concentration for three-dimensional spreading of cells, harvesting protocols, and sectioning strategies is presented. In conclusion, the described strategy can be used for high-throughput antibody-based proteomics.
Materials and Methods
Design of TMAs
For comparison of the staining quality between cells and tissues, TMAs containing tissue samples were designed and generated as previously described (Kampf et al. 2004). The cell TMA block was designed to contain 46 cell lines including 27 adherent (A), 18 suspension (S), and one with mixed adherent and suspension growth pattern as well as 10 cell aspirates from leukemia/lymphoma patients (four distinct different CLLs, two CMLs, two AMLs, one B-ALL, and one T-ALL) and two samples consisting of peripheral blood mononuclear cells (PBMC) from healthy blood donors (one male and one female) (Table 1). All cell samples were represented in duplicate. Leukemia/lymphoma cell lines were selected to represent different phenotypes representing fundamental stages of differentiation in the hematopoietic system. A majority of selected cell lines were acquired from commercial distributors: German Collection of Microorganisms and Cell Cultures, DSMZ (http://www.dsmz.de/), and ATCC (http://www.lgcpromochem-atcc.com). A selection of cell lines was obtained from academic research groups. These were KM-3, U-937, U-266/70, U-266/84, and Karpas-707 (Prof. Kenneth Nilsson, Dept. of Genetics and Pathology, Uppsala University, Uppsala, Sweden); U-251MG (Westermark et al. 1973) and WM-115 (Prof. Bengt Westermark, Dept. of Genetics and Pathology, Uppsala University); HaCaT (Boukamp et al. 1988) [Prof. Norbert E. Fusenig, German Cancer Research Center (DKFZ), Heidelberg, Germany]; HMC-1 clone 1:2 (Butterfield et al. 1988) (Prof. Gunnar Nilsson, Dept. of Genetics and Pathology, Uppsala University); HTh 83 (Dahlman et al. 2000) (Assoc. Prof. Nils-Erik Heldin, Dept. of Genetics and Pathology, Uppsala University); TIME (Venetsanakos et al. 2002) (Prof. Lena Claesson-Welsh, Dept. of Genetics and Pathology, Uppsala University and Prof. Martin McMahon, UCSF Comprehensive Cancer Center, San Francisco, CA).
Cell Culture and Handling of Cell Aspirates and PBMCs
Cell lines were cultured in 37C in a humidified atmosphere with 5% CO2 and in accordance with recommendations from the respective distributor (Table 1). Cell lines were grown and harvested in proliferation phase, i.e., 16-24 hr before harvest confluent cells were split 1:2. All cell lines were tested for mycoplasma infection by inoculation on indicator cells and Hoechst staining (SVA; Mycoplasma Laboratory, Uppsala, Sweden) (Chen 1977).
To obtain the best cell morphology, two strategies for detachment of adherent cells were tested: 0.5% trypsin or a rubber policeman. Adherent cells were washed with 1X PBS, 2 mM EDTA prior to detachment. After detaching the cells from the plastic surface using either trypsin or a rubber policeman, cells were resuspended in 8 ml (Petri dish, 10 cm in diameter) of respective medium with the addition of 2% fetal bovine serum (Gibco, Invitrogen, Life Technologies; Paisley, Scotland). Cell suspension was then removed from the plate and transferred to a 250-ml centrifugation flask and centrifuged at 400 × g at 4C. Following removal of the supernatant, the cell pellet was resuspended in ice-cold 1X PBS and transferred to a 50-ml tube where the volume was readjusted to 50 ml with 1X PBS. Finally, cells were counted and the centrifugation step was repeated.
Suspension-grown cell lines were started at a concentration of 0.2 × 106 cells/ml and harvested on ice by centrifugation as above in 250-ml centrifugation flasks at 400 × g at 4C, followed by the washing process in PBS as described for the adherent cell lines.
Patient cells and blood donor cells were collected with permission from the local Ethics Committee, and informed consent was obtained, respectively. Patient leukemia cells stored frozen in fetal bovine serum and 10% DMSO were thawed and washed twice in 1X PBS. In the second wash, cells were counted and viability was tested with trypan blue in a Bürker chamber. PBMCs were isolated from buffy-coat preparations with Ficoll Paque Plus according to the manufacturer's recommendation (GE Healthcare, Amersham Biosciences; Uppsala, Sweden), with the addition of lysis of the red blood cells.
Antibodies included in the study showing one example of positive immunostained cell line and tissue for each antibody
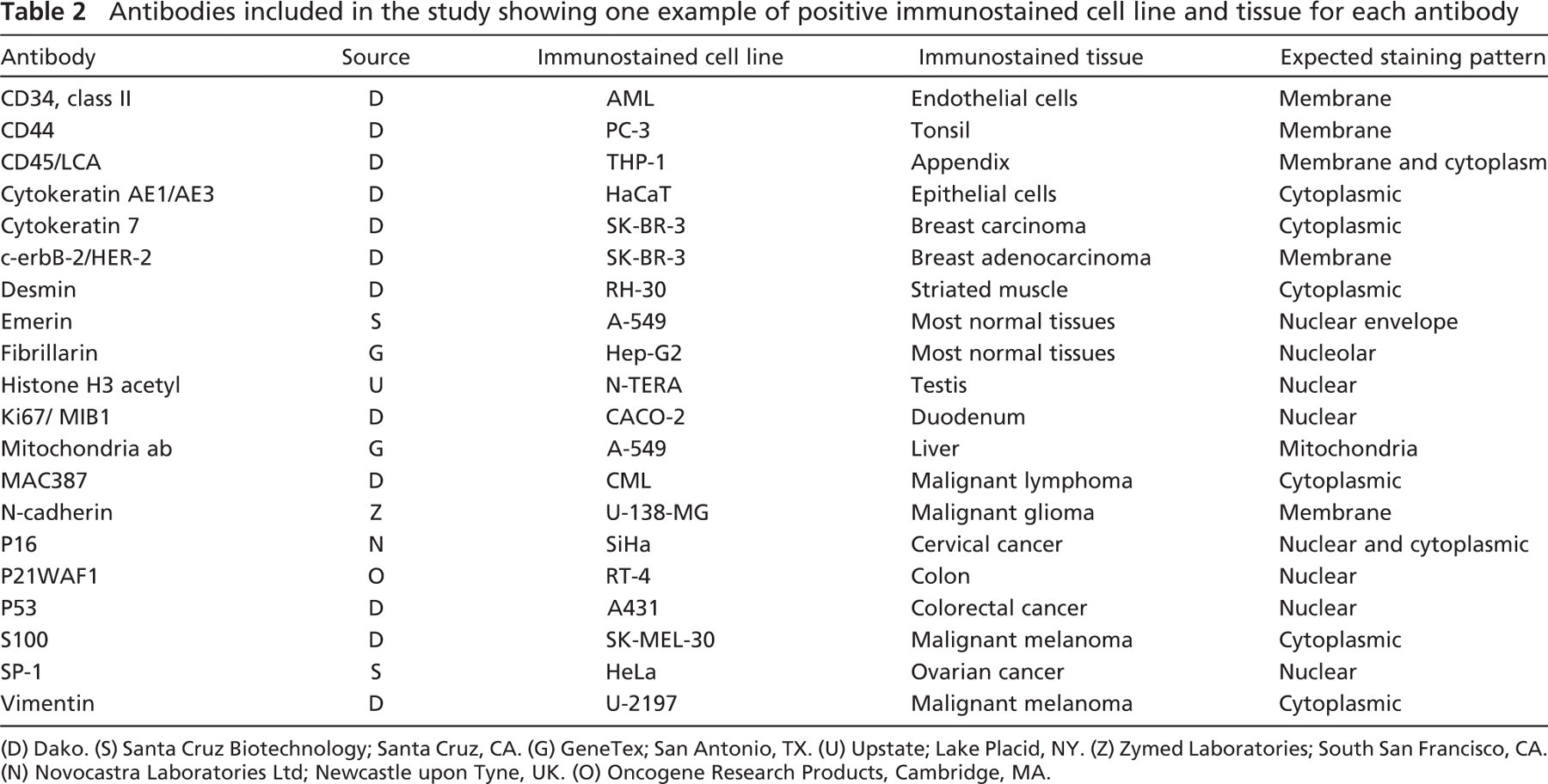
(D) Dako. (S) Santa Cruz Biotechnology; Santa Cruz, CA. (G) GeneTex; San Antonio, TX. (U) Upstate; Lake Placid, NY. (Z) Zymed Laboratories; South San Francisco, CA. (N) Novocastra Laboratories Ltd; Newcastle upon Tyne, UK. (O) Oncogene Research Products, Cambridge, MA.
Preparation of the Agarose Cell Gel
Each cell pellet was suspended in 10 ml of 4% neutral-buffered formaldehyde and fixed for 60 min as a minimum and 16 hr as a maximum (Wester et al. 2000). Cells were centrifuged 5 min at 400 × g at 4C and the supernatant was removed. To achieve equal temperature, agarose cells were resuspended in 750 μl 4% formaldehyde and placed in a 65C water bath. An equal volume of 5-6% Sea plaque agarose (FMC BioProducts; Rockland, ME) in 1X PBS was added, and cells were gently suspended with a pipette and transferred into a plastic 24-well cell culture plate with a well diameter of 16 mm (Costar; Cambridge, MA). Agarose was allowed to polymerize before the plate was transferred to 4C. Prior to histoprocessing, the cell-agarose gel was fixed in 4% formaldehyde for 1 to 4 hr. Histoprocessing was performed using a vacuum infiltration processor (Ventana Medical Systems; Tucson, AZ) according to standard procedures for tissue processing at the Department of Pathology, Uppsala University Hospital. Samples were initially fixed in 4% formaldehyde (3 hr), dehydrated in graded alcohols (8 hr) and xylene (3.5 hr), paraffin impregnated, and finally embedded in paraffin. To assess an optimal cell density and distribution of cells, cell gels were prepared at four different concentrations: 10, 25, 50, and 100 × 106 cells/ml.
TMA Production
TMAs were created essentially as previously described (Kampf et al. 2004). In brief, donor blocks were prepared from normal and tumor tissues, and hematoxylin-eosin-stained sections were used to select appropriate areas for the TMA construct. Using an automated tissue arrayer (Beecher Instruments; Silver Spring, MD), two 1.0-mm diameter punches were taken from each donor block and transferred to the recipient block. For construction of the cell TMA, the same technique as above was used, but here punches with a diameter of 0.6 mm were taken and placed in the recipient block with a core-to-core distance of 1.3 mm in all directions. For recipient blocks, three different brands of paraffin were tested: Histowax (Histolab; Gothenburg, Sweden), Medite tissue wax (Medite Histotechnic; Burgdorf, Germany), and J.T. Baker Ultrapar (Deventer, The Netherlands). The melting point for the different paraffin brands was 56-58C, 54-56C, and 56-58C, respectively. Two microtome models were used in the study: (1) a standard rotary microtome (RM2165; Leica, Nussloch, Germany; and (2) a rotary microtome designed for waterfall-sectioning technique (Microm; Walldorf, Germany). For immunostaining, 4-μm sections were cut, placed onto Super Frost/Plus slides (Menzel-Glaser; Braunschweig, Germany), and baked at 40C overnight.
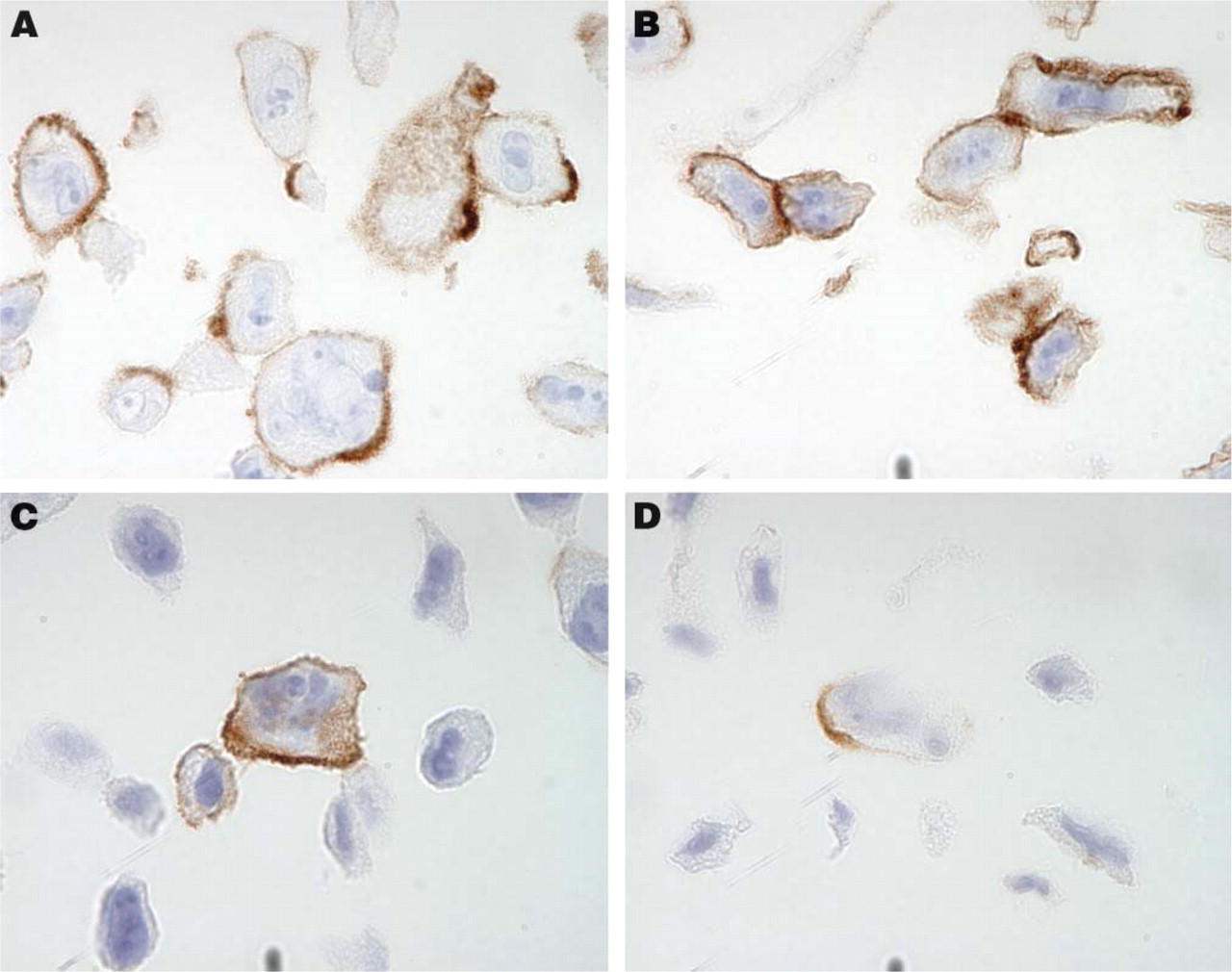
Immunohistochemical staining using antibodies directed toward the transmembrane glycoproteins CD44 (
IHC
After deparaffinization in xylene and hydration in graded alcohols, heat-induced epitope retrieval (HIER) was performed using a Decloaking chamber (Biocare Medical; Walnut Creek, CA) where slides were immersed in target-retrieval solution (TRS, pH 6.0; Dako, Glostrup, Denmark) and boiled for 7 min for the cell TMA and 10 min for the tissues, at 125C (Norton et al. 1994). TMA slides were stained with 20 commercially available antibodies (Table 2) in an automated immunostaining instrument, Autostainer Plus (Dako), using EnVision detection kit (Dako). Slides were manually counterstained in Harris hematoxylin (Sigma; St Louis, MO). Finally, slides were dehydrated through graded alcohols to xylene and mounted in organic mounting medium (Pertex; Histolab).
Image Acquisition and Image Analysis
For evaluation of the three-dimensional cell distribution in the cell gel, one section at every 100-μm depth was taken from the paraffin-embedded cell gels and stained with hematoxylin. The 756 × 572 pixel color images with 3 × 256 gray levels were captured using a color video camera (DXC-151; Sony, Tokyo, Japan) attached to a standard microscope (BH-10; Olympus, Tokyo, Japan) using a X10 objective. This resulted in a final magnification of X25 and a pixel size of 1.6 μm for a wavelength of 550 nm. A semi-automatic color-based quantification method was used to count the cells (Ranefall et al. 1997).
Results
Cell Culture and Handling of Cell Aspirates and PBMCs
Suspension-grown cell lines appear ideal for cell gel preparations, as no manipulation, e.g., trypsinization, is needed prior to fixation and histoprocessing. For adherent cell lines, there was no visible morphological difference if cells were harvested using trypsin or a rubber policeman. Both methods provided intact immunogenic properties of tested membrane proteins (Figure 1). Because the protocol using trypsin was demonstrated to be more efficient for detaching the cells, it was used for harvesting all adherent-grown cell lines to be included in the cell TMA. Both suspension- and adherent-grown cell lines embedded in agarose showed excellent cell morphology. For each cell line that tested positive for mycoplasma infection, a new negative batch was cultured, which together with its positive counterpart was included in the array.
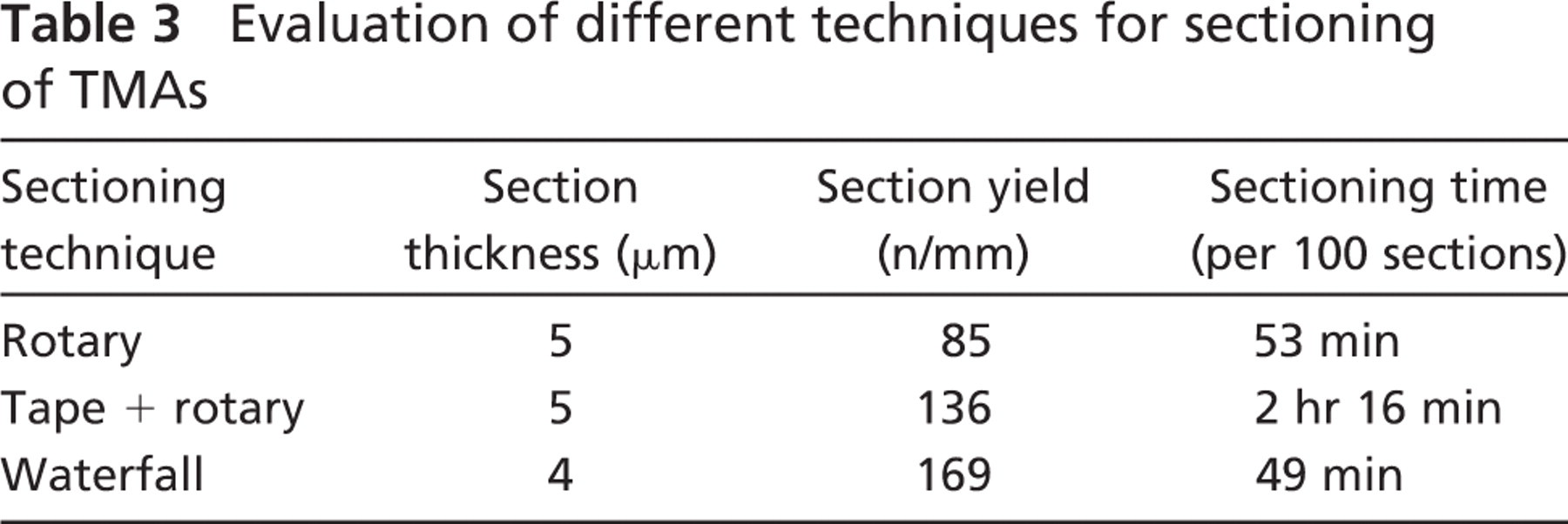
Evaluation of different techniques for sectioning of TMAs
Preparation of the Agarose Cell Gel
Three-dimensional distribution of cells in the cell gel was evaluated by quantifying hematoxylin-stained 4-μm sections taken at 100-μm intervals. This was done using cell concentrations of 25 and 50 × 106 cells/ml from the U-937 cell gel. Results from image analysis showed that the mean number of cells/mm2 in 28 sections was 311 (95% CI 288-336) and 884 (95% CI 720-1048), respectively. As visually observed, cells were homogeneously distributed in both concentrations.
Based on visual examination of immunostained slides from all four cell concentrations (5, 25, 50, and 100), a cell concentration of 100 × 106 cells/ml agarose was selected for cell gel production. This resulted in a representation of ∼450 cells/0.6-mm diameter spot, equivalent to ∼1600 cells/mm2. Considering that cell lines are fairly homogeneous (clonal), the selected concentration allowed for an accurate assessment of protein expression using IHC for each given cell line in comparison with the other cell lines used in the cell TMA. Due to a lower number of cells in the clinical cell samples, a lower cell concentration had to be accepted for these cell gels. For these samples we accepted a range of cell concentrations spanning from 25 × 106 to >100 × 106 cells/ml.
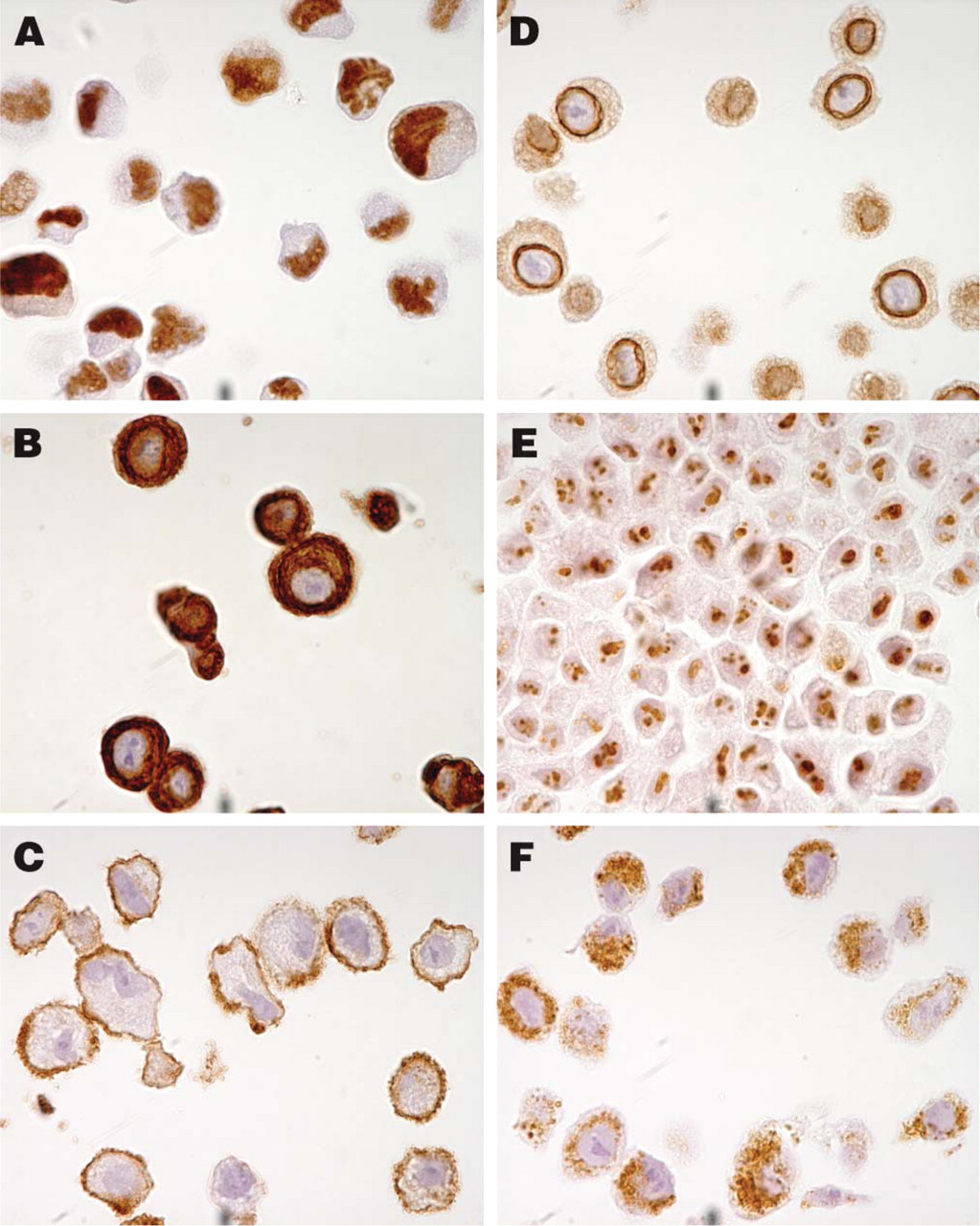
Examples of staining patterns using six antibodies toward antigens in specific subcellular compartments. The antibody toward the nuclear phosphoprotein p53 shows distinct positive nuclei in the epidermoid carcinoma cell line A431 (
TMA Production
The core yield from one cell gel (16 mm in diameter before histoprocessing) was ∼122 cores (0.6 mm in diameter), allowing for a production of ∼60 TMA blocks using duplicate samples. The paraffin with the best characteristics was Histowax (Histolab), which was used for production of both cell TMA recipient and donor blocks. For sectioning of cell TMAs, a waterfall microtome (Microm GmbH) was preferred because it provided a high yield of high-quality paraffin sections. The selection of strategy for sectioning was based on section yield and sectioning time from sectioning of TMAs (Table 3). From each cell TMA block, 350-400 paraffin sections (4 μm) could be extracted as compared with TMA blocks containing tissue cores that yield ∼250-275 sections. Cell TMA sections showed a high representation of cell gel cores. The mean loss of spots after 150 sections was 1, after 250 sections 2, after 300 sections 10, and after 350 sections 20. TMAs containing tissues show a higher rate of loss with a mean loss of spots after 100 sections being 6 and after 150 sections 12. The higher rate of loss in tissue-containing TMAs is due to differences in the thickness of various specimens used as templates.
IHC
The original protocol used for HIER on tissues was 10 min. This was adjusted to 7 min for cells to enable a strong and distinct hematoxylin staining in the cell nuclei. To evaluate the quality and morphology of the cells in the cell TMA, 20 well-characterized antibodies with known immunostaining patterns were selected (Table 2). As shown in Figure 2, antibodies toward antigens in six specific subcellular compartments (nucleus, cytoplasm, plasma membrane, nuclear membrane, nucleolus, and mitochondria) successfully recognized their respective antigen in sections from the cell TMA. For further validation, immunostaining of the cell TMA was compared with staining in the corresponding tissue sections. In all cases where such a comparison was applicable, the staining pattern was in agreement with published data on the expected protein distribution. This is exemplified by the anti-LCA (CD45) antibody, which was positive in a vast majority of leukemia/lymphoma cell lines and also showed positivity in lymphoid cells in the tissue sections (Figures 3A and 3B). The anti-desmin antibody recognized myocytes in striated muscular tissue as well as in the corresponding rhabdomyosarcoma-derived cell line RH-30 (Figures 3C and 3D). Congruent staining was also evident using the HER-2 antibody, which showed strong membranous staining in breast carcinomas and the cell line SK-BR-3 with known high Her-2 expression (Figures 3E and 3F).
Discussion
Statistical significance of TMA technology, i.e., using core biopsies as representatives for large tissue biopsies, has been evaluated in earlier studies, and it has been shown that duplicate or triplicate biopsies provide a high degree of concordance (Camp et al. 2000; Hoos and Cordon-Cardo 2001). TMA technology offers advantages in several aspects, including a minimal consumption of valuable biological samples as well as being time and cost effective, the latter due to a reduced amount of required reagents for a comprehensive experiment. Another important benefit is that all included tissue samples are analyzed simultaneously with the same protocol, thereby reducing the variability in IHC. This allows for controlled comparative studies of a multitude of tissues assembled in the same TMA. Additionally, this permits a robust and reproducible strategy when comparing differences in protein expression in various samples from the same patient or from tissues sampled at different time points during an experiment. Furthermore, TMAs provide an excellent platform to study different modes of fixation and other variables in histoprocessing. The assembly of in vitro cultured cells in a TMA also opens up possibilities to analyze protein expression in a range of different experimental settings where cells have been manipulated prior to fixation, e.g., technologies using transfections and RNAi.
It may be necessary to limit the number of cells in a study when large-scale efforts including several different experiments are to be performed. However, cells dispersed at relatively low cell densities in a cell gel reduce the amount of cells needed as compared with a cell pellet. This is advantageous because it also allows extensive studies on scarce patient samples from fine needle biopsies where only a limited number of cells are available. One requirement for a suitable cell gel is a viscosity at 65C that facilitates mixing of the cells and subsequent transferal into a plastic well. Furthermore, the cell-embedding medium must endure fixation, histoprocessing, and paraffin embedding with minimal shrinkage and without fragmentation. It must also preserve acceptable cell morphology and be easy to section in a microtome. This was true for the described cell gel preparation technique.
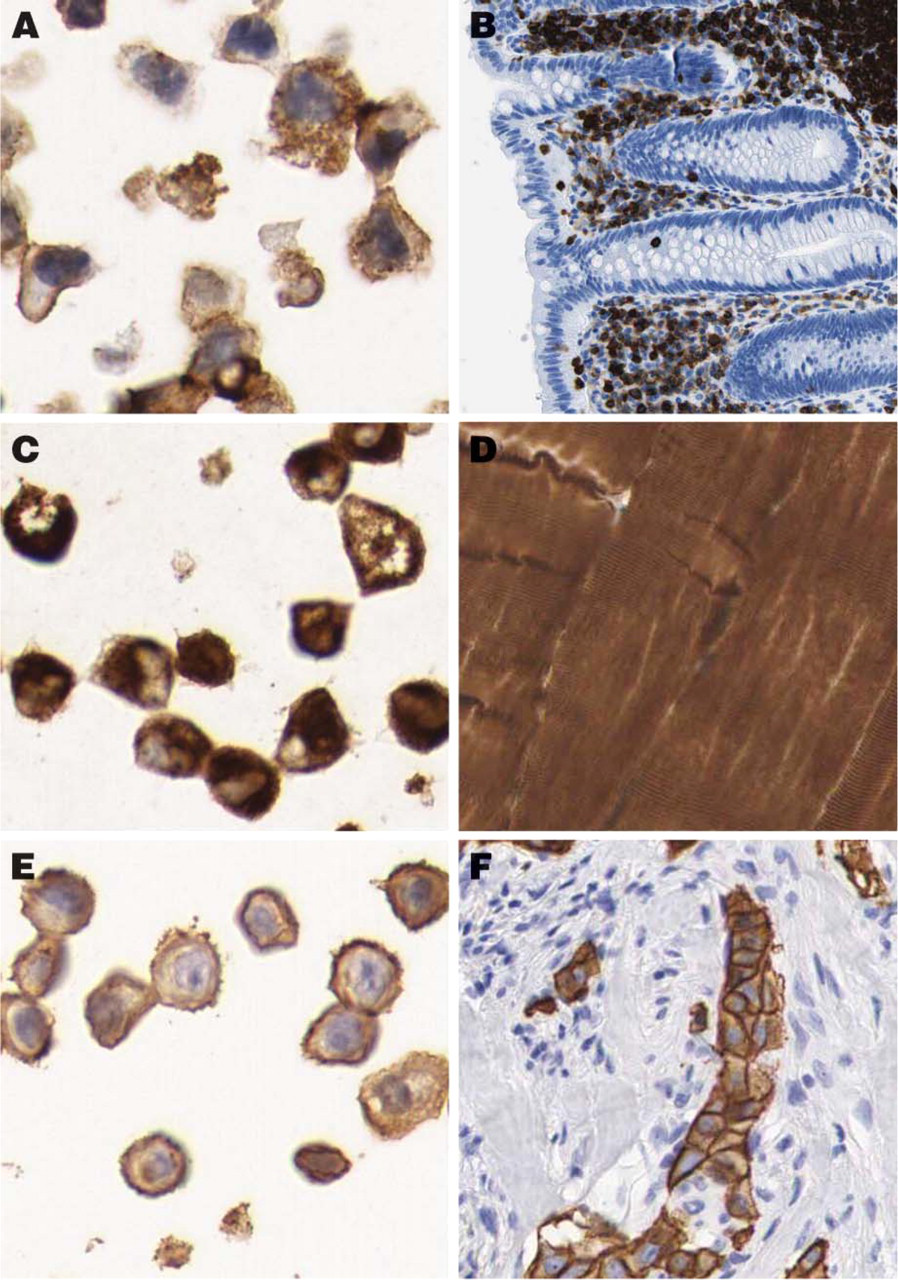
Examples of congruent staining patterns in tissue and corresponding cell line. Immunostaining with the anti-LCA (CD45) antibody recognizing the CD45 glycoprotein shows strong positivity in the THP-1 cell line (
The use of IHC is hampered by the lack of standardized protocols for fixation, antigen retrieval, and antibody-based targeting of proteins. IHC is, at best, semiquantitative; however, in a complex tissue context, data obtained from a single tissue including different cell populations can be of significant value. Such qualitative data are often impossible to assess using other methods. Although IHC using enzyme-based protocols results in a relatively narrow dynamic range of staining intensity, appraisal of immunoreactivity in tissues and cells allows for an outstanding method to perform a relative mapping of protein expression. The inborn mixture of different cells in a tissue enables the analysis of immunoreactivity in a given cell type as a relative measure of protein content, as compared with other cells in the same tissue. This is of particular importance when antibodies generated toward proteins of unknown tissue distribution and cellular localization are analyzed.
Selection of cells used in the described cell TMA includes cell lines from both hematopoietic cell lines and solid tumors, of which eight cell lines also are included in the NCI-60 cell line panel used for anticancer drug screening (Monks et al. 1991; Stinson et al. 1992). Despite the fact that cell lines do not represent normal cells, and sometimes not even the tumor cell type they originated from, they represent a clone of cells with defined characteristics and are possible to grow in large quantities. Selection of leukemia/lymphoma cell lines includes stages of hematopoiesis, enabling analysis of both hematological malignancies and normal differentiation steps. Of the 17 leukemia/lymphoma cell lines, 12 have previously been recommended as reference lines (Drexler and MacLeod 2003). Well-known and frequently used cell lines have been selected to represent solid tumors from the most common forms of human cancers. We have also included several cell lines for representation of specific phenotypical expression, e.g., sarcoma (RH-30, U-2 OS, U-2197), melanoma (SK-MEL-30, WM-115), neuroblastoma (SH-SY5Y), endothelial cells (TIME), etc. Selection and inclusion of >50 different cell types in the cell TMA makes it more comprehensive as compared with previously established cell TMAs consisting of a limited number of adherent-grown cell lines (Ferrer et al. 2005; Waterworth et al. 2005), thus ideal for screening of protein expression in cell lines. Also shown here is that cells from leukemia patients and healthy blood donors are suitable to embed in agarose and subsequently in paraffin, allowing TMA constructions of scarce patient cell material.
In conclusion, we have provided a strategy for using cells in a TMA format that enables high-throughput IHC as a method to screen for protein expression levels and distribution in a large number of cell samples. This approach could well be integrated in existing antibody-based proteomic strategies utilizing tens of thousands of antibodies. A comprehensive cell atlas can be envisioned as a complement to the existing protein profiling in human tissues and cancer (www.proteinatlas.org).
Footnotes
Acknowledgements
This work was financed by a grant supporting the Swedish Human Proteome Resource project from the Knut and Alice Wallenberg Foundation, Stockholm, Sweden.
We thank Prof. Kenneth Nilsson, Prof. Bengt Westermark, Prof. Gunnar Nilsson, Assoc. Prof. Nils-Erik Heldin, Prof. Lena Claesson-Welsh, Prof. Martin McMahon, and Prof. Norbert E. Fusenig for discussions and for permission to use specific cell lines cited in the manuscript.