
Abstract
Adult skeletal muscle possesses remarkable regenerative capacity that has conventionally been attributed to the satellite cells. These precursor cells were thought to contain distinct populations with varying myogenic potential. Recently, the identification of multipotent stem cells capable of new myofiber formation has expanded the general view on the muscle regenerative process. Here we examined the characteristics of turkey skeletal muscle-derived cell (MDC) populations that were separated according to their adhesion abilities. We sought to determine whether these abilities could be a potential tool for separating cells with different myogenic commitment. Using the preplate technique, we showed that MDCs display a wide range of adhesion ability, allowing us to isolate a marginal fraction with initial adhesion defect. Methodological investigations revealed that this defect represents an intrinsic and well-established biological feature for these cells. In vitro behavioral and morphological analyses showed that late adherent cells (LACs) share several primitive cell characteristics. Phenotypic assessment indicated that LACs contain early stage myogenic cells and immature progenitors of satellite cells, whereas early adherent cells consist mainly of fully committed precursors. Overall, our findings demonstrate for the first time in an avian model that differential MDC adhesion properties could be used to efficiently purify cells with varying myogenic commitment, including immature progenitor cells. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
Several studies have reported evidence that populations of adult stem cells may be isolated from muscle tissue or other tissues and still achieve muscle regeneration (Ferrari et al. 1998; De Angelis et al. 1999; Gussoni et al. 1999; Jackson et al. 1999; Galli et al. 2000; Young et al. 2001a, b; Jiang et al. 2002). Muscle-and bone marrow-resident side population (SP) cells, defined by their ability to exclude the fluorescent dye Hoechst 33342, have been shown to actively participate in the formation of skeletal muscle fibers during regeneration in the mdx mouse (Gussoni et al. 1999; Jackson et al. 1999). In Pax7−/- mice, which exhibited a complete absence of satellite cells but exhibited the presence of normal SP cells, it has been shown that SP cells are distinct from satellite cells (Seale and Rudnicki 2000). Additionally, bone marrow SP cells have been reported to participate in myogenesis when injected IV into injured mouse muscle (Ferrari et al. 1998) or mdx mouse muscle (Bittner et al. 1999). Finally, other studies have indicated that progenitor cells isolated from the neuronal compartment (Galli et al. 2000), the embryonic vasculature (De Angelis et al. 1999), and various mesenchymal tissues (Young et al. 2001a, b; Jiang et al. 2002) can also differentiate into myogenic lineage. A technique consisting of serial platings of muscle-derivedcells (MDCs) has demonstrated that mice MDCs can be separated according to their adhesion properties (Qu et al. 1998; Torrente et al. 2001; Jankowski et al. 2002b; Qu-Petersen et al. 2002). This technique, named prelate technique, was originally developed to preferentially select myogenic cells and eliminate other non-myogenic tissue from the culture on the basis of the cell type having differing propensities to adhere to collagen-coated culture flasks (Richler and Yaffe 1970). Recently, it has been presented as an effective tool for isolating delayed adhesion cells able to significantly improve both cell survival after IM injection (Qu et al. 1998) and efficiency of regeneration of mdx muscles (Qu-Petersen et al. 2002). Together these results suggest that preliminary selection of cell fractions could have considerable impact on the effectiveness of new muscle fiber formation.
Given this background, we separated MDCs based on their adhesion criteria in the avian model and explored the properties of the sorted subpopulations. With a slight adaptation of the preplate technique, we showed that the MDCs extracted from turkey Pectoralis major muscle displayed a wide, continuous range of adhesion ability. For the first time, we demonstrated that the initial adhesion defect exhibited by the late adherent cells (LACs) was not related to the experimental procedure or was not eliminated by the presence of muscle cells that were proliferating or differentiating. Additionally, we determined that LACs could not be generated from the earliest adherent cell (EAC)-derived primary cultures. In vitro, LACs displayed an initial mitotically quiescent status followed by atypical proliferation modalities and a specific ability to differentiate. Using morphometrical analysis, we found that LACs contained large numbers of small, round cells. We established, with myogenic stage markers, that LACs were composed of cells at an early stage toward the myogenic differentiation process and immature progenitor cells, whereas EACs were fully committed precursors. Overall, our data show that the initial adhesion defect of a marginal MDC fraction is a shared biological parameter between mouse and turkey, and that the cell adhesion function could be used to enrich myogenic cell fractions with immature progenitor cells.
Materials and Methods
Animals
Male turkeys (Meleagris gallopavo) were produced from a medium-heavy commercial BUT-T9 strain (British United Turkey Limited; Warren Hall, Broughton, Chester, UK). Animals were kept in a conventional animal facility of the National Veterinary School of Nantes, according to animal care guidelines. The French National Institute for Agricultural Research guide for the care and use of laboratory animals was followed.
MDC Isolation
MDCs were isolated from Pectoralis major muscle of at least 20 7-day-old turkeys, as previously described (McFarland et al. 1988; Rouger et al. 2004). In brief, muscles were removed under sterile conditions, cut into 1–2 cm2 sections, and digested for 1 hr at 37C with 0.12% Pronase E (Sigma; St Louis, MO) dissolved in 199 medium (M199; VWR, Strasbourg, France) in a shaking water bath. The mixture was centrifuged at 150 × g for 5 min and the supernatant discarded. The pellet was washed with PBS and submitted to successive centrifugation (300 × g, 20 min) and sequential filtering through 100-, 70-, and 40-μm pore-diameter nylon mesh (BD Biosciences; San Jose, CA) to allow separation of the MDCs from the muscle debris. Cells were resuspended in a proliferation medium (88% M199, 10% fetal calf serum; Sigma), 1% penicillin streptomycin fungizon (PSF; Sigma), and 1%
MDC Fractioning Based on Adhesion
The muscle cell suspension was plated on the culture flasks using a modified version of the technique described by Qu et al. (1998). A flow chart is presented in Figure 1A. Freshly extracted MDCs were plated on 0.1% gelatin-coated flasks (Sigma) for 1 hr. Floating cells found in the supernatant were then centrifuged, counted, and transferred to other gelatin-coated flasks, and adherent cells (ACs) were discarded. ACs, which rapidly adhered, were highly enriched in fibroblast cells as previously described (Rando and Blau 1994; Qu et al. 1998). After 24 hr, floating cells were collected, centrifuged, and plated on new flasks with a proliferation medium. In parallel, the ACs (named AC1) were trypsinized (0.1% trypsin/EDTA; Sigma), centrifuged, resuspended in proliferation medium, and tested for their viability using trypan blue exclusion test. Finally, they were plated at 10,000 viable cells/cm2 on fresh gelatin-coated wells. This procedure was repeated at 24-hr intervals for AC2 and AC3 isolation. It took the last floating cells, obtained after 72 hr of experimental procedure, an additional 3 days without medium change to attach to gelatin-coated flasks. These cells were named AC4. This process resulted in four cultures of ACs plated at the same density. Cultures were routinely grown at 37C in a 5% CO2 atmosphere. Medium was replaced every 2 days. This experiment was independently repeated five times.
Methodological Investigations of the Preplate Technique
To ensure that AC4 has not been artificially selected by the experimental procedure (cell detachment during rinses), MDCs obtained after initial plating of 1 hr (e.g., depleted from most fibroblastic cells) were plated in primary cultures and maintained at 37C for 72 hr (e.g., equivalent time to isolate AC4 in original protocol) without any manipulation. After that, floating cells in supernatants were centrifuged, counted, and plated on new flasks with a proliferation medium. These cells were maintained for 3 days without medium removal to allow their adhesion. Cells were named AC4-like (Figure 1B). Cultures were routinely grown at 37C in a 5% CO2 atmosphere. Medium was replaced every 2 days. This experiment was independently repeated four times.
Furthermore, to determine whether AC4 could be generated by the EACs or were derived only from the tissue at the time the primary culture was established, MDCs depleted from most fibroblastic cells were submitted to the modified protocol allowing AC4-like isolation, as presented above. After the supernatant removal at 72 hr, floating cells were treated as described above, whereas the flasks containing ACs were divided into two parts: (i) one-half received a new proliferation medium and were maintained at 37C for 72 hr without any manipulation (Figure 2A); (ii) cells from the second half were trypsinized, centrifuged, and resuspended in proliferation medium. They were then plated at 10,000 viable cells/cm2 on fresh gelatin-coated cultureware and also maintained for 72 hr without any manipulation (Figure 2B). In both conditions, supernatants were collected, centrifuged, and residual population of floating cells was counted. Medium change or trypsinization was done a second time. The experiment was independently repeated two times.
MDC Differentiation
The capacity for muscle differentiation of cells from each preplate step was studied daily in primary culture based on cells' morphology and expression profile for the embryonic/adult fast myosin heavy chain (MHC). Embryonic/adult fast MHC was detected using EB165 monoclonal antibody (MAb), which reacts with all embryonic MHC isoforms and adult fast MHC (Cerny and Bandman 1987; Bandman and Bennett 1988). EB165 MAb was a kind gift from Everett Bandman (University of California; Davis, CA). This MAb that is specific for chicken MHC also reacts with turkey myosin isoform (Maruyama and Kanemaki 1991). Cells were fixed in 4% cold paraformaldehyde (PFA) in 0.1 mol/liter phosphate buffer and treated with 0.5% Triton X-100/20% (w/v) goat serum in PBS, prior to incubation for 1 hr with mouse MAb to MHC (1:2500). Cells were washed in excess PBS and then incubated successively with the biotinylated goat anti-mouse Ig for 30 min (1:300; Dako, Glostrup, Denmark) and the streptavidin-horseradish peroxidase for 30 min (Dako) with washing between incubation with three changes of PBS. Peroxidase activity was revealed using the chromogen DAB (Dako) as substrate with a reaction time of 10 min. Control staining was performed in an identical fashion with omission of the primary antibody. Syncytia with at least three nuclei were considered to be myotubes.
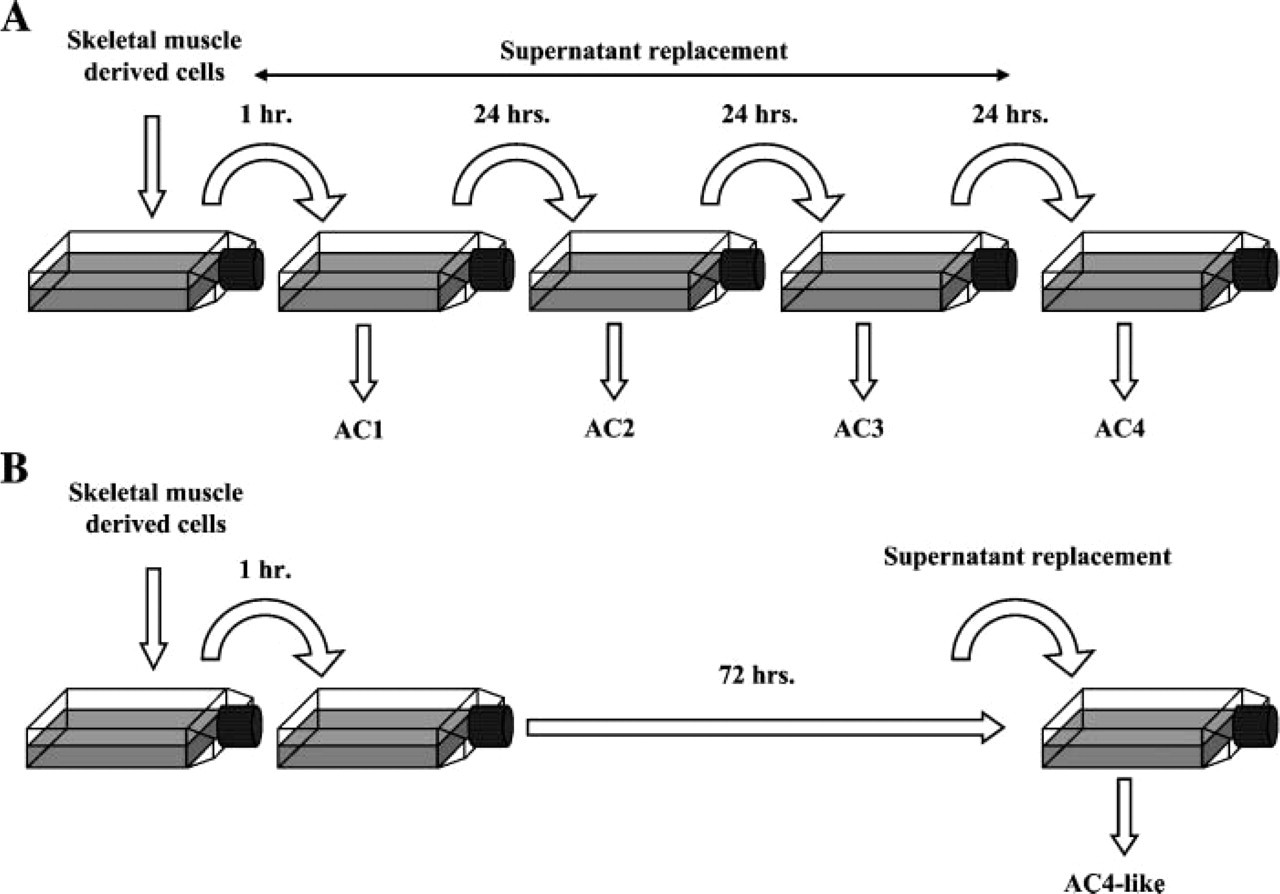
Muscle-derived cell (MDC) subpopulation isolation using differential adhesion properties. (
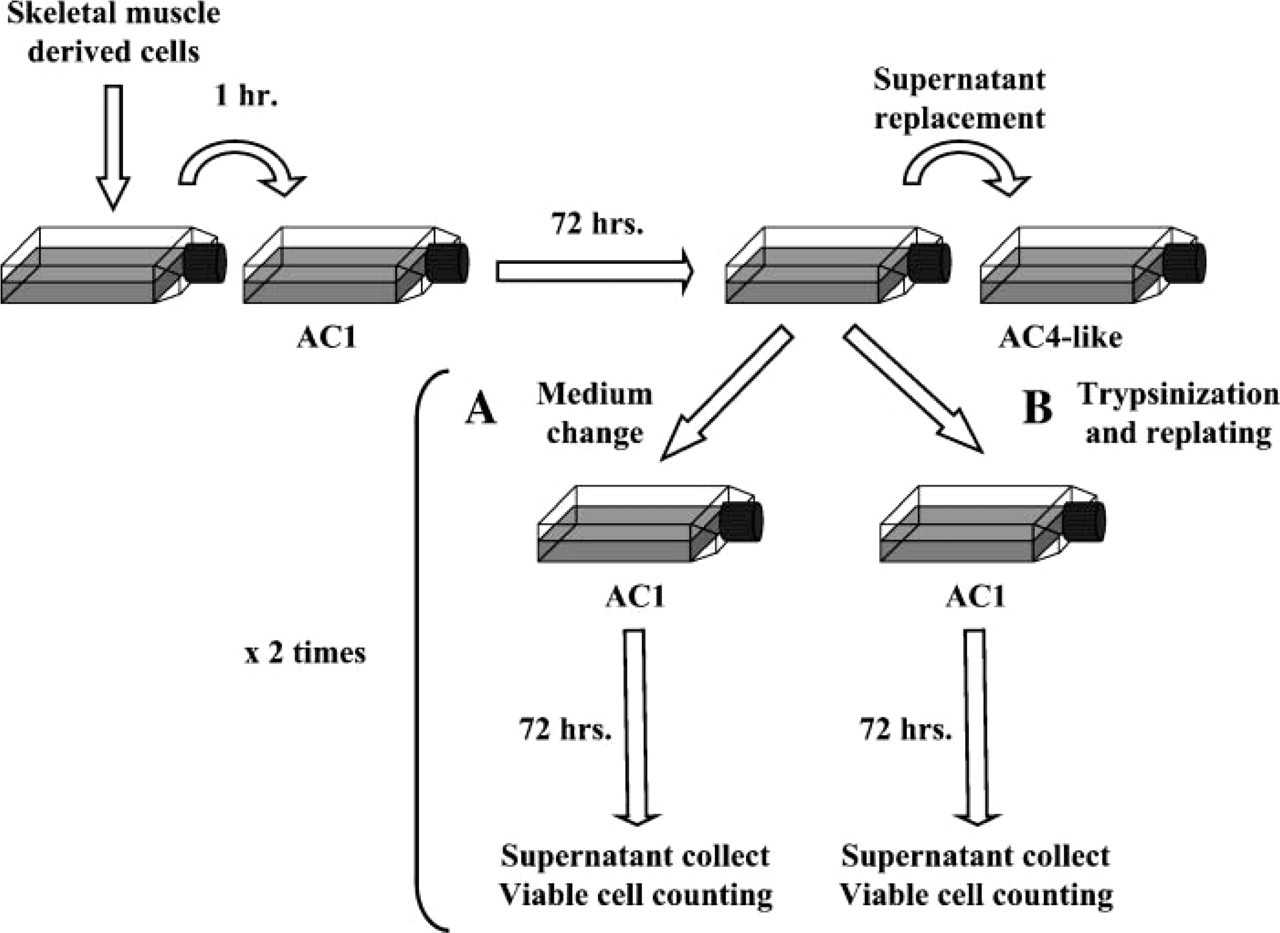
Investigation of the late adherent cell (LAC) origin. After major depletion on fibroblastic cells, MDCs were maintained at 37C for 72 hr without any manipulation. At that time, floating cells collected in the supernatant were seeded on new culture flasks and maintained for 3 days without medium removal, to allow their adhesion. In parallel, flasks containing ACs were divided into two parts and submitted to different procedures. (
MDC Proliferation
Clonal cell cultures from each AC fraction were used. Clonally derived colonies were obtained by limiting dilution to a concentration of one cell per well in gelatin-coated 96-multiwell dishes (BD Biosciences). Medium was changed every 2 days. To select the clonally derived colonies of myogenic nature from those developed from fibroblastic cell, all cultures were fixed in 4% cold PFA at day 11 and initially tested for their expression of desmin, a myogenic lineage-restricted intermediate filament protein (Carlsson and Thornell 2001). Desmin was detected with mouse MAb (1:50; Dako), in the same way as described above. A control of the antibody reactivity with turkey equivalent protein has been done using a 1:50 dilution (See online Supplementary Materials, Figures SF1 and SF2). Proliferation was then evaluated in AC-derived myogenic colonies. The cell number per well was not high enough to allow a [3H] thymidine incorporation procedure. Therefore, the proliferation rate of each AC type was evaluated by counting the number of nuclei in each myogenic clonally derived colony previously stained with Giemsa.
Morphometric Analysis of MDCs
The image analysis system consisted of a digital camera (DXM1200; Nikon, Champigny Sur Marne, France), a microscope (DMIRB; Leica, Gennevilliers, France), a digitizer tablet with an optic pencil (Wacom; Stagnum, France), and a software program system for image processing and analysis (LUCIA G 4.51; Laboratory Imaging, Prague, Czech Republic). Cells were detached from the gelatin-coated flasks, and at least five microscopic fields were randomly selected to explore >300 cells from each AC fraction (n>4, independent experiments). Captured images were submitted to LUCIA G (Laboratory Imaging) image analysis system that allowed automatic measure of the cell size. The minimal ‘Feret's diameter’, defined as the minimum distance between parallel tangents at opposing borders of the muscle cell, was selected to express cell size. This geometrical parameter was frequently used for morphometrical analysis (Dolapchieva et al. 2000; Briguet et al. 2004; Nguyen et al. 2005) and allows for reliable measure of cell cross-sectional size.
Evaluation of the Myogenic Commitment of MDCs
Each AC fraction was evaluated for QH1 expression to determine the possible contamination by endothelial and hematopoietic cells. Indeed, QH1 is a MAb that essentially recognizes these two cell types (Pardanaud et al. 1987). M-cadherin, Pax7, and desmin expression were determined to gauge the respective progression of cells toward end-stage myogenic differentiation. Cells were centrifuged at 470 × g for 5 min and resuspended with PBS to a final concentration of 4 × 106 cells/ml; 40,000 cells were deposited on Polysin microscope slides (Menzel-Glaser; Braunscheig, Germany) that were dried at 37C for 30 min and stored at −20C until further processing (cytospin preparation). The following antibodies were used: mouse MAb to quail QH1 (1:40, supernatant; Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit polyclonal antibody to human M-cadherin (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), mouse MAb to chicken Pax7 (1:10, supernatant; Developmental Studies Hybridoma Bank), and mouse MAb to human desmin (1:50; Dako). Preliminarily, QH1 MAb reactivity with turkey cells was validated, using positive and negative controls (see online Supplementary Materials, Figures SF3 and SF4). Also, reactivity of the M-cadherin and Pax7 antibody in turkey animal was controlled (see online Supplementary Materials, Figures SF1, SF5 and SF1, SF6, respectively). Specificity of these three antibodies was preliminarily tested using the dilutions indicated above (see online Supplementary Materials). Slides were treated for 30 min with 0.5% Triton X-100 and incubated for 20 min with a blocking solution (2% goat serum, 5% dog serum, and 2% BSA diluted in PBS) prior to primary antibody incubation. Anti-mouse Alexa Fluor-488 secondary antibody and anti-rabbit Alexa Fluor-488 secondary antibody (1:100; Molecular Probes, Eugene, OR) were used. Coverslips were mounted using Vectashield containing DAPI nuclear dye (Vector Laboratories; Burlingame, CA), and slides were viewed using a con-focal laser scanning microscope (CLSM; Nikon). For each immunolabeling, >200 cells were considered from each AC fraction (n = 4, independent experiments).
CLSM
Slides were viewed by CLSM using a TE2000 Nikon inverted microscope equipped with ×20 water-immersion objective. Alexa Fluor-488 was excited using an argon ion laser with a bandpass of 515–530 nm. Propidium iodide was excited at 543 nm using helium-neon laser with a bandpass of 565–605 nm. Each image was recorded separately in two different channels (red and green). Overlaying of the recorded images allowed simultaneous visualization of the immunolabeling and the counterstained nuclei.
Statistical Analysis
All reported data were compared among different cell fractions with ANOVA followed by Fisher's protected least significant difference test using statistical software (Stat View, Brain Power; Calabasas, CA). Proportions were checked with the χ2 test. Data were reported as mean ± SD. A value of p<0.05 was considered statistically significant.
Results
MDC Distribution Based on Adhesion Ability
Immediately after their extraction, at least 1 × 108 MDCs underwent a modified version of the preplating technique. MDC distribution was assessed by determining the proportion of viable cells that adhered to the substrate on each plating (Table 1, five independent experiments). After 24 hr, 90.5 ± 4.7% of whole MDCs were adherent to gelatin-coated cultureware and named AC1; 7.7 ± 3.6% (AC2) and 0.8 ± 0.6% (AC3) of the initial extracted cells were adherent to the gelatin matrix 48 hr and 72 hr later, respectively. Finally, 1.0 ± 0.9% (AC4) of all MDCs were adherent after 6 days. MDCs showed variable adhesion ability, enabling us to separate them into four fractions over 6 days; 98.2% of all MDCs were adherent during the first 2 days.
To ensure that the presence of AC4 was not due to a methodological artifact (cell detachment during rinses), some of the freshly extracted MDCs were seeded in primary culture and maintained at 37C for 72 hr without manipulation, e.g., the time necessary to allow adhesion of AC1 to AC3 in the other protocol. After three additional days, repeated enumeration of ACs revealed that 4.0 × 0.8% of the total initially seeded cells were adherent (four independent experiments). These cells were named AC4-like. The number of AC4-like cells was greater than AC4 cells (p<0.001). This demonstrates that the delayed adhesion attributed to the AC4 fraction was not inherent to the experimental procedure and was not modified by the presence of proliferating or differentiating muscle cells.
Distribution of MDCs based on adhesion abilities
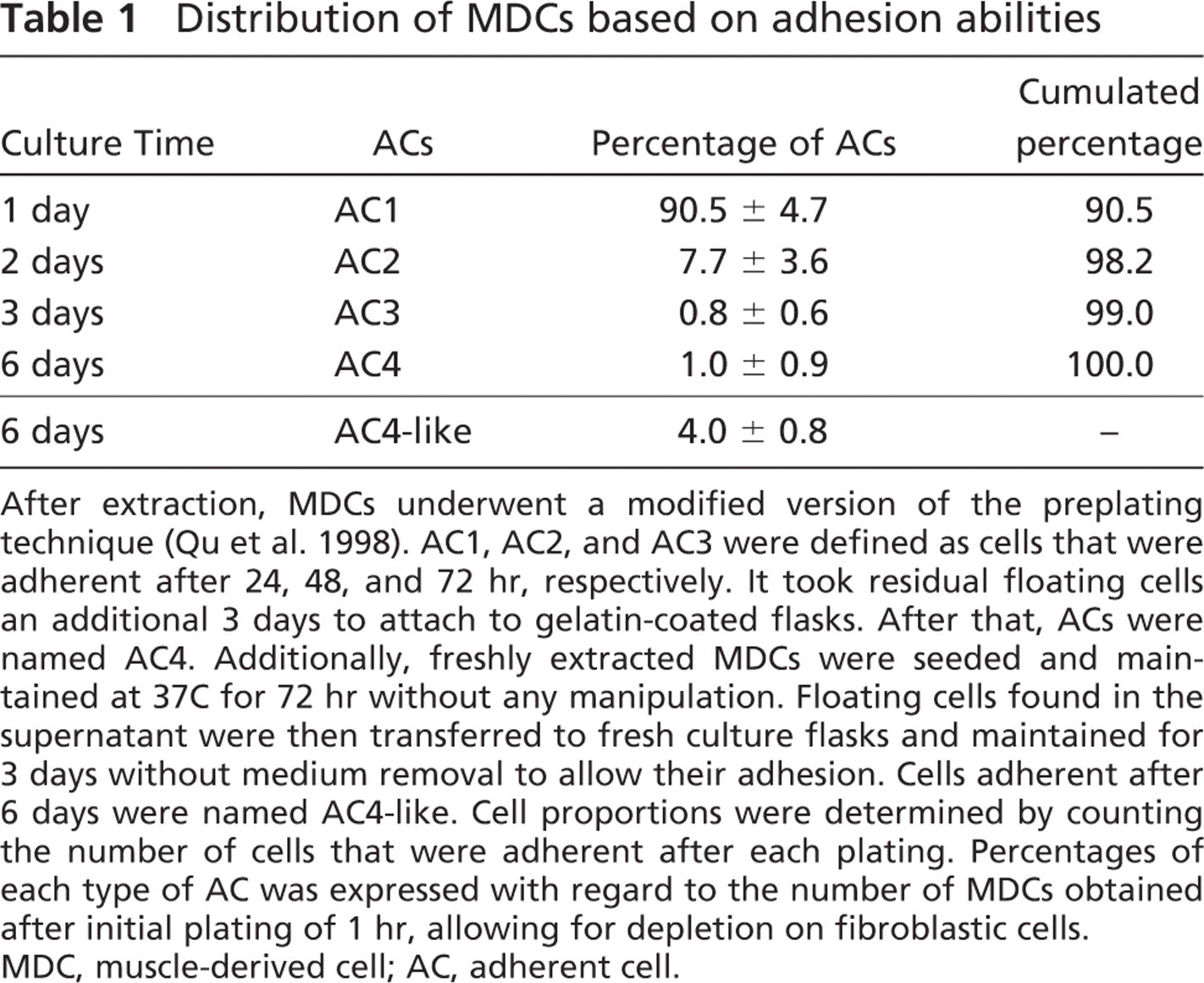
After extraction, MDCs underwent a modified version of the preplating technique (Qu et al. 1998). AC1, AC2, and AC3 were defined as cells that were adherent after 24, 48, and 72 hr, respectively. It took residual floating cells an additional 3 days to attach to gelatin-coated flasks. After that, ACs were named AC4. Additionally, freshly extracted MDCs were seeded and maintained at 37C for 72 hr without any manipulation. Floating cells found in the supernatant were then transferred to fresh culture flasks and maintained for 3 days without medium removal to allow their adhesion. Cells adherent after 6 days were named AC4-like. Cell proportions were determined by counting the number of cells that were adherent after each plating. Percentages of each type of AC was expressed with regard to the number of MDCs obtained after initial plating of 1 hr, allowing for depletion on fibroblastic cells. MDC, muscle-derived cell; AC, adherent cell.
Furthermore, to gain data on the AC4 origin, MDCs were maintained at 37C for 72 hr without manipulation, after which culture supernatants were collected to isolate the AC4-like cells, as described above. Parallel flasks containing ACs received new proliferation medium, or cells from other flasks were trypsinized and re-seeded on fresh gelatin-coated flasks. In both procedures, culture supernatants were collected after 72 hr without manipulation, and viable cells were counted and transferred to gelatin-coated flasks. This was repeated two times. Although AC4-like cells were enumerated at day 6, any viable cell was found in supernatants collected from both experimental procedures (two independent experiments). This revealed that AC4 derived only from the tissue at the time the primary culture was established and clearly were not generated by the early AC-derived cultures. Moreover, this enabled us to precise that the AC4 isolation after several days in vitro could not be assimilated to a cell culture artifact, as it could not be reproduced from two independent cell culture contexts.
In Vitro Evolution of MDCs
To investigate whether the differential adhesion criteria demonstrated with the preplating technique could be associated with varying proliferation and differentiation abilities, cells from each preplating step, as well as AC4-like cells, were analyzed in primary culture.
Within 1–2 days, many AC1 cells were fusiform and began to develop cytoplasmic extensions. After 4–5 days, cultures displayed a 50%–60% confluence and were composed of numerous multinucleated myotubes identifiable by their tubular form. Three days later, the cell number had increased and some large, stellate myotubes with numerous additional nuclei were observed (Figure 3A). At day 12, the myotubes were strongly positive for embryonic MHC (Figure 3D). AC2- and AC3-derived cultures displayed the same time course of proliferation, as well as the formation of identical myotubes. Surprisingly, AC4 cells were round and refringent within the first days of culture. After 4 days, they were still mononucleated without morphological change, similar to cells in quiescent state (Figure 4A). They then began to grow and take on an atypical appearance, forming microspheroid colonies composed of joined cells (Figure 3B) that occasionally appeared superimposed (Figure 4B). Once the colonies displayed >20 cells, the cells scattered and adopted a spindle shape like classical myoblasts (Figures 4C and 4D). After 12 days, small myotubes positive to embryonic MHCs were observed (Figure 3E). They were thinner than those observed in the cultures derived from cells that more rapidly adhered and also contained a smaller number of nuclei. AC4-like-derived cultures showed the same behavior as those derived from AC4. Indeed, an initial quiescent-like state followed by specific proliferation modalities defined by the formation of microspheroid colonies was also noted (Figure 3C). The exclusive presence of thin myotubes was similarly observed (Figure 3F).
Together these results showed that cells initially defined by their different adhesion abilities could be split into two cell fractions with distinct behavioral characteristics: (i) cells that were adherent during the first 72 hr (e.g., AC1, AC2, and AC3) and that displayed in vitro a typical muscle cell differentiation and (ii) AC4 cells (or AC4-like) that had not yet adhered at that time and that later displayed a specific pattern of proliferation and differentiation into myotubes. The first were referred to as EACs and the second, LACs.
Proliferation Ability of MDCs
To assess their proliferation abilities, EACs and LACs were seeded in clonal culture. After 11 days, 645 clonally derived colonies were identified and tested for their desmin expression to select those with a myogenic origin: 531 clones were composed of desmin positive (desmin+) cells, and 114 other clones were composed of desmin negative (desmin−) cells. Myogenic clones were distributed as follows: 313 in EACs and 218 in LACs (Table 2). The average number of nuclei per colony was 224 ± 270 for EACs and 158 ± 185 for LACs. The standard deviation revealed considerable variation in colony sizes. Proliferation ability of EACs was 1 1/2 times that of LACs (p<0.001). These data confirmed our results in primary culture, indicating that LACs have a lower capacity of proliferation over the first few days compared with EACs. All clonally derived colonies were classified according to the number of nuclei (Table 3): 38.1% of all LAC clonally derived colonies vs. 28.1% of EAC clonally derived colonies displayed <64 nuclei. Also, 17.0% of LAC clonally derived colonies had >256 nuclei vs. 26.5% of EAC clonally derived colonies. The number of poorly proliferative cells was higher in LACs than EACs (p<0.001).
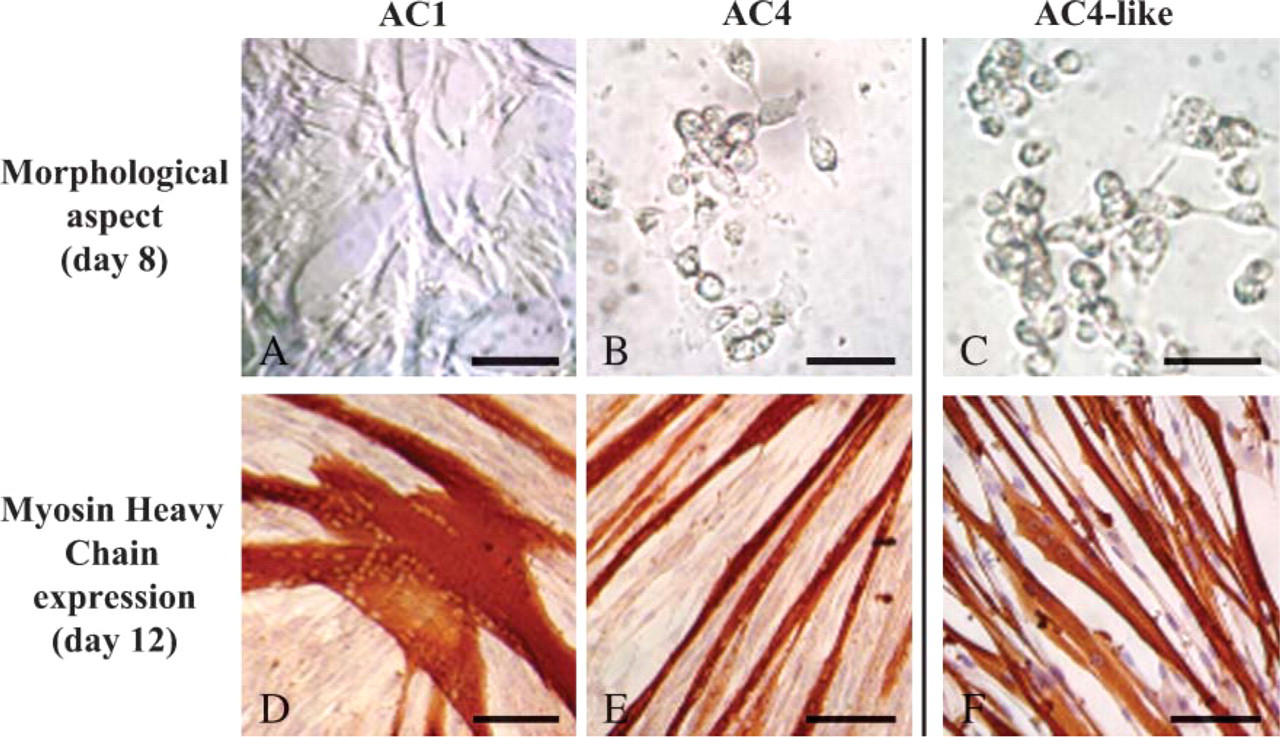
Evolution over time of AC-derived cultures. ACs of each preplate step were detached from gelatin-coated cultureware, seeded in primary culture, examined daily in phase contrast with regard to their morphology (
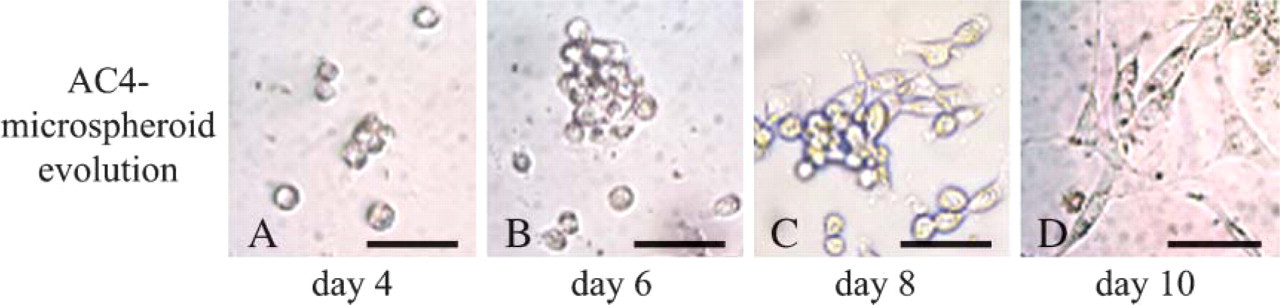
Atypical proliferation modality of LACs. AC4 cells that were adherent after 6 days were detached from gelatin-coated cultureware, seeded in primary culture, and examined daily in phase contrast. After 4 days, they were round, refringent, and remained mononucleated without morphological change (
Morphometrical Analysis of MDCs
To determine whether LACs could also be identified using specific morphometrical characteristics, EAC and LAC diameters were measured by LUCIA imaging software (Laboratory Imaging) (Figure 5A). As shown in Figure 5B, both cell fractions displayed a wide range of sizes between 5 μm and 12 μm but showed a clearly different cell distribution based on this morphological parameter. Whereas 37 ± 7% of EAC had a diameter of <6.5 μm, 36 ± 7% had a diameter ranging between 6.5 and 7.5 μm, and 27 ± 6% measured >7.5 μm in diameter. With respect to LACs, diameters were as follows: 73 ± 6% measured <6.5 μm, 19 ± 4% were between 6.5 μm and 7.5 μm, and 8 ± 2% were >7.5 μm. Cell mean diameter was 7.5 ± 1.7 μm and 5.0 ± 0.2 μm for EACs and LACs, respectively. LAC mean diameter was lower than that of EAC due to a significantly high proportion of small cells (χ2, p<0.001).
Proliferation ability of EACs and LACs
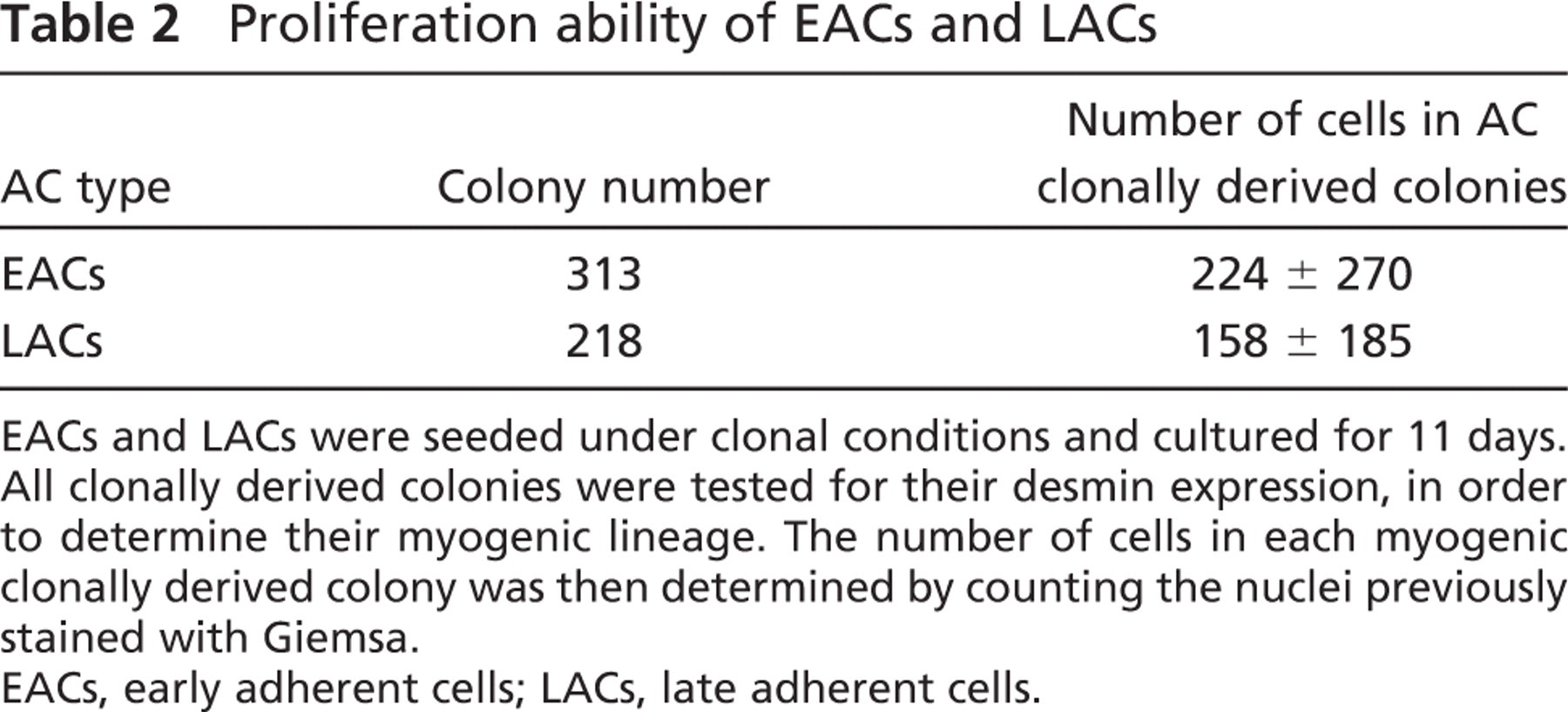
EACs and LACs were seeded under clonal conditions and cultured for 11 days. All clonally derived colonies were tested for their desmin expression, in order to determine their myogenic lineage. The number of cells in each myogenic clonally derived colony was then determined by counting the nuclei previously stained with Giemsa.
EACs, early adherent cells; LACs, late adherent cells.
Determination of Myogenic Commitment of MDCs
To assess the percentage of cells expressing some proteins in relation to myogenic differentiation status, immunolabeling for M-cadherin, Pax7, and desmin was performed on EAC and LAC fractions immediately after the cells were detached from the gelatin matrix during the preplating technique (Figure 6A). Using QH1 protein expression, possible contamination of both AC fractions by endothelial and hematopoietic cells was also determined (Figure 6B). Immunolabelings revealed that the three muscle proteins were all expressed by both EACs and LACs, but in distinct proportions: indeed, M-cadherin, Pax7, and desmin were observed in a large majority, a low proportion, and ~50% of EACs, respectively. In contrast, M-cadherin+ and Pax7+ cells represented the majority of LACs, whereas LACs expressing desmin were few. M-cadherin+ and desmin+ cells seemed to be more represented in EACs compared with LACs, whereas Pax7+ cells appeared much more numerous in LACs. Concerning the endothelial/hematopoietic marker, EACs and LACs expressing QH1 were rarely observed. As shown in Figure 7, many differences in the number of cells expressing proteins specific to myogenic lineage were indeed confirmed between EACs and LACs (χ2, p<0.001). EACs contained the following percentage of cells: 83.2 ± 2.2% M-cadherin, 11.6 ± 3.5% Pax7, and 41.8 ± 2.9% desmin. In contrast, LACs were characterized as displaying the following phenotype: 62.7 ± 4.8% M-cadherin, 50.0 ± 3.7% Pax7, and 24.4 ± 0.7% desmin. Compared with EACs, LACs showed a lower percentage of cells expressing M-cadherin, a satellite cell-specific marker, but a higher percentage of cells expressing Pax7, a marker limited to cells in the early stages of myogenesis (p<0.001). Also, cells positive for desmin expression, a marker of committed myoblasts, were less numerous in LACs than EACs (p<0.001). The percentage of QH1+ cells was 5.6 ± 1.9% (EACs) and 4.7 ± 0.9% (LACs), indicating that both AC fractions contained few endothelial cells and hematopoietic cells. Moreover, this showed that LACs did not contain many more of these two cell types than EACs. These data show that EACs and LACs corresponded to cell fractions composed of cells that differed by their commitment level in the myogenic pathway.
Distribution of AC clonally derived colonies (%)
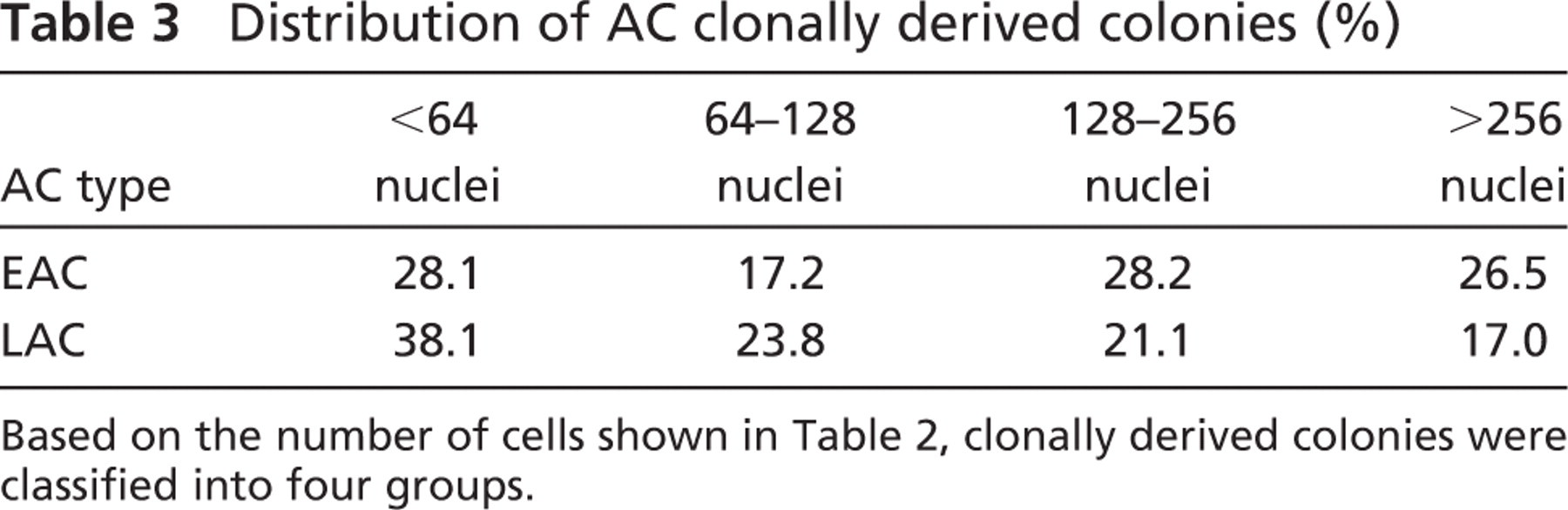
Based on the number of cells shown in Table 2, clonally derived colonies were classified into four groups.
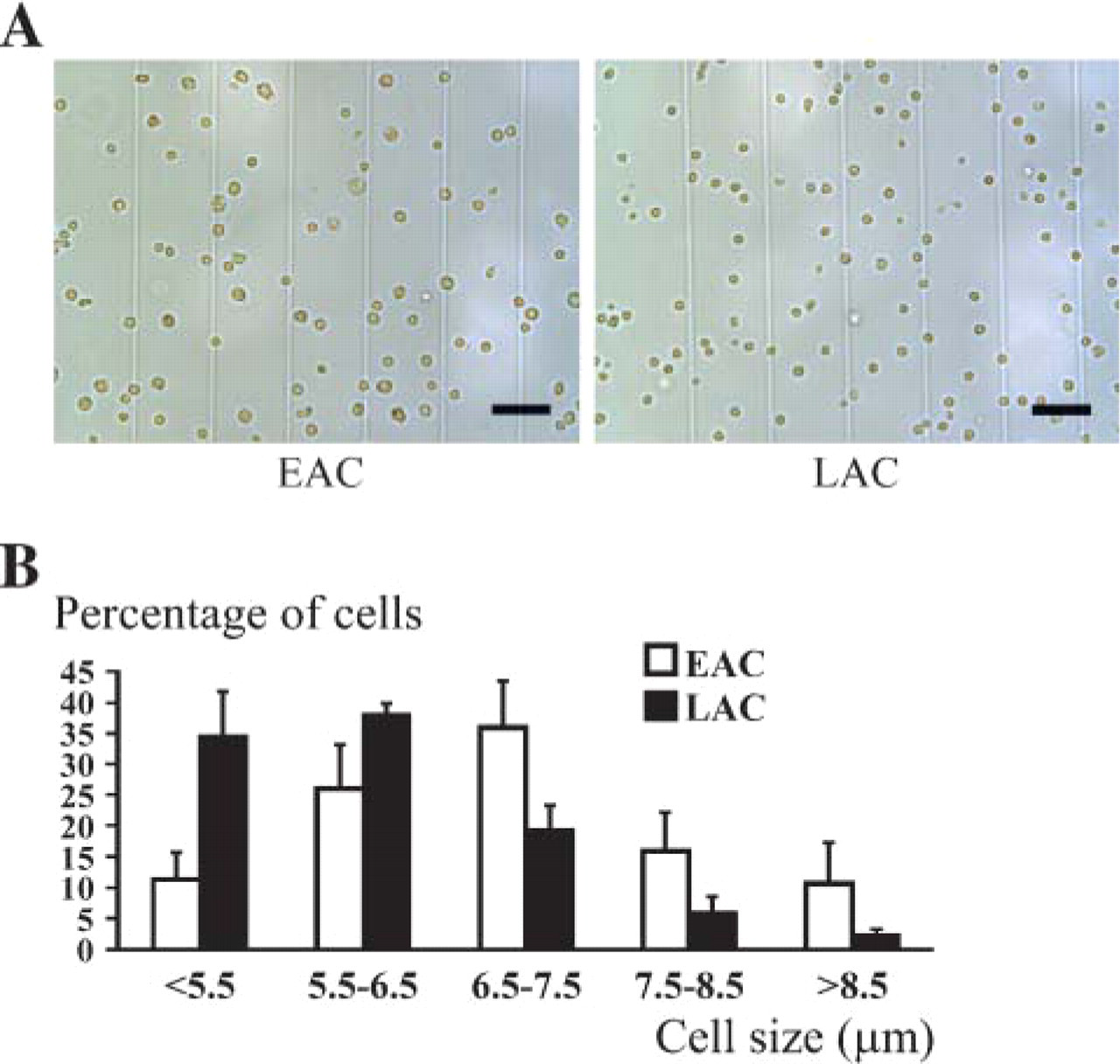
Size analysis of EACs and LACs. Both AC types were observed under a light microscope (
Discussion
In this study we compared the characteristics of avian skeletal MDC subpopulations separated according to their adhesion properties, including in vitro behavior, morphometry, and phenotype. We sought to determine whether this kind of cell sorting could be used in avian models to separate cells displaying varying degrees of myogenic commitment. We also attempted to investigate whether the delayed adhesion might allow us to isolate progenitor cells from MDCs. Using the preplate technique, we found that skeletal MDCs displayed different adhesion properties, allowing us to isolate a marginal cell fraction with clearly delayed adhesion. For the first time, we demonstrated that this initial adhesion defect was not modified by the presence of muscle cells, revealing that it was a major and intrinsic feature of these cells. Moreover, we determined that LACs were not generated from the EAC-derived primary cultures, implying that this marginal cell fraction derived only from the skeletal muscle tissue and could not be assimilated to the descendants of activated satellite cells. Using automated morphometry, we established that LACs contained high numbers of small, round cells. We found that LACs displayed an initial quiescent status followed by atypical proliferation modalities with microspheroid colonies formation, and that these cells were only able to form thin myotubes. Using immunocytochemistry, we showed that LACs were composed of cells at an early stage toward the myogenic differentiation process and had high numbers of progenitor cells. Together these results demonstrate that the delayed adhesion of a marginal fraction of MDCs is a shared biological parameter between mouse and turkey and could be used to isolate cell fractions composed of progenitor cells and cells with limited myogenic differentiation from whole MDCs.
We reported here that LACs could be identified among turkey skeletal MDCs, using the preplate technique developed by Qu et al. (1998). The LAC population, which was finally isolated after 6 days, represented ~1% of the whole extracted mononucleated cells. This result is similar to results published in a previous study performed in mice (Qu-Petersen et al. 2002). An interesting observation was that LACs could also be harvested from the supernatant of MDC-derived primary culture seeded 3 days prior. In this case, LACs represented ~4% of the initially seeded MDCs; this could be due to a less significant cell loss with the simplification of the experimental procedures. The fact that LACs could also be isolated without successive plating clearly shows that they could not be assimilated to detached cells during the repeated steps of the experiment. This means that they represented a constitutive cell fraction of MDCs. Moreover, this result indicated that the adhesion defect was not modulated by the presence or absence of muscle cells. This shows that the initial ineffectiveness of the LACs in adhering was not modified by the paracrine factors secreted by the ACs that were proliferating or differentiating. Additionally, LACs could not be isolated from the EAC-derived primary cultures, which reinforced the notion that these cells derive only from skeletal muscle tissue. Also, this evocated that LACs could not correspond to the descendants of activated satellite cells, like “reserve” cells resulting from their asymmetrical division (Yoshida et al. 1998), or that specific signals/stimuli would be required to generate LAC from satellite cells.
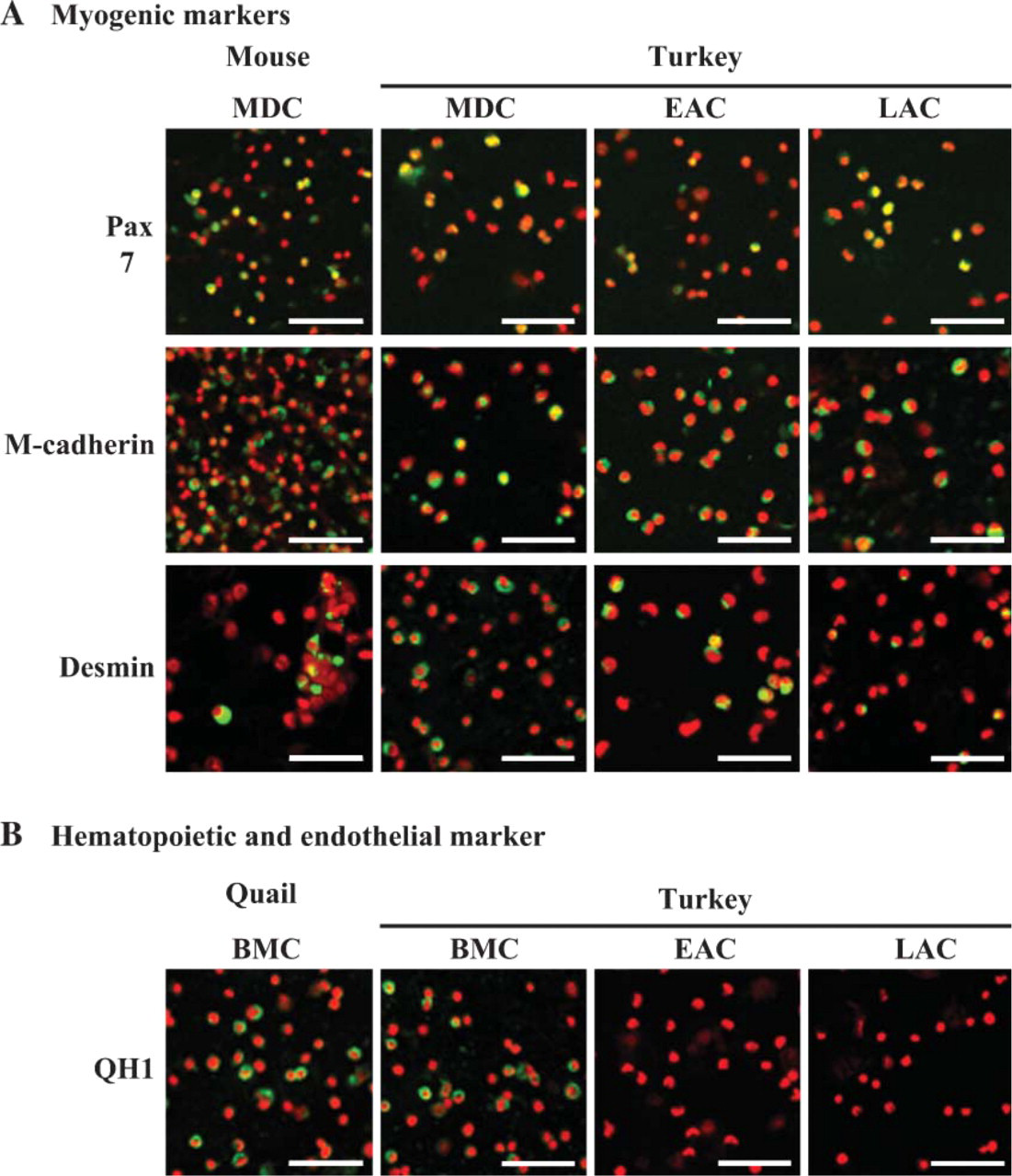
Immunofluorescent labeling of EACs and LACs. Following isolation, cells were deposited on slides (cytospin preparation), and their expression for specific markers of myogenic cells (e.g., M-cadherin, Pax7, and desmin) (
Immediately after their adhesion, LACs were characterized as highly enriched in small cells compared with EACs. They did not show any proliferation during the first week. During the following week, LACs slowly began to atypically proliferate as round cells and formed few microspheroid colonies of mononucleated cells, as previously described for some mice MDC fractions (Tamaki et al. 2003). They then showed a specific ability to differentiate into myotubes, as noted by the formation of very thin myotubes after 2 weeks. Interestingly, myosphere formation (e.g., clusters of cells) was also recently described from mice slow adherent myogenic cells that were maintained in suspension. Also, when myospheres grown in gelatin-coated plates are left for several days, many adhered as single rounded cells and formed very thin myotubes (Sarig et al. 2006). Our findings, which were consistent with analyses performed in mice (Rando and Blau 1994; Qu et al. 1998; Lee et al. 2000; Qu and Huard 2000), indicated that the late preplate cells displayed a smaller diameter and tended to be morphologically round, compared with the early preplate cells. Moreover, the late preplate cells were distinguished from early plate cells by slow division and poor ability to fuse (Torrente et al. 2001; Qu-Petersen et al. 2002). Additionally, it is clear that muscle-derived SP cells, a small fraction of adult stem cells purified using Hoechst 33,342 staining/FACS methods, also were small and spherical (Gussoni et al. 1999; Majka et al. 2003; Benchaouir et al. 2004). They remained mononucleated without displaying any proliferation activity for over a week and showed poor, delayed muscle differentiation, whereas the non-SP cells were fully differentiated into myotubes after 1 week. Together these comparative results suggested that the LACs shared several morphometrical and proliferation/fusion characteristics with identified primitive cells.
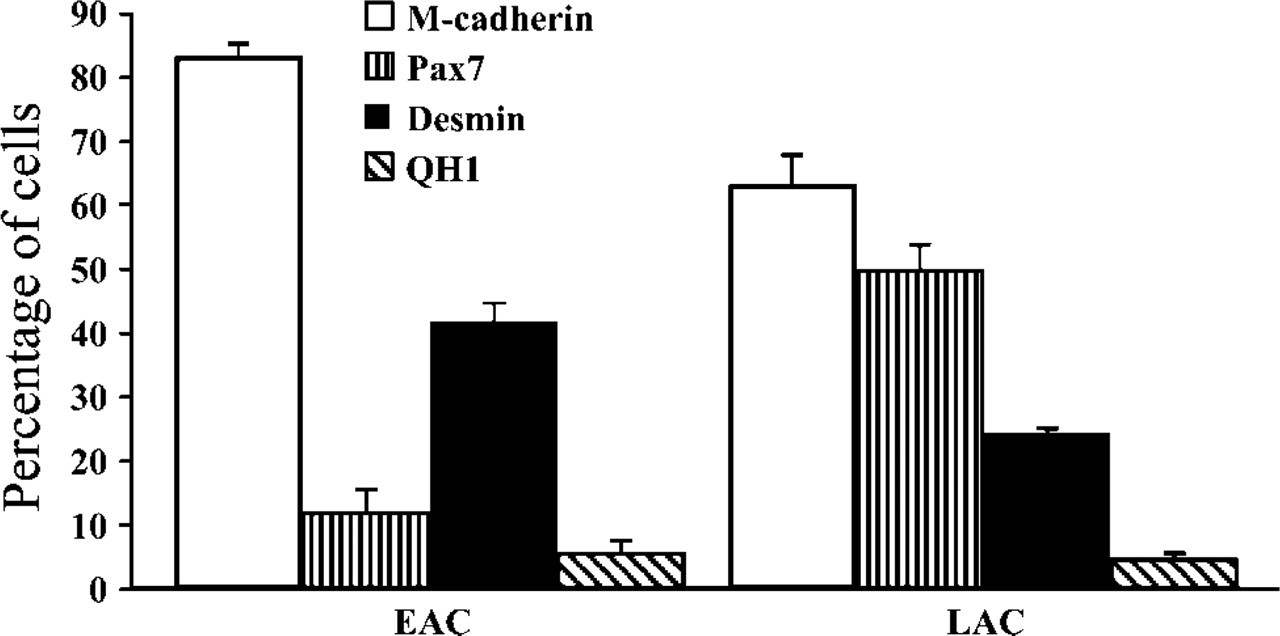
Phenotypic characterization of EACs and LACs. Both AC types were immunolabeled with Ab specific to M-cadherin, Pax7, desmin, and QH1 protein, and the proportion of positive cells for each marker was determined. Results are expressed as a mean of at least 200 values, and error bars represent SD values (n = 4 experiments for EACs and LACs). LACs displayed 62.7 ± 4.8% of M-cadherin+ cells compared with 83.2 ± 2.2% in EACs. LACs were characterized by 50.0 ± 3.7% of Pax7+ cells, which is considerably higher than in EACs where 11.6 ± 3.5% of cells expressed this transcription factor. Inversely, the proportion of desmin+ cells in LACs (24.4 ± 0.7%) was much smaller than observed in EACs, which represented 41.8 ± 2.9%. These findings showed clear-cut differences in the expression for myogenic markers between EACs and LACs (p<0.001). EACs were enriched in activated myogenic cells, whereas LACs contained non-myogenic cells and cells with limited myogenic commitment. In EACs and LACs, the percentage of QH1+ cells was 5.6 ± 1.9% and 4.7 ± 0.9%, respectively, indicating that both AC fractions contained only a few endothelial cells and hematopoietic cells.
Phenotype characterizations of late preplate cells revealed that they contained cells at different stages of differentiation. Concerning myogenic marker expression, important differences between studies were noted about desmin and M-cadherin proteins. Initial research (Qu et al. 1998; Lee et al. 2000; Deasy et al. 2001; Jankowski et al. 2001) indicated that the sequential preplates were enriched in their content of myogenic precursors based on expression of desmin, a marker of committed myoblasts (Cornelison and Wold 1997; Sabourin et al. 1999). In contrast, Qu-Petersen et al. (2002) showed that, although >95% of cells in expanded cell cultures expressed desmin, only 30%–40% were found to be desmin+ at an earlier passage. Additionally, the late preplate cells were defined either by >90% of M-cadherin-expressing cells (Jankowski et al. 2002a) or by a relatively low percentage of the same cells (Lee et al. 2000; Qu-Petersen et al. 2002), as only 5%-30% of cells expressed this satellite cell-specific marker (Moore and Walsh 1993; Irintchev et al. 1994). Using different markers exhibited by the subpopulations of hematopoietic stem cells (Gussoni et al. 1999; Ziegler et al. 1999), several studies reported the existence of stem cells among the late preplate cells (Lee et al. 2000; Jankowski et al. 2001; Qu-Petersen et al. 2002; Sarig et al. 2006). Then, part of the late preplate cells, which were myogenic lineage−, were identified as Sca1+, CD34+/-, CD45−, and c-kit−. Here we have attempted to define the LAC fraction by labeling different proteins according to myogenic commitment, to gauge their respective progression toward end-stage myogenic differentiation. In agreement with the results obtained in mice (Lee et al. 2000; Qu-Petersen et al. 2002), we noticed that LACs displayed a higher proportion of M-cadherin− cells and desmin− cells compared with EACs. This revealed that LAC fraction was enriched in myogenic lineage− cells. We were able to observe that endothelial and hematopoietic cells were poorly represented in the LAC fraction, and that a considerable proportion (~40%) of non-myogenic cells in LACs could not be attributed to an increase in both cell types. In light of this, it might be suggested that the LAC-derived myogenic lineage− cells are essentially progenitor cells and a much smaller proportion are quiescent satellite cells and, as described, some do not express M-cadherin and desmin (Cornelison and Wold 1997). Taking into account the similarities noted invitro among the LACs and the late preplate cells selected in mice, we may hypothesize that the myogenic lineage− cells may contain some muscle-derived stem cells. Unfortunately, absence of antibodies against hematopoietic stem cells markers for our animal model did not allow us to determine whether some LACs showed markers common to stem cells. The percentage of M-cadherin+ cells obtained in the LAC fraction appeared higher than that reported in previous studies performed in mice, suggesting that our fraction was not as purified in immature progenitors of satellite cells.
Pax7 has been reported to play an essential role in satellite cell biogenesis by restricting alternate developmental programs (Seale et al. 2000; Oustanina et al. 2004). This paired box transcription factor was expressed in both quiescent and activated satellite cells, as well as in proliferating primary myoblasts. However, it was inhibited after myogenic differentiation (Seale et al. 2000; Halevy et al. 2004). For the first time, its expression was assessed in freshly isolated cell fraction resulting from the preplate technique. We showed that ~50% of LAC were Pax7+ compared with <15% in EACs, which confirmed that these two cell fractions contained some subpopulations displaying distinct degrees of myogenic commitment. Similarly, ×70% of cells forming myospheres expressed Pax7 (Sarig et al. 2006). The low percentage of Pax7+ cells in EACs would support the hypothesis that this cell fraction contained a significant proportion of late myogenic precursors that entered into terminal differentiation. Also, it appeared that nearly all M-cadherin+ LACs were Pax7+, which was consistent with the results of our proliferation/fusion studies and supported the notion that LAC fraction was partly composed of myogenic cells at an early stage.
In conclusion, our observations with regard to the MDC separation resulting from the preplate technique provide evidence that differential adhesion properties can be used in avian models to purify different myogenic cell subpopulations. This approach enables not only the separation of cells based on their ability to proliferate and differentiate, but also the extraction of immature progenitors of satellite cells from whole MDCs. Further experiments are required to explore the degree of interaction of these progenitor cells with satellite cells and the biological signals that control their behavior in muscle niche.
Footnotes
Acknowledgements
This work was supported by grants from the Association Française contre les Myopathies.
The authors thank P. Guyot for assistance with animal care and facilities. We are grateful to E. Bandman and P.Y. Rescan for their generous supply of antibodies. Monoclonal antibodies specific for Pax7 (PAX7), vimentin (AMF-17b), and endothelial cell surface (QH1) developed here by us were obtained from the Developmental Studies Hybridoma Bank under the auspices of the National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD and maintained by the Department of Biological Sciences, University of Iowa, Iowa City, IA.