
Abstract
The purpose of this study is to analyze the time-dependent molecular states of rhodopsin (Rho) phosphorylation in the specimens originating from eyeballs cryoimmobilized in situ in living animals. Whole eyeballs of living mice under various dark- and light-exposure conditions were quickly frozen using the in vivo cryotechnique with isopentane-propane cryogen cooled down in liquid nitrogen (−196C). The frozen whole-mount eyeballs were freeze substituted in acetone containing paraformaldehyde and embedded in paraffin wax. Deparaffinized sections were immunostained with anti-phosphorylated 334Ser Rho (P-Rho334) antibody. Immunoreactivity of P-Rho334 was specifically recognized in the outer segments of mouse retinas exposed to daylight. In the 12-h dark-adapted retinas, P-Rho334 immunoreactivity was completely eliminated. Moreover, in other retinas dark adapted for 12 or 36 hr and then exposed under the safety red light for 2 min, it was still barely recognized. Even in the eyeballs exposed to strong visible light for 10 sec, it was not detected. However, after 30, 60, and 180 sec of visible light exposure, P-Rho334 immunoreactivity was definitely recovered, similar to that under daylight condition. This is a new immunohistochemical approach to visualize the time-dependent Rho phosphorylation of living mice using the in vivo cryotechnique, in which changes could be detected within seconds following exposure to light.
T
It is generally accepted that rhodopsin (Rho) is a photoreceptor quickly changing its molecular structure by light stimulation (Menson et al. 2001). An active form of Rho is further inactivated by two sequential processes: first the Rho phosphorylation and then binding of arrestin to the Rho (Fain et al. 2001). During these processes, Rho phosphorylation is considered to play a general role in a broader light spectrum for visual functions, ranging from the ability of rods to generate reproducible electrical responses to the other to adapt themselves to substantial darkness after light exposure (Arshavsky 2002). Therefore, Rho phosphorylation has provided an example of the ubiquitous regulatory pattern of specific kinases downregulating the activity of G-protein-coupled receptors (Maeda et al. 2003). We have also reported that some main sites of Rho amino acids to be phosphorylated are 334Ser, 338Ser, and 343Ser near the carboxyl terminus (Ohguro et al. 1996). Concerning Rho phosphorylation after light adaptation, according to the biochemical analysis with mass spectrometry, the phosphorylation reaction is started more quickly to accommodate itself to various light conditions by the quick treatment of resected mouse retinas (Kennedy et al. 2001). However, it has not been directly visualized by routine immunohistochemistry of conventionally prepared tissues (Hicks and Barnstable 1987; Adamus et al. 1988) because of technical problems in preserving such rapid changes of the molecular structure obtained from dead or anesthetized animal retinas. Recently, several specific antibodies against major phosphory-lated rhodopsin (P-Rho) molecules were produced and well characterized by immunological analyses, by comparing the duration of positive immunoreactivity of P-Rho after the dark adaptation of retinas in normal rat and those of the Royal College of Surgeons (Ohguro et al. 2003). In the present study, we have examined whether IV-CT with the FS method is immunohistochemically applicable to examining time-dependent molecular changes of Rho to P-Rho under various light-illumination conditions in living mouse retinas.
Materials and Methods
The present study was approved by the University of Yamanashi Animal Care and Use Committee and performed in accordance with the guidelines governing animal experiments at the University of Yamanashi.
Preparation of Phosphorylated Rhodopsin (P-Rho)
After overnight dark adaptation, freshly prepared mouse Rho protein (5 mg) (Ohguro et al. 1995) was dissolved in 5 ml of buffer solution A (100 mM Na-phosphate buffer, pH 7.2, 5 mM MgCl2, 100 mM KCl) containing 1 mM ATP and incubated from a distance of 20 cm under a 150-W lamp at 30C for 10 min. Phosphorylation was terminated by the addition of buffer solution B (200 mM Na-phosphate buffer, pH 7.2), containing 5 mM adenosine, 100 mM potassium fluoride, and 200 mM EDTA. The P-Rho pellet was obtained by centrifugation at 13,000 × g for 5 min.
Production of Anti-P-Rho334 Antibody
An amount of specific antiserum against phosphorylated Rho at the 334Ser residue was obtained from rabbits by immunization of the phosphorylated authentic peptide, P-Rho334, as described previously (Ohguro et al. 2003). In addition, affinity-purified anti-P-Rho334 antibody was subjected to Western blot analysis to endoproteinase-, Asp-N, treated (enzyme:substrate = 1:1000 for overnight at room temperature) or untreated P-Rho. The endoproteinase AspN cleaves off C-terminal 19 amino acids containing all possible sites of phosphorylation (Palczewski et al. 1991). The pellet was dissolved in 50 μl SDS-PAGE sample buffer and analyzed with SDS-PAGE using 12.5% gel. Both primary and secondary antibodies were used at 1:2000 dilutions, and their specific antibody/antigen binding was visualized by chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ).
Dot-blot Analysis with the Quick-freezing Method
Dot-blot analysis, especially with both the quick-freezing (QF) and FS methods was performed as shown in Figure 1. To attach peptides strongly by chemical covalent reaction onto glass slides, 3-aminopropyl triethoxysilane (Figure 1A) followed by glutaraldehyde (Figure 1B) were previously coated on them, as reported previously (Ohno et al. 1993; Terada et al. 1997). Five μl of 334Ser-phosphorylated (DDEApSA-TASK) or unphosphorylated (DDEASATASK) peptide was attached to the silane-glutaraldehyde-coated glass slides (Figure 1C). The glass slides with both types of peptides were treated with 5% BSA (Sigma; St Louis, MO) in PBS, pH 7.4, for 1 hr (Figure 1D) and quickly frozen in liquid isopentane-propane cryogen (−193C) precooled in liquid nitrogen (−196C) (Figure 1E). Frozen samples were then freeze substituted in acetone containing 2% paraformaldehyde. Some were treated with xylene, graded alcohol with distilled water, and finally with PBS. They were immunostained with anti-P-Rho334 antibody and then with Alexa Fluor 488-conjugated anti-rabbit IgG antibody (Molecular Probes; Eugene, OR). Some glass slides with peptides were treated with albumin and immunostained in a similar manner without both QF and FS steps. Immunostained dots were observed under a fluorescence microscope (BX-61; Olympus, Tokyo, Japan).
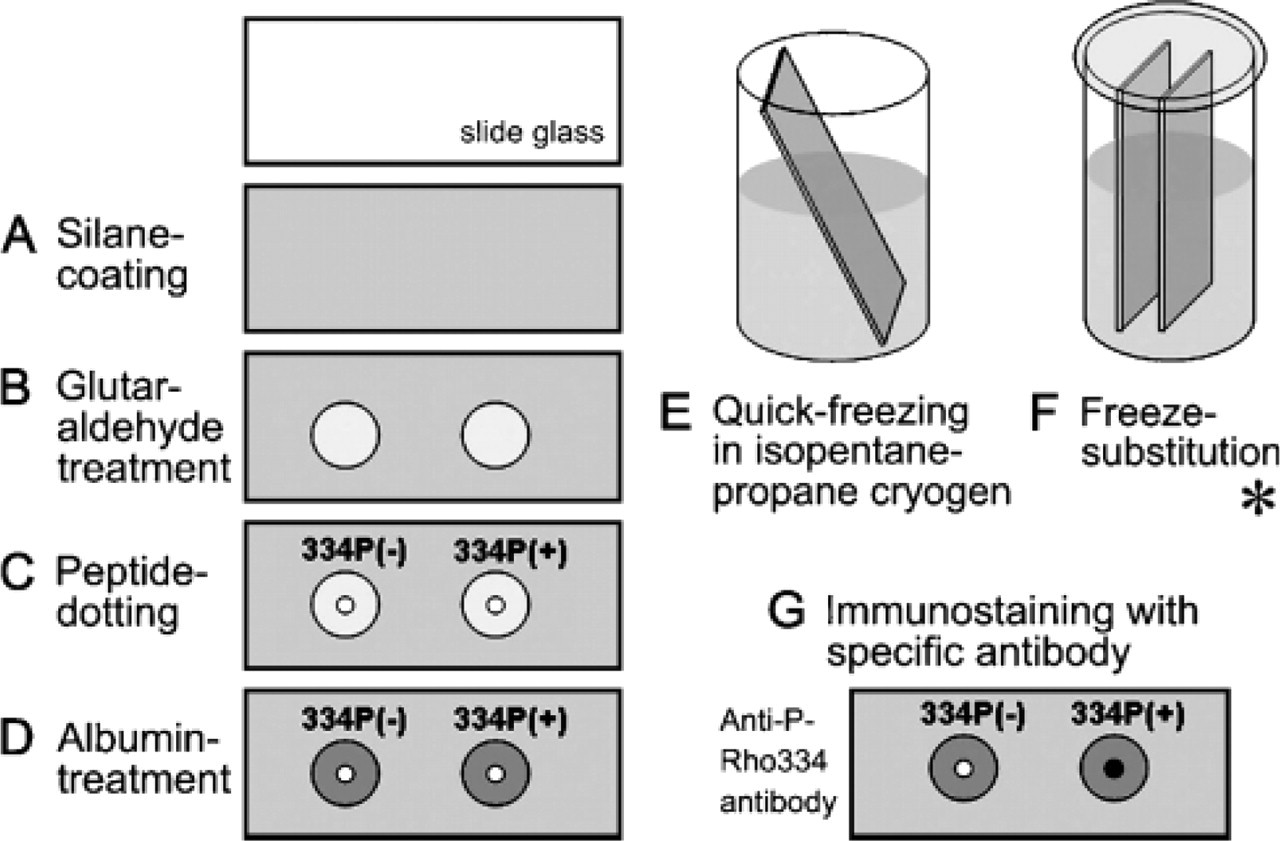
Dot-blot analysis with the quick-freezing (QF) method. A glass slide is coated with 3-aminopropyl triethoxysilane (
IV-CT of Mice under Illumination Conditions
Twenty adult C57BL/6 mice weighing 20-30 g were used for the present experiment. Two mice were analyzed for each experimental condition as follows. IV-CT was performed against left eyeballs of mice under deep chloroform anesthesia, which were kept under different illumination conditions (Figure 2A). To treat the eyeballs more easily than those in the previous cornea experiment with IV-CT (Chen et al. 1995, 1997, 1998), palpebral skins were slightly cut around them (Figure 2B). The eyeballs were then immediately frozen in vivo by pouring 50 ml liquid isopentane-propane cryogen (−193C), which was cooled in liquid nitrogen just before pouring (Figure 2C) as reported in detail (Chen et al. 1995). They were always under normal physiological conditions with their hearts beating, as previously described (Chen et al. 1995, 1997, 1998; Ohno et al. 1996). Intensity of light illumination was measured with an illuminator (Digital Illuminator #8581; Sato-Shoji, Yokohama, Japan) at each experimental time-point three times. We set up the daylight condition with natural light coming through the windows (1434 ± 14 lux) to perform the IV-CT of living mice under normal daylight. For complete darkness, after the 12-h dark adaptation the IV-CT was performed under infrared light for 3 min by directly observing the mice with an infrared camera (SONY DCR-PC120 (night shot mode); SONY, Tokyo, Japan) (Figure 2A). The camera's viewfinder, not a television monitor, was always used to obtain visible images through the camera, to avoid light stimulation against the living mouse retina. For the condition under the safety red light, IV-CT was performed under the light shielded by a red-color plate for 2 min (1.61 ± 0.04 lux) (Figure 2A) after 12 or 36 hr dark adaptation. To obtain light-illuminated mouse retinas, the dark-adapted eyeballs were continuously illuminated with a flashlight (8182 ± 76 lux) for 10, 30, 60, and 180 sec before performing IV-CT. They were then immediately frozen in a similar way with the liquid cryogen and maintained in liquid nitrogen at a low temperature. Mice were killed under deep anesthesia by bathing the whole body in liquid nitrogen. Frozen eyeballs were finally removed with a pair of nippers in the liquid nitrogen and then processed for the next freeze-substitution step.
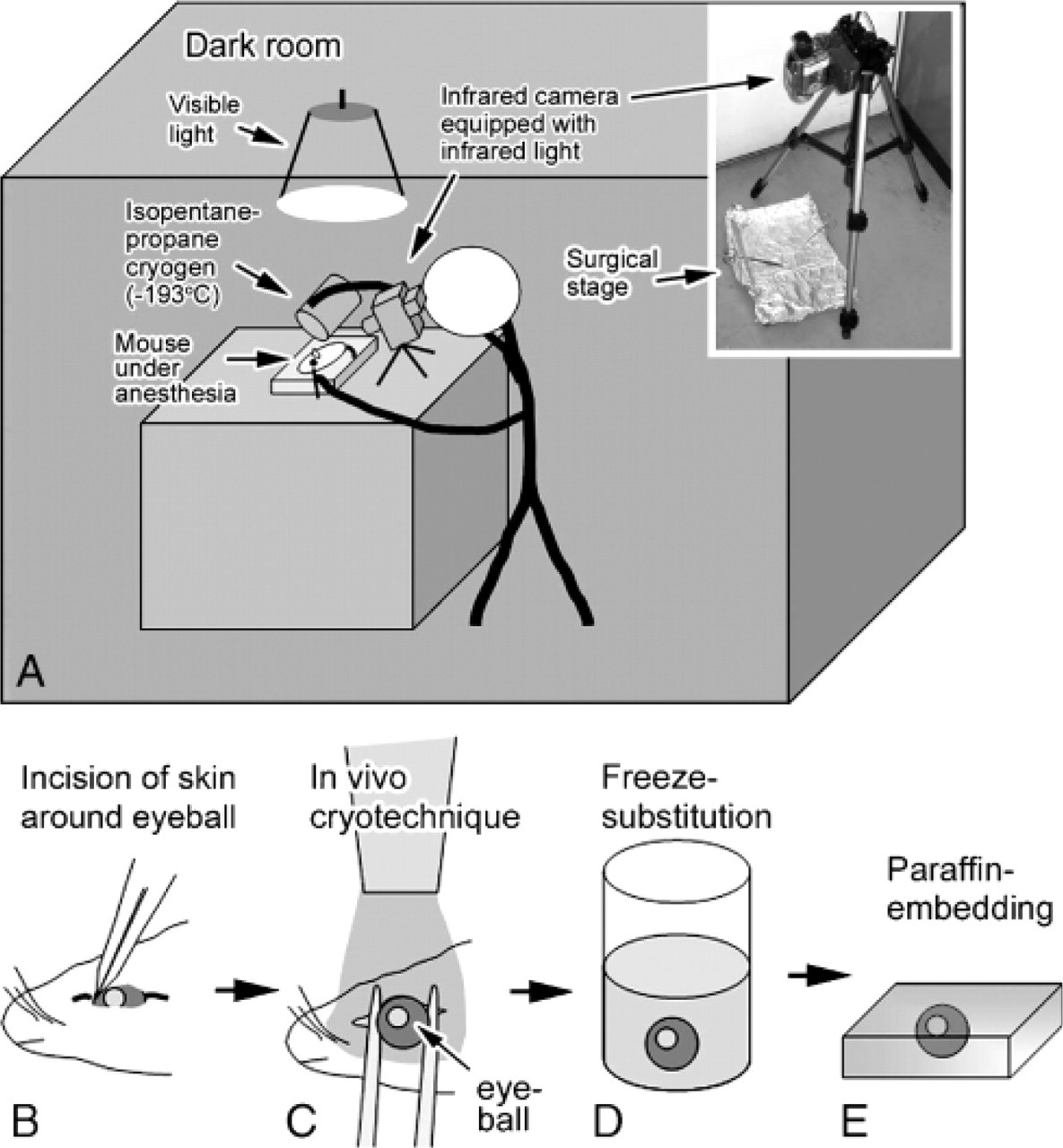
Schematic representation of in vivo cryotechnique (IV-CT) for whole eyeballs of living mice. In a dark room they were anesthetized, and IV-CT was performed (
To indicate more precisely the affect of light stimulation, Rho regeneration was determined by the photospectrometric analysis, as described previously (Ohguro et al. 1995). Briefly, living mice were exposed to different illumination conditions as described above and then subjected to the dark adaptation. At different time points of the dark adaptation, mice were anesthetized and their eyeballs were enucleated and cut into two parts, anterior and posterior segments. Posterior segments were homogenized with a glass homogenizer in 10 mM Hepes buffer, pH 7.5, containing 10 mM dodecyl β-maltoside and 20 mM hydroxylamine. Samples were centrifuged at 20,000 × g, and their spectra were recorded before and after complete bleaching. Percent of photolyzed rhodopsin (R∗) was finally calculated (Ohguro et al. 1995).
Freeze Substitution and Following Tissue-preparation Steps
FS solution usually consisted of absolute acetone containing 2% paraformaldehyde, which was prepared by using a molecular sieve (Type 3A; Nakarai Tesque, Kyoto, Japan) for complete dehydration, as reported previously (Zea-Aragon et al. 2004; Li et al. 2005; Ohno et al. 2005). Frozen eyeballs were kept at ~-80C for 24 hr (Figure 2D) and then put into a deep freezer at −30C and −10C for 2 hr each, and finally placed in a refrigerator at 4C for 2 hr. They were warmed at room temperature for 1 hr, washed in pure acetone, immersed in xylene, and routinely embedded in paraffin wax (Figure 2E).
Immunostaining for Phosphorylated Rhodopsin (P-Rho)
Paraffin-embedded eyeball tissues were cut at 5-um thickness and routinely deparaffinized with xylene and a graded series of ethanol. Some sections were commonly stained with hematoxylin-eosin (HE) for pure morphology. For immunostaining, others were incubated with 1% hydrogen peroxide in PBS for 60 min. They were then washed in PBS, incubated with 10% normal goat serum for 60 min, and individually incubated with avidin and biotin (Vector Laboratories; Burlingame, CA) in PBS for 1 hr each. Furthermore, they were immunostained with the rabbit anti-P-Rho334 antibody (1:1000 dilution) at 4C for 12 hr. Immunocontrol sections were incubated with PBS or normal rabbit serum instead of the primary antibody. They were incubated with biotinylated goat anti-rabbit IgG antibody (Nichirei Co.; Tokyo, Japan) at room temperature for 1 hr and with horseradish peroxidase (HRP)-conjugated avidin-biotin complex (ABC) for 1 hr. They were routinely visualized using the peroxidase enzyme reaction with metal-enhanced DAB (Pierce; Rockford, IL) in the buffer solution containing 0.02% hydrogen peroxide for 5 min (ABC-DAB method), as previously reported (Terada et al. 2004). Finally, they were incubated in 0.04% osmium tetroxide solution for 2 min to increase the contrast of immunoreaction products.
Results
Antibody Specificity by Western Blot and Dot-blot Analyses for Quick-frozen Peptides
Immunological specificity of anti-P-Rho334 antibody was already determined by using serially diluted phosphorylated rhodopsin with Western blot in the previous report (Ohguro etal. 2003) and reconfirmed by Western blot analysis for Asp-N-treated Rho (Figure 3A). Without the Asp-N treatment, the 70-kDa or 35-kDa band showed the dimer or monomer Rho, respectively (Figure 3B). After the Asp-N treatment, the residual 35-kDa band was immunostained with anti-P-Rho334 antibody, but the 33-kDa band, showing the major cleaved Rho at C terminus with 334Ser, was not immunostained (Figure 3B). Moreover, to examine whether the phosphorylated peptide was still immunoreactive after the QF method, another dot-blot analysis was performed, as described in Materials and Methods. As common nitrocellulose was easily melted in acetone, and the polyvinylidene difluoride membrane did not absorb peptides, the silane-glutaraldehyde-coated glass slides were more useful for attaching the peptides to their surface (Figures 3C-3F). After their preparation with both QF and FS methods, specific immunoreactivity with anti-P-Rho334 antibody was detected for the phosphorylated 334Ser peptide (Figure 3F), in the same manner as without the QF method (Figure 3D). Even after xylene treatment of the peptide, the same results were obtained (Figures 3G-3J). These findings indicate that phosphorylated and non-phosphorylated features of the peptides were well preserved to be immunostained even after the QF and FS methods.
Immunostaining of Retina for P-Rho under Daylight Condition
In the eyeballs prepared with IV-CT followed by the FS method, whole retinal tissue layers were clearly detected on HE-stained sections (Figure 4). The eyeballs were always frozen from the scleral outside with the poured cryogen, and so the rod and cone layer was well preserved at a light microscopic level (Figures 4A, 4C, 4F, 4I). Under daylight condition (1434 ± 14 lux; percentage of photolyzed rhodopsin, R∗ = 40%), the rod and cone layer was specifically immunostained with the anti-P-Rho334 antibody (Figures 4E, 4H, and 4K). Immunostained area was in all parts of the mouse retina (Figures 4E and 4H). At higher magnification, immunostaining was not obtained near the outer nuclear layer, indicating that the immunopositive part was not in the inner segment of the rod and cone layer (Figures 4I-4K). Specific immunoreactivity was eliminated without the primary antibody on immunocontrol sections (Figure 4K, inset). This finding indicates that the retinal rod and cone layer can be analyzed on paraffin sections prepared by IV-CT for immunostaining.
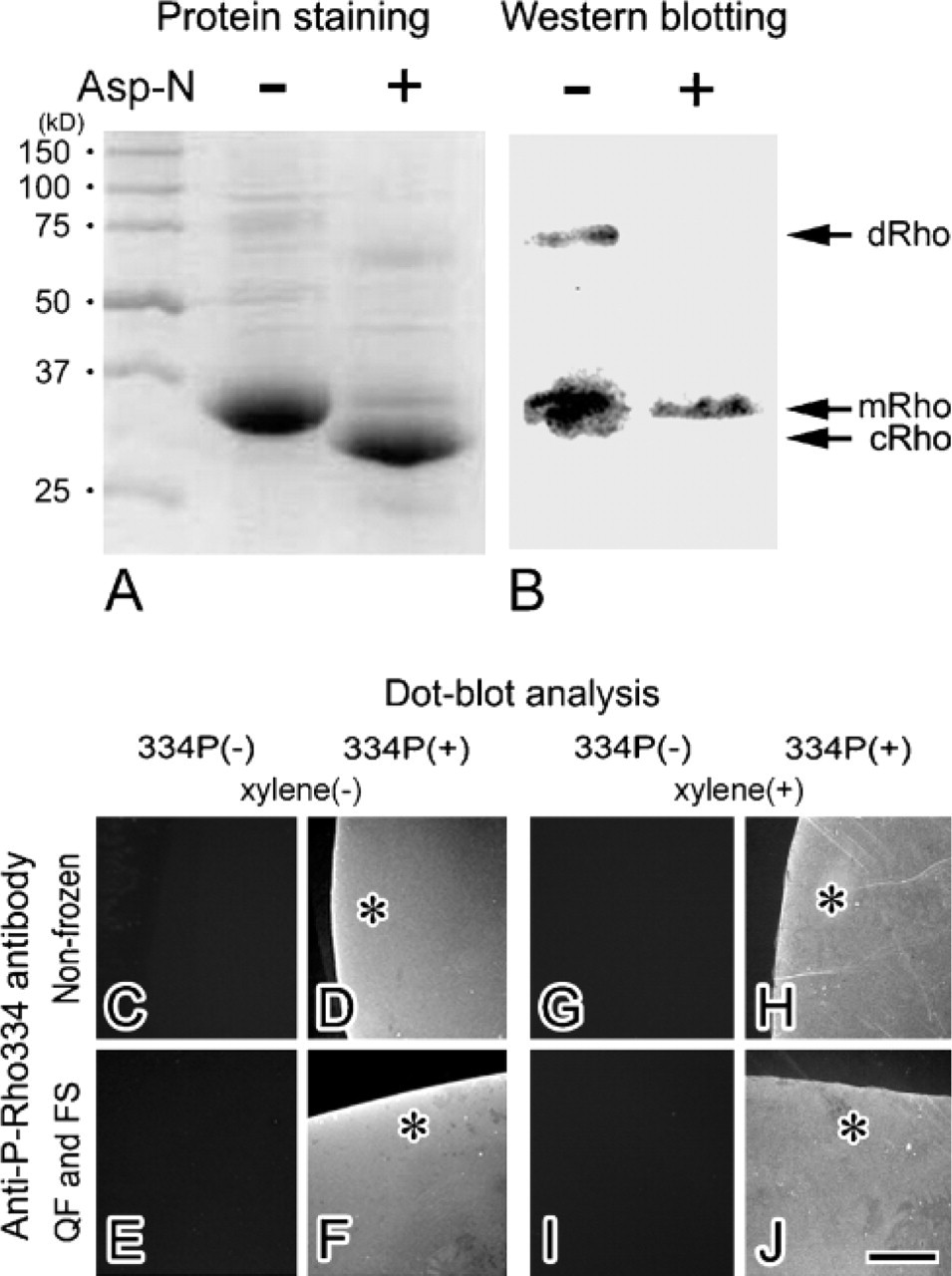
Characterization of anti-P-Rho334 antibody by Western blot and dot-blot analyses. The purified native and Asp-N-cleaved rhodopsin proteins were stained with Coomassie blue. (
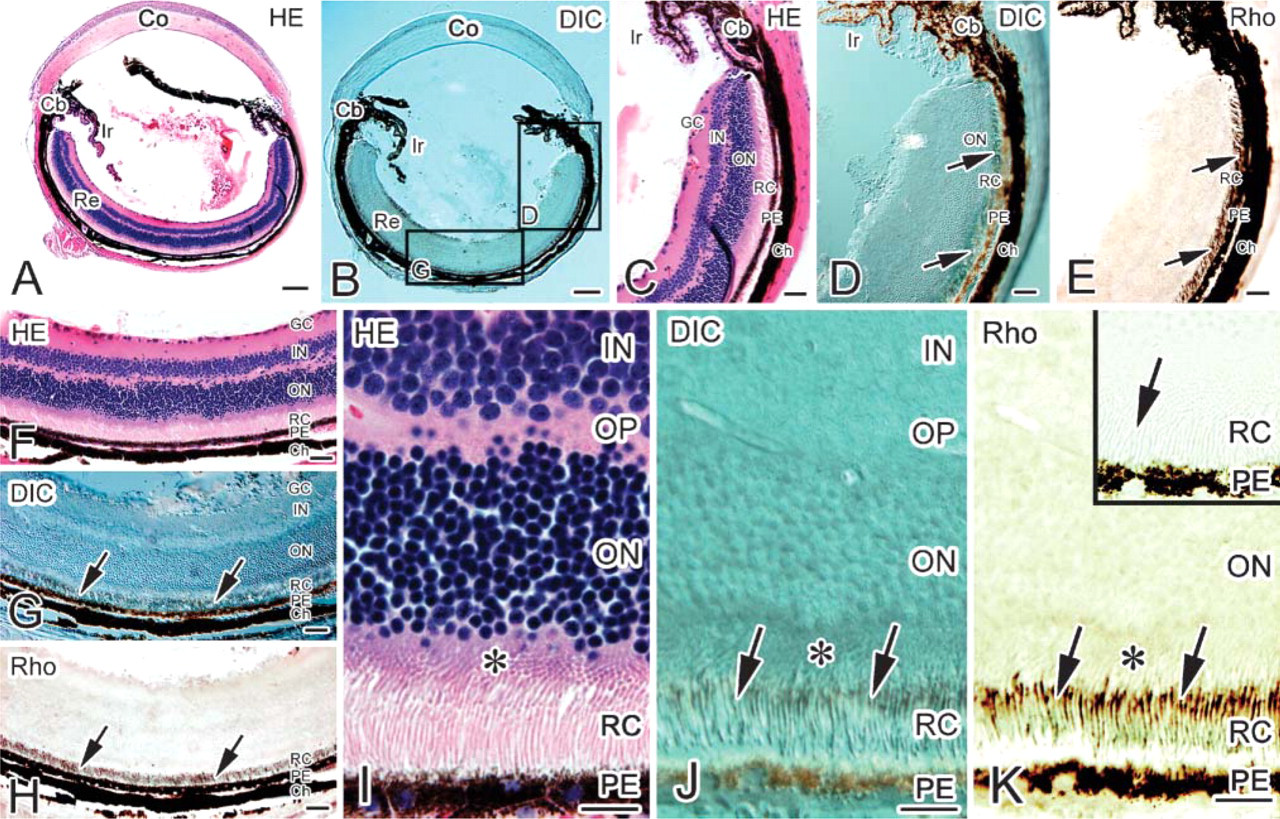
Light microscopic immunohistochemistry of the whole-mount eyeball exposed to daylight. By hematoxylin-eosin (HE) staining (
Loss of P-Rho334 Immunoreactivity after Dark Adaptation
To examine the immunoreactivity of P-Rho334 in the dark-adapted retina without any exposure to light (Figures 5C and 5D), IV-CT was performed using the infrared camera equipped with infrared light, as described in Materials and Methods (Figure 2A), and were compared with those under daylight condition (Figures 5A and 5B). Using the infrared camera with the infrared light, it was easy to perform IV-CT while looking through the viewfinder (Figure 2A). The isopentane-propane cryogen (−193C) cooled in liquid nitrogen was easily carried into the dark room. In the living mouse retina under condition of complete darkness, P-Rho334 immunoreactivity was not detected (Figures 5C and 5D). These findings indicate that 334Ser of rhodopsin is not phosphorylated during dark adaptation.
To further examine the specific P-Rho334 immunoreactivity in the living mouse retina under the safety red light after dark adaptation, some mice were kept in complete darkness for 12 hr, and then IV-CT was performed within 2 min under the safety red light (1.61 ± 0.04 lux) (Figures 5E and 5F). In the mouse retina prepared under such an illumination condition, specific P-Rho334 immunoreactivity was not yet detected (Figure 5F). To confirm whether the immunoreactivity loss continued after a longer dark-adaptation time, other mice were also kept in darkness for 36 hr, and then IV-CT was similarly performed under the safety red light (Figures 5G and 5H). P-Rho334 immunoreactivity was also not detected under the 36-h darkness condition (Figure 5H). These findings indicate that 334Ser of the rhodopsin is not phosphorylated even under the safety red light after dark adaptation.
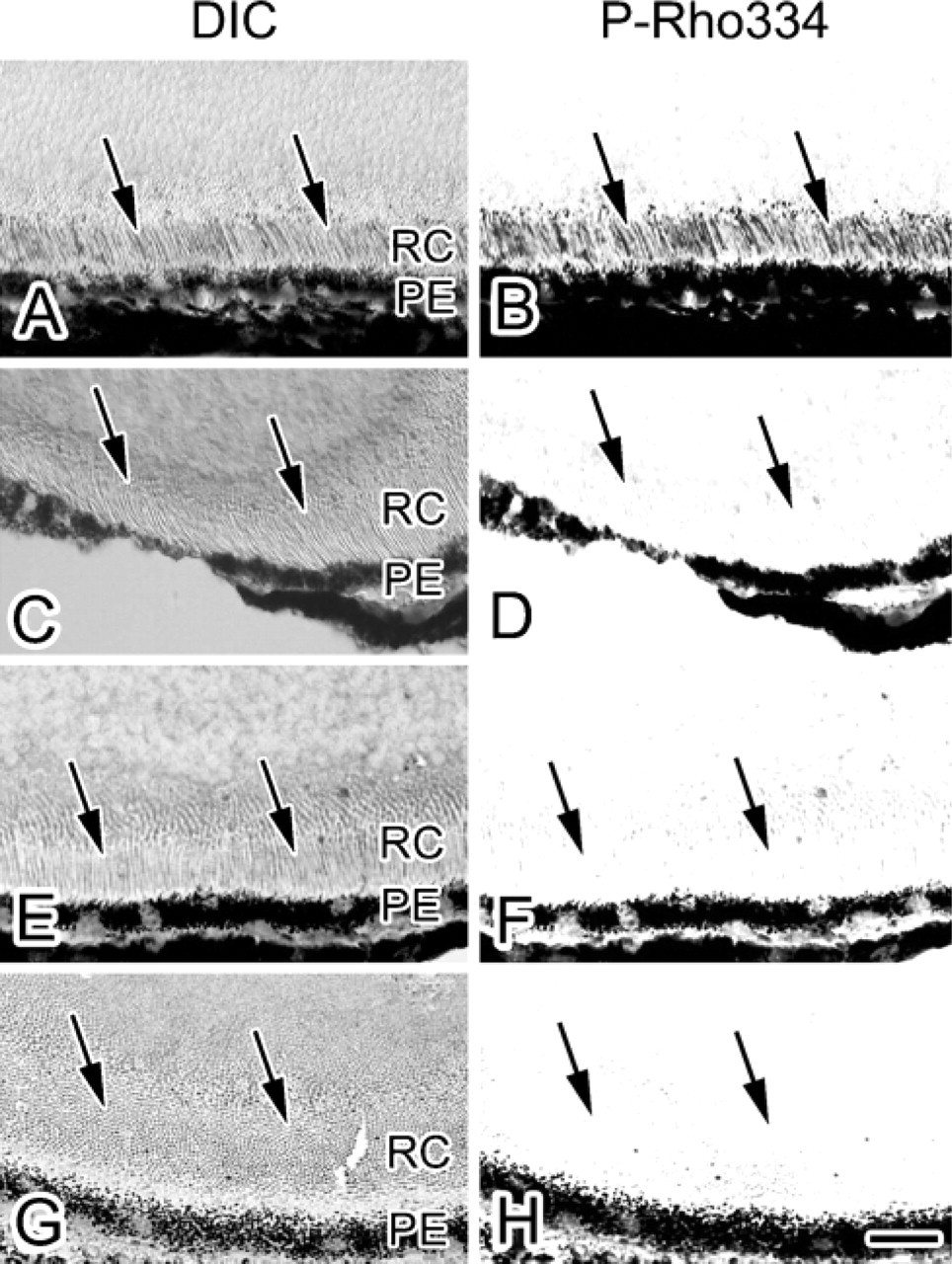
Light micrographs of DIC (
Recovery of P-Rho334 Immunoreactivity after Light Exposure
Under daylight condition P-Rho334 immunoreactivity was always detected, but it was completely eliminated after the dark adaptation as described previously. To further determine how long it takes to recover P-Rho334 immunoreactivity in the dark-adapted retina again under the visible light, the eyeballs dark adapted for 12 hr were continuously exposed to visible light (8182 ± 76 lux) for 10, 30, 60, and 180 sec (Figure 6). Using liquid cryogen, IV-CT was accurately performed at each time point after starting the light exposure (Figure 2A). P-Rho334 immunoreactivity was not detected in the living mouse retina after the 10-sec light exposure (Figure 6B). In other retinas exposed to visible light for >30 sec (R∗ = 10%) and more (i.e., 60 and 180 sec), P-Rho334 immunoreactivity was definitely recovered (Figures 6D, 6F, 6H). This finding shows that 334Ser of rhodopsin in retinal tissues of living mice is generally phosphorylated between 10 and 30 sec after visible light stimulation.
Discussion
In the living animal retina, phototransduction has been thought to proceed in time courses of milliseconds to seconds (Menson et al. 2001). To capture such a highly temporal resolution of images, QF is performed at microsecond order when vitreous ice is produced. Using the QF method, some cytoskeletal changes in squid photoreceptor microvilli were already detected after light exposure (Tsukita et al. 1988). In the present study, we extended an idea of QF for time resolution as follows. Although freezing is not accomplished as formation in the vitreous ice, stopping of proteins' structural changes by IV-CT must be much quicker than that by the conventional chemical fixation method.
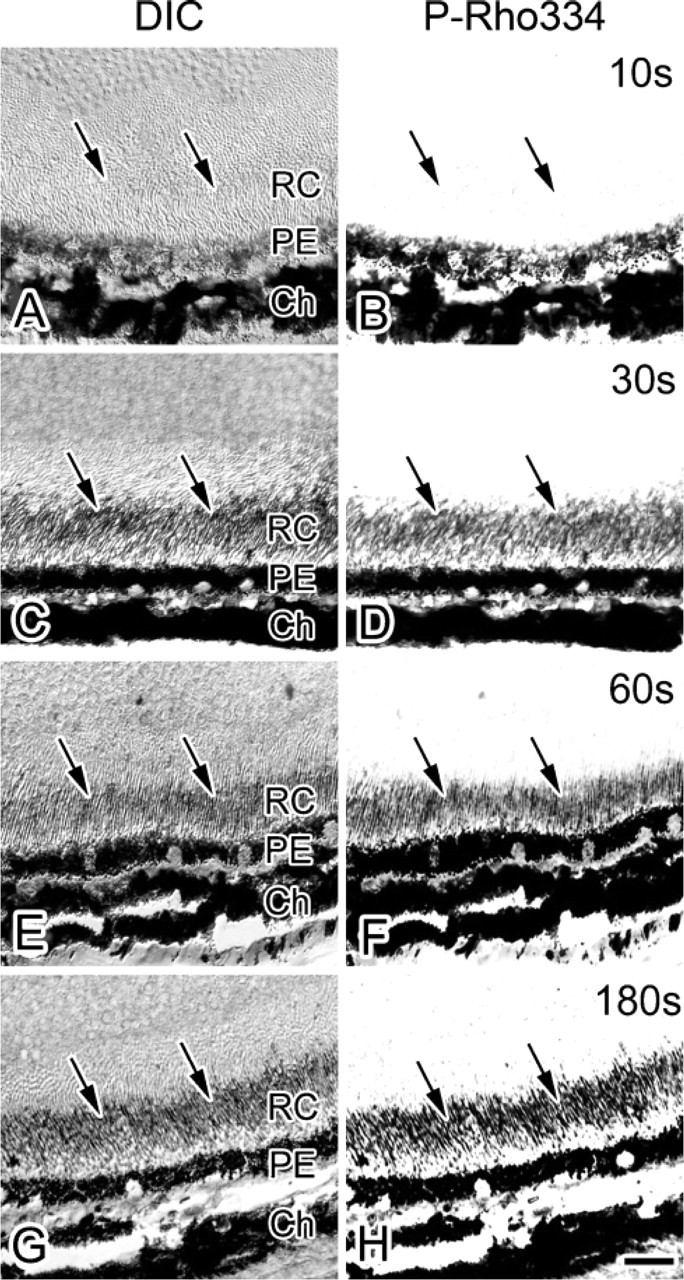
Light micrographs of DIC (
In a previous report, it was claimed that a necessary time for changes at the center of a 10-μl drop of water from 293K (20C) to 233K (−40C) was ~387 msec in propane cooled in liquid nitrogen (Barlow and Sleigh 1979). When the mouse eyeball (~18 mg) is similar to the water drop (~18 μl), it could be frozen within 1 sec. Moreover, stabilization of Rho phosphorylation was completed fast enough for this estimation due to the following two reasons, even though it is difficult to measure the actual temperature. First, the depth of the rod and cone layer in retina from the sclera is estimated to be ~200 μm. Second, Rho phosphorylation may be almost stopped when the temperature was decreased <0C because it needs enzyme reactions by the kinase to proceed (Menson et al. 2001). Therefore, for the purpose of immunohistochemistry for P-Rho, IV-CT is assumed to cryoimmobilize all components in the retina of anesthetized mice at a second scale after the starting time point of direct light illumination.
In addition to the time resolution, immunoreactivity with common cryofixation was also more clearly detected, as compared to that with conventional chemical fixation (Ohno et al. 2005). The diluted solution of anti-P-Rho334 antibody with conventional chemical fixation was 10 times more concentrated to obtain similar immunostaining intensity than that with the IV-CT followed by FS (data not shown). This may be due to exposure of the Rho molecular structures, which were easily hindered by the strong chemical fixation of proteins, because tiny ice crystal formation and solubilization of lipid components around the Rho molecules always occur during QF and FS procedures.
Multiple phosphorylation epitopes of the Rho molecule have been reported to play an important role for the rod photoreceptor dark adaptation because of the reduced ability of Rho to activate the phototransduction cascade processes (Fain et al. 2001) and the stabilization of Rho conformation after the interaction with arrestin (Gibson et al. 1998; Kisselev et al. 2004). In the serial Rho-phosphorylation steps, only 343Ser was assumed to be the most rapidly phosphorylated, and the other 338Ser and 334Ser were slower (Kennedy et al. 2001). In their experimental procedure, a mouse was sacrificed by cervical dislocation and its eyeballs were removed as quickly as possible while illuminated with a flashlight. Immediately after light stimulation, the computer system automatically activated a solenoid valve to open for 1 sec and allow their incubation with urea to analyze the phosphorylation sites with mass spectrometry (Hurley et al. 1998). In our experiments using the higher sensitivity of immunostaining with IV-CT, we demonstrated that the P-Rho334 rapidly appeared in the living mouse retina between 10 and 30 sec after the strong light exposure. There may be a time-lapse among the conformational change of retinol and the rhodopsin phosphorylation in rod cells. We recognize the time difference between the first reaction for light and the second accommodation against the extensive light. Recently, it was also reported that phosphorylation of rhodopsin in cone cells showed quicker photoresponses than in rod cells (Tachibanaki et al. 2005). For future study, IV-CT presented here is also applicable to other Rho phosphorylation and dephosphorylation sites under rapidly changing experimental conditions in living mouse retinas.
Footnotes
Acknowledgments
The authors thank Drs. T. Baba and Y. Fujii from the Department of Anatomy, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, for their constructive comments on this work.