
Abstract
Fibril-associated collagens with interrupted triple helices (FACITs) XII and XIV act as fibril organizers and assist in the maintenance of uniform fibril size. We investigated the spatial expression patterns of collagens XII and XIV in cryptogenic organizing pneumonia (COP)/organizing pneumonia (OP) and in idiopathic pulmonary fibrosis (IPF)/usual interstitial pneumonia (UIP) and compared them to normal human lung. Study subjects included 10 patients with COP/OP, 10 patients with IPF/UIP, and 8 control subjects. Immunostaining for collagens XII and XIV was carried out in paraffin-embedded human lung tissue sections. Picrosirius red histochemical staining for collagen I expression and electron microcopy to evaluate fibril diameter were also performed. In normal lung, collagens XII and XIV were expressed in perivascular and subpleural connective tissue. In COP/OP, both collagens showed intense staining in perivascular connective tissue, thickened alveolar septae, and subpleural areas. In IPF/UIP, XII and XIV were expressed in perivascular connective tissue, in areas of established fibrosis, and in areas of subpleural thickening. Only collagen XII was expressed in granulation tissue plugs in COP/OP and in fibroblastic foci in IPF/UIP. Collagen type I was overexpressed in fibrotic areas. Electron micrographs revealed obvious fibril diameter alteration and fusion in the same areas. FACITs XII and XIV are expressed in normal and fibrotic lung. Unlike collagen XIV, collagen XII was expressed in granulation tissue plugs in COP/OP and in fibroblast foci in IPF/UIP. This may suggest a possible distinct role for both collagens in the modulation of the extracellular matrix during the onset of fibrotic process.
Keywords
T
Because a large group of proteins of the extracellular matrix has been thought to be important as immune effectors to constituents of the interstitium (Reynolds et al. 2005), a group of collagens has been targeted as potentially useful immune effectors (Negri et al. 2000; Rozin et al. 2005). Among these, types XII and XIV have shown promise (Walchli et al. 1994). Collagen types XII and XIV belong to a subgroup of non-fibrillar collagens termed fibril-associated collagens with interrupted triple helices (FACITs). FACITs XII (Gordon et al. 1987) and XIV (Gordon et al. 1991; Gerecke et al. 1993) have one domain that anchors the molecule to the surface of the fibril and three finger-like domains (Gordon and Olsen 1990). These collagens, whose chains and molecules are schematically represented in Figures 1A and 1B), sit on the surface of collagen I fibrils (Figure 1C) (Keene et al. 1991; Young et al. 2000) and appear to alter the interactive properties and organization of the fibrils (Young et al. 2002). Whereas collagen XII is thought to stabilize collagen fibril structure in tissues during development or during rapid remodeling (Chiquet et al. 1996; Marchant et al. 2002), collagen XIV is found to limit fibril diameter by preventing lateral fusions of adjacent fibrils (Young et al. 2000).
We have recently reported the first data characterizing the spatial and temporal expression pattern of FACITs XII and XIV in the lung during bleomycin-induced pulmonary fibrosis in mice by immunohistochemistry and in situ hybridization (Tzortzaki et al. 2003). Our results showed that the expression of collagens XII and XIV was low until 4 weeks after bleomycin treatment, collagen XII expression was greatest between 4 and 8 weeks, and expression of collagen XIV persisted from 4 to 12 weeks, which suggests that these two proteins may play distinct roles in the fibrotic process, modulating the biochemical properties of fibrils (Tzortzaki et al. 2003).
To validate the importance of collagen types XII and XIV and to explore the relationship between this factor and fibrosis, as well as the relationship between collagen types XII and XIV and parenchymal remodeling, we studied this collagen in lungs of patients with IIPs and report our results.
Materials and Methods
Patients and Study Design
Our study included tissue samples from 28 patients: 10 patients with final histopathological diagnosis of IPF/UIP according to the American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias criteria (2002), with ages ranging from 59 to 74 years (65.43 ± 5.5); 10 patients with final diagnosis of COP/OP according to ATS/ERS criteria (2002), ages 57 to 71 years (64.8 ± 5.76); and 8 control subjects, ages from 61 to 70 years (64 ± 6.0) and who all underwent surgery for a solitary lung nodule. Samples were obtained from patients undergoing video-assisted thoracoscopic surgery or open lung biopsy for diagnostic purposes. Written informed consent was obtained from each patient and the Committee on Medical Ethics approved the protocol.
Preparation of Lung Sections and Immunohistochemistry for Collagens XII and XIV
Tissue samples were fixed in 10% buffered formalin and embedded in paraffin. Four-μm-thick serial tissue sections were obtained and mounted in Superfrost/Plus glass slides (Fischer Scientific; Pittsburgh, PA). The sections were stained immunohistochemically. Anti-collagens XII and XIV rabbit sera antibodies were used and have been previously described (Keene et al. 1991; Lunstrum et al. 1991). The primary antibody was incubated for 1 hr at room temperature at a dilution of 1:100, followed by reaction with a labeled streptavidinbiotin peroxidase kit (LSAB kit; DAKO, Carpinteria, CA). The sections were faintly counterstained blue with hematoxylin. Positive and negative controls were used. Signal intensity was graded as mild (grade I), moderate (grade II), and intense (grade III). Two independent pathologists (AK and KD), unaware of the antibody used, evaluated and graded the slides in a light microscope.
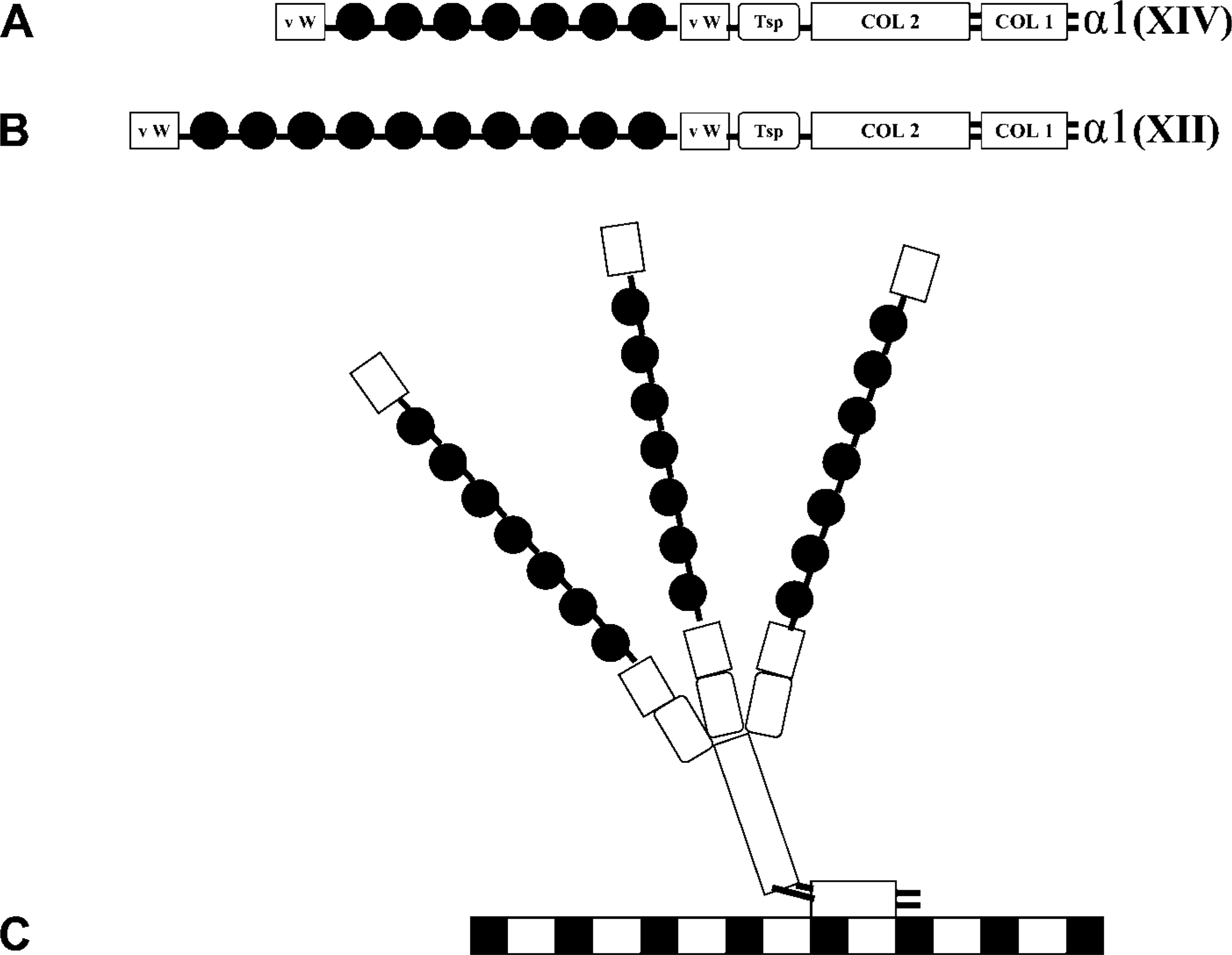
Schematic representation of collagens XII and XIV. The modular structure of the short-form individual α chains of collagens XII and XIV are shown (
Semiquantitative expression analysis of collagens XII and XIV in normal human lung, in COP/OP, and in IPF/UIP
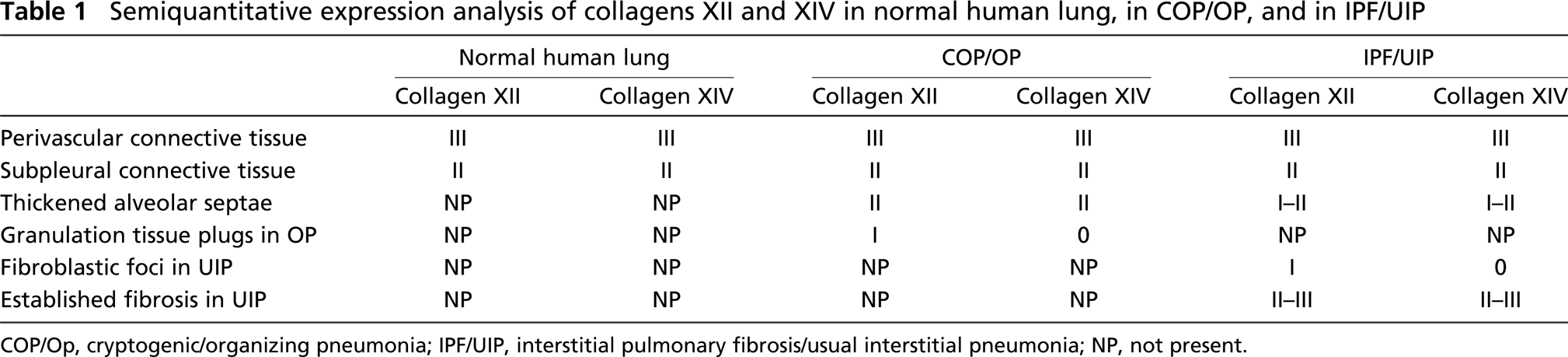
COP/Op, cryptogenic/organizing pneumonia; IPF/UIP, interstitial pulmonary fibrosis/usual interstitial pneumonia; NP, not present.
Picrosirius Red Staining Procedure
Slides were dewaxed and rehydrated in distilled water. They were then incubated in 0.2% phosphomolybdic acid (Sigma-Aldrich; St Louis, MO) for 2 min, rinsed with distilled water, and incubated at room temperature for 110 min in picrosirius red (0.6 g in 600 ml of saturated picric acid; Sigma-Aldrich). The slides were then incubated in 0.01 N hydrochloric acid (Sigma-Aldrich) for 2 min, dehydrated, cleared in xylene, and mounted. Slides were examined with a polarized light microscope.
Transmission Electron Microscopy
Serial electron microscopy lung sections (70-90 nm) were collected on pioloform-coated copper slot grids. Images were obtained using a Jeol-100 CX transmission electron microscope (JEOL; Tokyo, Japan) operating at 80 KV.
Results
Table 1 summarizes a semiquantitative expression analysis of collagens XII and XIV in different tissue areas in normal lung, in COP/OP, and in IPF/UIP.
Expression of Collagens XII and XIV in Normal Human Lung
In normal lung, collagen XII showed intense staining (grade III) in the perivascular region (Figure 2A). Collagen XIV showed intense staining (grade III) in the perivascular region and moderate staining (grade II) in the subpleural connective tissue (Figure 2B). Weak positivity (grade I) was observed in peribronchial smooth muscle cells for both collagens XII and XIV in normal lung parenchyma.
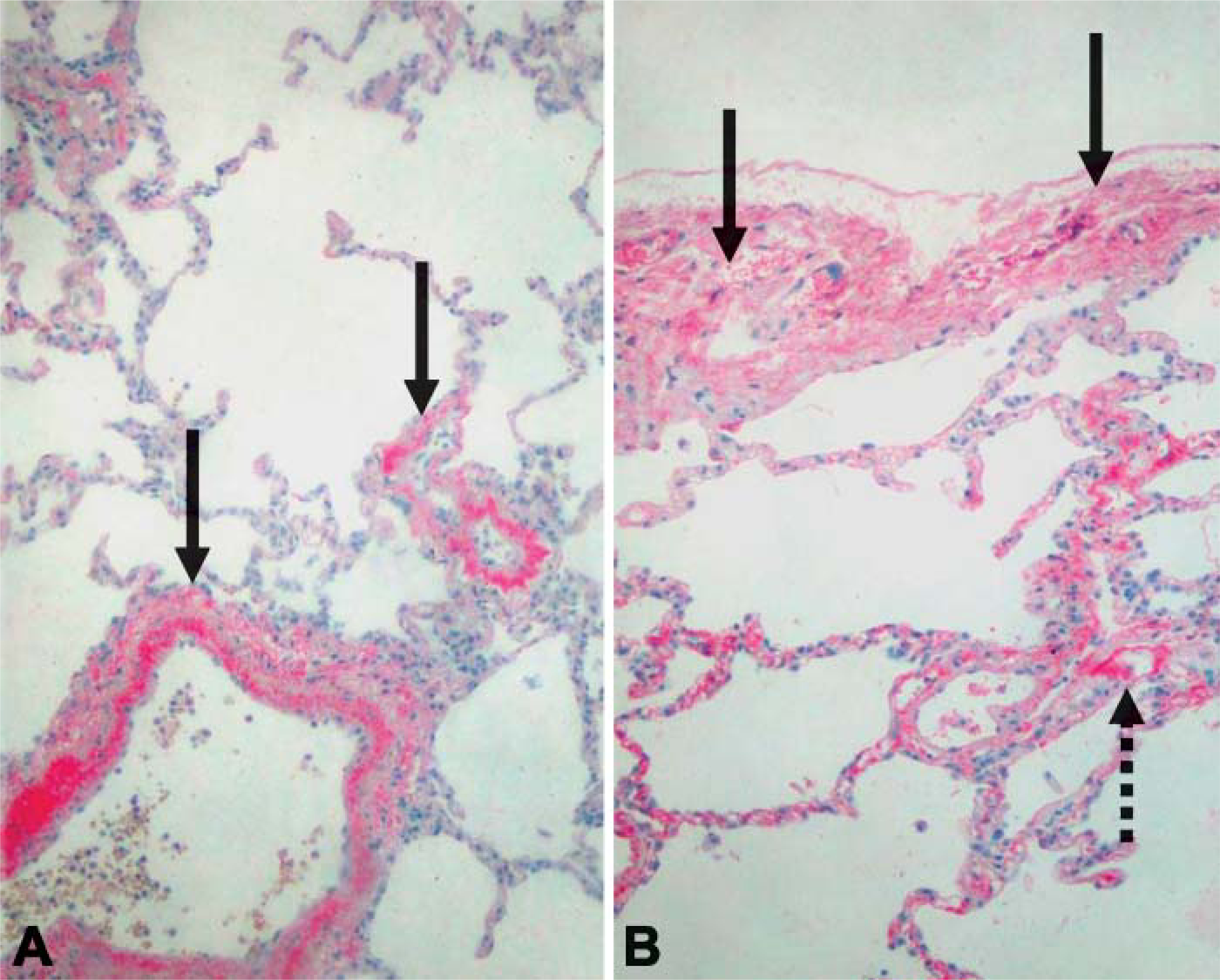
Expression of collagens XII (
Expression of Collagens XII and XIV in COP
When compared with normal lung (Figure 2A), the COP/OP collagen XII showed the same intense staining pattern (grade III) in the perivascular connective tissue but had a slightly more obvious staining (mild, grade I) in the subpleural area of the COP/OP sample (Figure 3A). However, unlike normal lung tissue, fibrosis was apparent in the COP/OP section. Collagen XII had mild staining (grade I) in the granulation tissue plugs and moderate staining (grade II) in thickened alveolar septae (Figure 3A). In contrast, collagen XIV demonstrated intense staining (grade III) only in the perivascular connective tissue with moderate staining (grade II) in both the subpleural connective tissue and in thickened alveolar septae (Figure 3B). Contrary to collagen XII, the COP/OP sample had no collagen XIV signal in the granulation tissue plugs (Figures 3A and 3B). Bronchial epithelial cells, pneumocytes, and peribronchial smooth muscle cells exhibited positive signals for both collagens XII and XIV (grades I-III) and acted as positive internal controls (Gerecke et al. 1997) for both collagens.
Expression of Collagens XII and XIV in IPF/UIP
In IPF/UIP patients, levels of expression for collagen XII ranged from mild to intense (grades I-III). Collagen XII was observed in the perivascular connective tissue (grade III), in areas of established fibrosis (grades II and III), and in areas of subpleural thickening (grade II) (Figure 4A). Mild positivity was observed in fibroblast foci (Figure 4A). Myocytes (peribronchial smooth muscle cells and areas of smooth muscle cell hyperplasia) and epithelial cells showed mild positivity (Figure 4A). Expression pattern for collagen XIV in the IPF/UIP sample was identical to collagen XII with one exception. There was no signal for collagen XIV in the fibroblast foci (Figure 4B).
Picrosirius Red Histochemical Staining
Tissue samples stained with picrosirius red exhibited overexpression of collagen type I in fibrotic areas, using a polarized light microscope (Figure 5).
Transmission Electron Microscopy
Electron microscopy in fibrotic tissue samples revealed fibrillar diameter alterations, disorganization, and fusion (Figure 6).
Discussion
Despite the abundance of publications related to the pathobiology of pulmonary fibrosis, the exact pathogenetic mechanisms are still undetermined. Many questions remain regarding the identity of mediators of the pathological process and the mechanisms involved in pulmonary structural remodeling. For instance, one of the major manifestations of pulmonary fibrosis is an increase in lung extracellular matrix (Selman et al. 2001). This is particularly true for type I collagen, a main structural component of lung. Maintenance of collagen I fibril structure is a requirement for healthy lung function (Crouch 1990). During onset of pulmonary fibrosis, collagens accumulate and contribute to reduced lung compliance, resulting in the pathology of the disease (Shahzeidi et al. 1994; American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias 2002). How the specific collagen I fibril structure is maintained is presently unknown but most likely involves separate proteins that sit on the surface of the collagen I fibrils. Two of these are FACITs XII and XIV (Keene et al. 1991; Shaw and Olsen 1991; Nishiyama et al. 1994; Walchli et al. 1994).
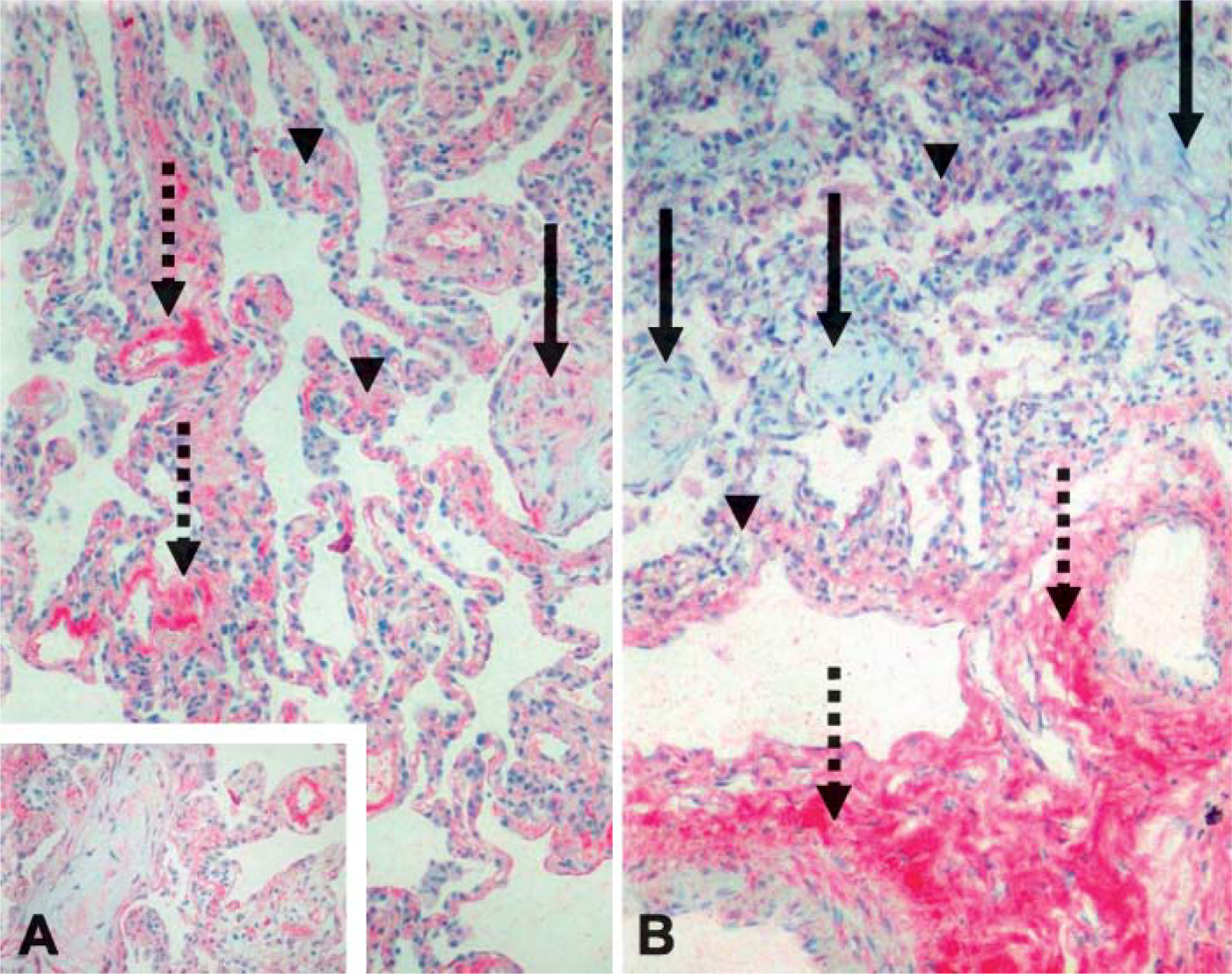
Expression of collagens XII (
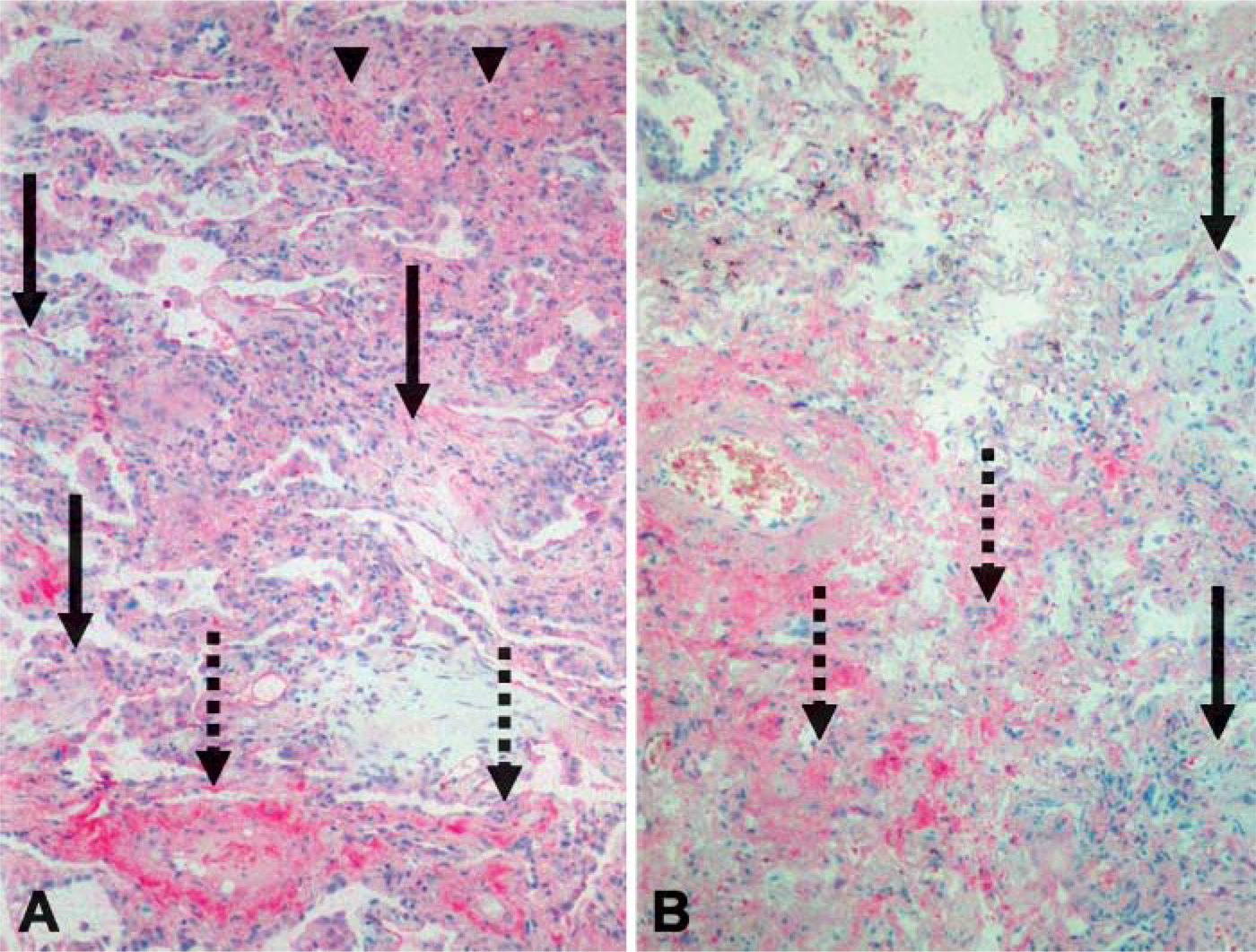
Expression of collagens XII (
It has been well established that there is an extracellular matrix accumulation manifested as either granulation tissue plugs (intraluminal fibrotic buds) in COP/OP or fibroblast foci or areas of established fibrosis in IPF/UIP (American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias 2002). In COP/OP, the process is patchy with relatively normal lung adjacent to zones of organization that may center on a bronchiole. The organizing fibrosis consists of anastomosing, intraluminal plugs of loose connective tissue that occlude bronchioles, alveolar ducts, and surrounding alveoli. All the connective tissue appears about the same age and consists of polypoid intraluminal plugs of loose organizing fibrosis. The architecture of the lung is preserved with no remodeling from dense fibrosis or honeycomb changes. The intraluminal polypoid plugs of connective tissue are mucopolysaccharide rich and lack the abundant collagen fibers seen in dense collagen (American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias 2002). In IPF/UIP there is patchy interstitial fibrosis, often in a subpleural and/or paraseptal distribution, alternating with areas of normal lung. The fibrosis is temporally heterogeneous with two major types: the dense scarring and honeycombing and the fibroblastic foci scattered at the edges of the dense scars. The fibroblast foci consist of a loose type of fibrosis that contains myofibroblasts within a stroma that has few collagen fibers (American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias 2002).
Our previous study on bleomycin-induced mouse lung fibrosis showed that collagen XII is expressed in early stages of fibrosis, and collagen XIV is observed in the later stages (Tzortzaki et al. 2003). In the present study we have chosen cases of COP/OP and IPF/UIP to extend the findings further and to examine if collagens XII and XIV follow the temporal heterogeneity in the human fibrotic process of these two pathologies as well. We also compared our findings to normal human lung. Little is known about the expression and distribution of FACITs in normal lung or in either of the above diseases. This is the first study to report the spatial expression pattern of these collagens in human normal and fibrotic lungs.
In this study we have demonstrated that collagens XII and XIV were expressed in both normal and fibrotic human lung. As components of normal human lung we expect that these two collagens may act as collagen I fibril stabilizers to maintain the normal extracellular matrix architecture (Crouch 1990; Keene et al. 1991; Shaw and Olsen 1991; Nishiyama et al. 1994; Shahzeidi et al. 1994; Walchli et al. 1994). Under abnormal lung conditions, as found in COP/OP and IPF/UIP, both FACITs XII and XIV mimic the previously reported collagen I distribution accumulating and overexpressing mainly in areas of established fibrosis (Tzortzaki et al. 2003). More specifically, in the fibrotic lungs collagens XII and XIV colocalized exactly where type I collagen was expected (e.g., collagen I is abundant in perivascular and subpleural connective tissue and in areas of established fibrosis) (Figure 5). However, in addition to this colocalization, collagen XII, but not collagen XIV, was expressed in granulation tissue plugs/fibroblastic foci in both COP and IPF/UIP, respectively.
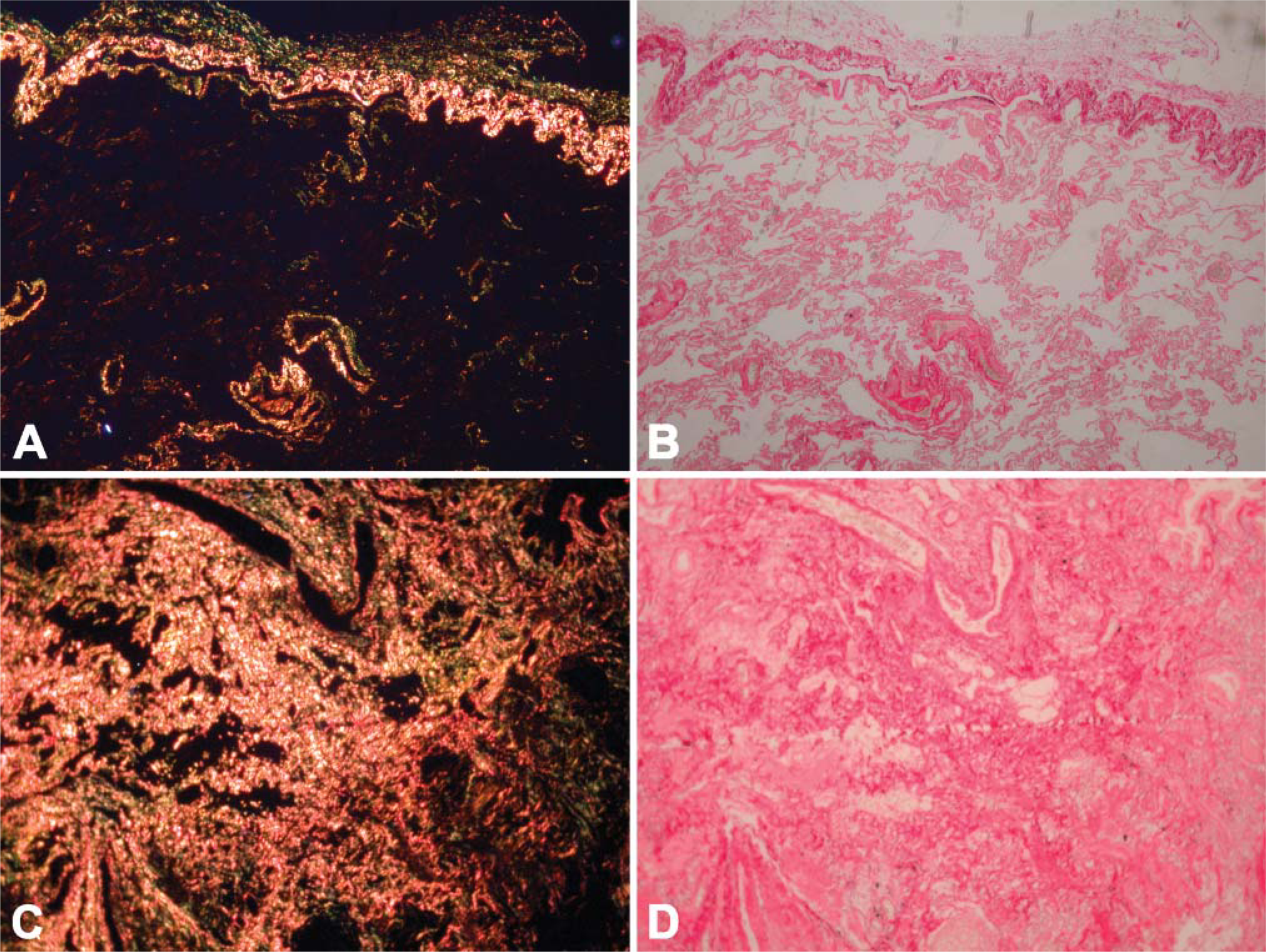
Staining for collagen I using picrosirius red in normal tissue (
We have further demonstrated that collagen XII, but not XIV, was expressed in the granulation tissue plugs/fibroblastic foci. Our interpretation of this difference is that the plugs/foci contain an overabundance of collagen I fibrils. Collagen XII also becomes upregulated to temporarily stabilize collagen I fibrils and keep them apart so they do not permanently crosslink. To date, these data all support the fact that collagen XII is up-regulated in areas of stress, and the function of collagen XII appears to be as a temporal stabilizer of fibrils in the initial stages of the fibrotic process (Chiquet et al. 1996; Jin et al. 2003; Tzortzaki et al. 2003). Because collagen XIV has been implicated as a modulator of fibril diameter, its absence in newly formed fibrosis (granulation tissue plugs/fibroblastic foci) was anticipated. We view the plugs/foci as the early stages of remodeling when the basic components of the extracellular matrix are being made, without the assembly and organization that is to follow. Because collagen XIV is more of an organizing molecule that helps the collagen I fibrils to be a uniform size, we expect its appearance to be slightly later than collagen XII. However, in the case of fibrosis, collagen XIV may appear too late to help modulate fibril diameter. Although collagen XII was expressed abundantly in the same locations as collagen I, we believe the expression of collagen I far outpaced that of collagen XII (Figure 5). This leads to a dilution effect where there is not enough collagen XII to temporarily stabilize the collagen fibrils and keep them from fusing to even larger fibrils. Moreover, it appears that collagen I fibrils have already fused and cross-linked, by the time collagen XIV is expressed (Figure 6). Electron micrographs showed fibrillar remodeling even in apparently ‘normal’ alveolar septae in UIP under light microscopy (Figure 6). This finding could be extremely important in clinical practice for early detection of this fatal disease. It would be interesting to look at our models to see if the appearance of lysyl oxidase, the major enzyme that permanently crosslinks collagen I fibrils, precedes the appearance of collagen XIV during the onset of fibrosis.
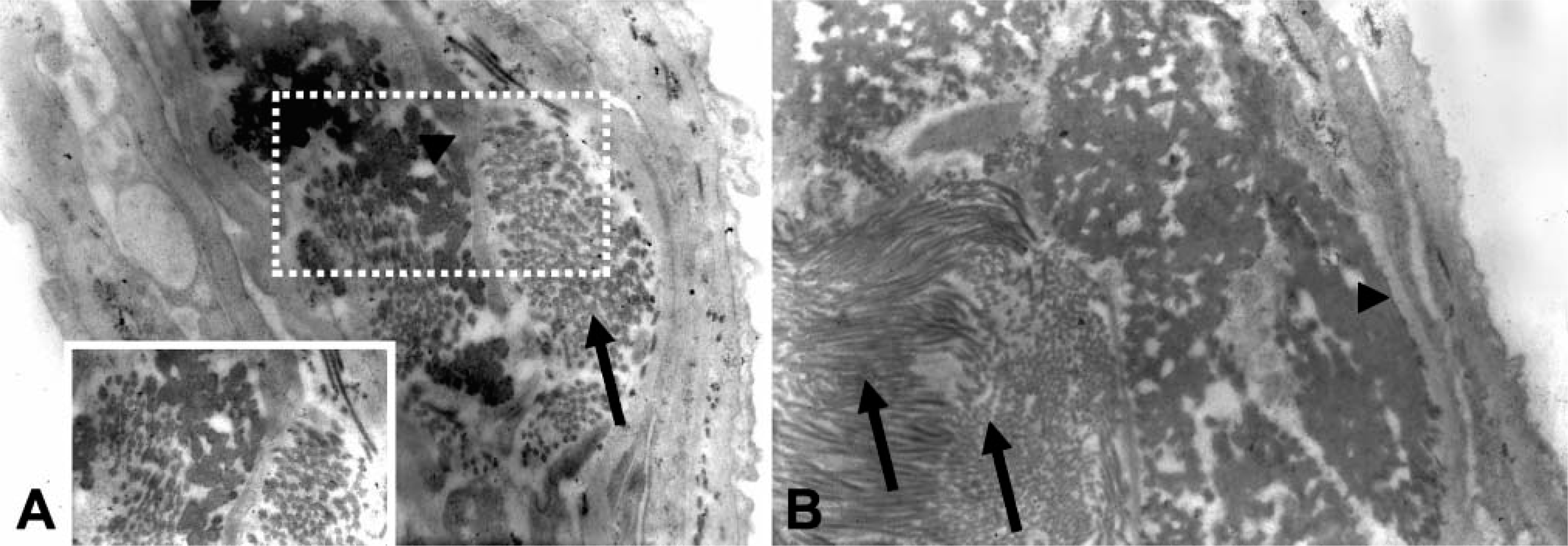
Transmission electron microscopy sections shows areas of increased fibrillar diameter, altered architecture, and fusion. (
There are numerous reports implicating TGF-β in promoting pulmonary fibrosis (Selman et al. 2001; Arai et al. 2002). It is proposed that TGF-β promotes fibroblast proliferation, which in turn contributes to collagen overproduction. The role of TGF-β on the expression of collagens XII and XIV is unclear. One report suggests that addition of TGF-β results in the upregulation of collagen XII and the downregulation of collagen XIV in a dose-dependent manner. Thus, TGF-β may be turned on when the granulation tissue plugs/fibroblastic foci appear, causing both collagens I and XII, but not XIV, to be made. Future studies will include the role of TGF-β in the progression of fibrosis.
Limitations of the Method
Evaluation of the precise role of FACITs XII and XIV in IIPs is still under investigation. This is a descriptive study on the expression of collagens XII and XIV in COP/OP and IPF/UIP. Further studies are required for quantitative evaluation (e.g., RT-PCR) of collagens I, III, XII, and XIV in IIPs. Moreover, it would be interesting to study the above collagens in different time points of disease progression to evaluate their precise role in the pathogenesis of IIP remodeling. Nevertheless, that may require serial invasive procedures to obtain tissue samples. Another limitation is the small number of samples tested. Because the above diseases are not very common in clinical practice, and tissue samples are not always available, we undertook this investigation with a limited number of samples.
Clearly, fibrosis is a multistep process involving not only collagen accumulation but also changes in molecules that modulate the biomechanical properties of fibrils (e.g., collagens XII and XIV) as well as other proteins that influence collagen interactions (e.g., lysyl oxidase and TGF-β). Better understanding of the roles and regulation of collagens XII and XIV in lung fibrosis may enable us to access new therapeutic targets interfering with fibril-stabilizing ability in the early stages of developing fibrosis.