
Abstract
The histological study of arteries with implanted metallic scaffolding devices, known as stents, remains a technical challenge. Given that the arterial response to stent implantation can sometimes lead to adverse outcomes, including the re-accumulation of tissue mass within the stent (or in-stent restenosis), overcoming these technical challenges is a priority for the advancement of research and development in this important clinical field. Essentially, the task is to section the stent-tissue interface with the least amount of disruption of tissue and cellular morphology. Although many methacrylate resin methodologies are successfully applied toward the study of endovascular stents by a variety of research laboratories, the exact formulations, as well as subsequent processing and sectioning methodology, remain largely coveted. In this paper, we describe in detail a methyl methacrylate resinembedding methodology that can successfully be applied to tungsten carbide blade, as well as saw and grinding sectioning methods and transmission electron microscopy. In addition, we present a comparison of the two sectioning methodologies in terms of their effectiveness with regard to morphological, histochemical, and immunohistochemical analyses. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
T
Methacrylate resins are successfully utilized as embedding media for hard biological tissues such as undecalcified bone samples, and these methods have recently been adapted or further modified to suit the study of stented vessels (Theuns et al. 1993). Currently, glycol methacrylate, methyl methacrylate (MMA), and a combination of MMA and n-butyl methacrylate (BMA) are the most widely used resins for the histological processing of endovascular stented material (Rogers et al. 1996; van Beusekom et al. 1996; Malik et al. 1998; Brasen et al. 2001). Although many of these methacrylate resin methodologies are employed by a variety of vascular research laboratories, the exact formulations, as well as the processing and sectioning methodologies, have largely remained coveted. In this paper, we describe in detail an MMA resin-embedding methodology that can be successfully applied to both tungsten carbide (TC) blade as well as saw and grinding (SG) sectioning methods. In addition, we present a comparative review of the two sectioning methodologies in terms of their effectiveness with regard to morphological, histochemical, immunohistochemical, and transmission electron microscopy (TEM) analyses. It is our intention that these resin-embedding and sectioning methodologies will serve as a template for other laboratories embarking on the study of intravascular stent pathology.
Materials and Methods
Stented human coronary arteries from patients were obtained at necropsy and processed as follows.
Tissue Processing
Coronary arteries were removed from the heart, fixed in 10% neutral buffered formalin (NBF) for >24 hr, decalcified in 10% formic acid, and subjected to radiography to localize the stent(s). Decalcification of the entire artery is routinely performed at The Ottawa Hospital to accurately assess adjacent atherosclerotic plaques at either end of the stent. The isolated stented vascular segments were then transferred to 70% ethanol before being processed and embedded in MMA resin (for schedule details, see Supplemental Table 1) (Erben 1997). The following solutions were used to make the resin mixture: Solution I [60 ml MMA (Marivac, Inc.; Montreal, Canada), 35 ml BMA (Marivac), 5 ml methylbenzoate, and 1.2 ml polyethylene glycol 400]; Solution II (100 ml of Solution I, 0.4 g benzoyl peroxide); Solution III (100 ml of Solution I, 0.8 g benzoyl peroxide), and a polymerization resin mixture (400 μl of
Infiltrated stented segments were carefully placed in an upright position in 20-ml polypropylene vials (Leica Instuments GmbH; NuBloch, Germany), which were completely filled with the polymerization mixture to exclude air, then tightly capped and polymerized at −18 to −20C. The presence of oxygen within the embedding vials can ultimately cause bubble formation, thereby inhibiting adequate polymerization and adversely affecting subsequent sectioning technique. This can, however, be prevented either by bubbling nitrogen gas into the tissue-embedded, resin-filled vials or by embedding the tissue segments in vials containing prepolymerized resin bases or plateaus (Erben 1997).
Depending upon specimen size, other suitable molds include glass vials (Fisher Scientific; Nepean, Ontario, Canada), polypropylene microcentrifuge tubes (Fisher Scientific), Eppendorf tubes (Fisher Scientific), or Beem capsules (Marivac). Following adequate polymerization, the specimens were removed by either cutting the ends off the polypropylene vials or, alternatively, breaking the glass vials and pushing the specimens out. The polymerized blocks were then shaped and trimmed of excess plastic with a grinder and sandpaper prior to sectioning.
Rotary Microtome and TC Blade Method of Tissue Sectioning
Tissue sections of a 4-5 μm thickness were generated on an automated Leica RM2255 rotary microtome utilizing a D-profile TC blade (Leica). Because MMA resin is hydrophobic, sectioning was facilitated by moistening both the specimen block and the knife edge with 30% methanol. In a 42C water bath, cut sections were picked up with forceps and first floated onto the surface of a solution of 30% methanol for stretching and then transferred onto the surface of a 0.1% aqueous Elmer's glue solution to promote adherence of the section to the slide (Figure 1A). The sections were then picked up on 3′aminopropyltriethoxysaline-treated slides (Sigma-Aldrich; Oakville, Canada), stretched again with chloroform vapors, covered with a thin plastic film, and gently flattened with a rubber roller (Figure 1B). Finally, the slides were carefully stacked, pressed together with a spring clamp, and dried in an oven at 42C for a period of 2 days (Figure 1C).
SG Method of Tissue Sectioning
A Buehler IsoMet 5000 high-speed precision saw (Buehler; Dusseldorf, Germany) was used to section the stented specimens at a cutting speed of 3200 rpm with a feed advance of 12 mm/min. Continual water cooling was employed throughout sectioning, and a cooling agent (Isomet Plus lubricant, at a ratio of 9:1) was added to reduce heat caused by friction. The surfaces of the resin blocks were prepared for sectioning by grinding on a Metaserve 2000 grinder (Buehler) using fine 600-grit water paper to remove irregular surfaces in the block. Clean, dry blocks were glued onto polysine slides using cyanoacrylate super glue by placing a small drop of glue on the block surface and holding it firmly in position on the slide for 20 sec (Figure 1D). Care must be taken to avoid irregular pressure, because this will result in the formation of bubbles underneath the section. Slides were secured in the slide holder of the Isomet saw, and 100-μm sections were cut (Figure 1E). A wafering diamond blade (12.7 mm × 0.4 mm; Buehler) was used for this purpose. A dressing stone was used to maintain a clean blade edge. Thick sections were ground on the Metaserve 2000 grinder (Buehler) using two grades of water paper (Buehler) (Figure 1F). Coarse 600-grit paper was used to obtain the required thickness, followed by a polishing with 1200-grit paper to remove any scratches. A fine residue of sanding grains was sometimes noted, and attempts to remove the residue using isotonic baths, mild detergents, or polishing were unsuccessful. A final thickness of 8-10 μm was achieved.

Equipment and procedure for cutting tungsten carbide (
Histochemistry
After undergoing deplasticization at room temperature (RT), tissue specimens sectioned using the TC blade method were stained with Gill 3 hematoxylin (Fisher) and eosin (H and E), Movat pentachrome, Masson trichrome, Azan, and Verhoeff's Van Gieson dyes according to conventional methodologies (Bancroft and Gamble 2001). For deplasticization, the sections were brought through solutions of 2-methoxyethylacetate for 20 min thrice, acetone for 5 min twice, and distilled water for 5 min twice.
In our experience, deplasticization of SG sections was often unsuccessful because of the reaction of the solvents with the cyanoacrylate glue used to affix the sections onto the slides, thereby resulting in the sections becoming detached from the slides. A limited number of stains were made possible without prior removal of the resin, and these included Mayer's H and E and Verhoeff's elastin. For H and E staining of SG sections, air-dried slides were placed on a hot plate at 60C and covered with Mayer's hematoxylin (Sigma-Aldrich; Steinheim, Germany) for 12 min, taking care not to let the slides dry out. Slides were then rinsed and blued in running tap water for 30 min before further staining with eosin/phloxine (1:1) on a hot plate at 60C for 1 min, rinsing in water, and finally mounting in Glycergel (DakoCytomation; Glostrop, Denmark). For Verhoeff's elastin staining, slides were immersed in Verhoeff's solution (20 ml alcoholic hematoxylin, 8 ml 10% ferric chloride, 8 ml Lugol's iodine) for 20 min at RT, rinsed in tap water, and mounted in Glycergel (Dako-Cytomation) (Figure 2).
Immunohistochemistry
Deplasticized TC sections were permeabilized in 1% Tween 20-PBS for 30 min, followed by incubation with primary antibodies diluted in 1% BSA at RT for 1 hr. The following primary antibodies were used: Factor VIII (DakoCytomation, 1:50), smooth muscle α-actin (SMA; 1:100, Research Diagnostics, Concord, MA), vimentin (DakoCytomation, 1:100), HAM 56 (DakoCytomation, 1:100), and proliferating cell nuclear antigen (PCNA; 1:100, Santa Cruz Biotechnology, Santa Cruz, CA). Following two washes with 1% Tween 20-PBS for 5 min, tissue sections were incubated with a secondary antibody (DakoCytomation; EnVision K4000, horseradish peroxidase-labeled polymer) for 30 min at RT. The standard peroxidase enzyme substrate DAB (DakoCytomation) was applied as the chromogen. Slides were dehydrated in graded alcohol, cleared in xylene, and mounted in entellan (Sigma-Aldrich) (Figure 3).
Combination of Immunofluorescence and Histochemistry in Same Tissue Section
Multilabel immunofluorescence detection is an invaluable tool for both research and diagnostic endeavors, dramatically advancing the study of cell- and tissue-specific expression of many proteins (antigens). In the tissue section, however, photographed immunofluorescence is difficult to interpret with respect to the morphological relationships among various cells and their environments. Here, we are the first to report a technique for presenting both immunofluorescence and histochemistry on the same section of MMA-embedded human coronary arterial stent tissue. Briefly, the deplasticized TC sections were incubated in 3% skim milk for 1 hr at RT, followed by incubation with mixed primary antibodies at 4C overnight. The following mixed primary antibodies were employed: rabbit anti-PCNA and mouse anti-SMA (DAKO, 1:100), rabbit anti-PCNA and mouse anti-macrophage (CD68; DAKO, 1:100). Following triplicate washes in PBS, the sections were incubated with mixed Texas red anti-mouse IgG (1:100, Vector Laboratories) and fluorescent anti-rabbit IgG (1:100, Vector Laboratories) secondary antibodies for 30 min at RT. Three washes with PBS were again performed before mounting in 50% glycerol in PBS. Photomicrographs were obtained using a fluorescence microscope (Olympus). The same slide was then washed with PBS and subjected to Masson's trichrome and H and E staining, and repeat photomicrographs were obtained (Figure 4).
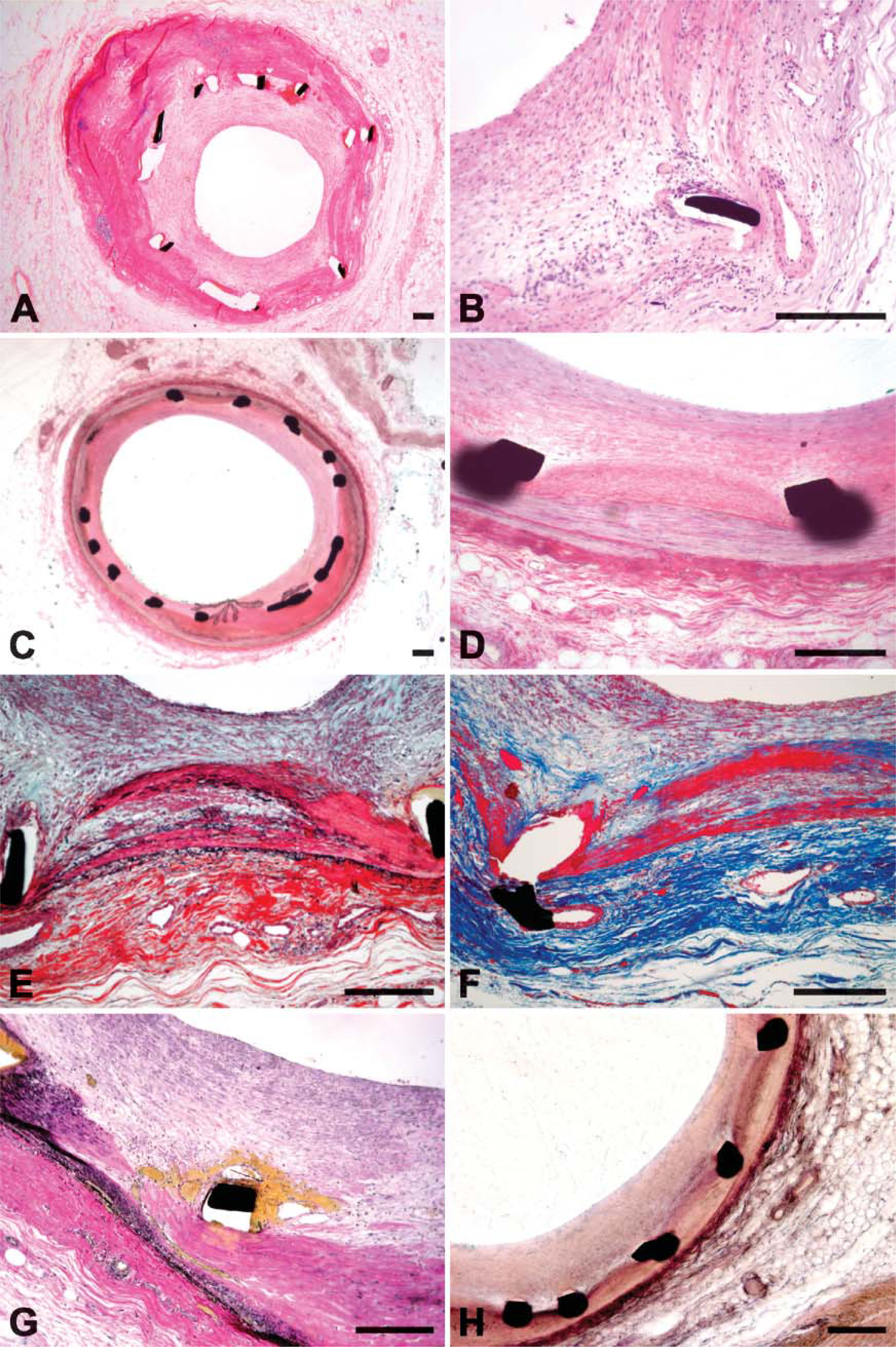
Histochemical staining. (
Immunohistochemical staining of SG sections was performed without prior deplasticization. SMA immunolabeling was done with overnight incubation of the primary antibody (1:100) at 4C. Following two washes in 0.1% Tween 20-PBS, alkaline phosphatase-conjugated (EnVision, DakoCytomation) anti-mouse secondary antibody was applied for 30 min at RT, and the interaction was visualized with nitroblue tetrazolium (Sigma-Aldrich) as the chromogen. Sections were mounted with Glycergel without prior dehydration through graded alcohol.
Transmission Electron Microscopy
Two 10% NBF-fixed human coronary artery specimens were processed in the outlined MMA resin mixture for both routine TEM and immuno-electron microscopy (IEM) analyses. Postfixation with 1% osmium tetroxide was omitted for the specimens designated for IEM. Thin tissue sections (60-90 nm) were cut on a Reichert Ultracut E ultramicrotome using a diamond knife (Diatome; Hatfield, PA) and mounted on formvar-coated nickel grids (Marivac). Tissue sections for routine TEM were counterstained with 3% aqueous uranyl acetate (Marivac) for 30 min and Reynolds' lead citrate (Marivac) for 5 min. In contrast, tissue sections for IEM analysis were placed onto drops of 1% BSA in PBS for 30 min at RT, and, without rinsing, the sections were placed onto drops of the primary antibody, polyclonal anti-SMA (DakoCytomation; 1:100, diluted in 0.1% BSA-PBS) before being incubated overnight at 4C. After rinsing in PBS, the sections were placed onto drops of a secondary 15 nm gold-conjugated goat antirabbit IgG (EY Laboratories, San Mateo, CA; 1:100) for 1 hr at RT. Gentle rinsing in distilled water was followed with counterstaining in aqueous 3% uranyl acetate and Reynolds' lead citrate. The stained sections were then examined by TEM (JEOL 1230; JEOL, Peabody, MA) equipped with software from Advanced Microscopy Techniques (Danvers, MA).
Results
Sectioning
Thin tissue sections were consistently produced with a TC knife. The main concern with regard to the TC knife method was the potential for blade damage and resultant scoring of tissue sections. In contrast, production of SG sections proved to be more laborious and technically challenging. Because the SG sections are considerably thicker, specimen depletion was also a concern. Typically, only one or two SG sections were produced per block.
Morphology
Although both sectioning methods yielded sections of acceptable morphological integrity, fine histological detail was more readily observed in the thinner TC sections (Figure 2). Section thickness proved variable with the SG method because of uneven section surfaces, as compared with consistently uniform thickness with the TC method. Sectioning artifacts such as scoring and folding were not present in SG sections but occurred with a noticeable frequency in TC sections. Stent struts consistently remained in situ for SG sections but were often displaced or lost within TC sections (Figure 2).
Histochemical Staining
The range of histochemical staining methods applicable to SG sections was limited because of the incompatibility of the deplasticization solvent and the glue utilized to fix the tissue sections to the slides. Staining was therefore accomplished without deplasticization and limited to H and E and Verhoeff's elastin stains (Figure 2). In contrast, TC sections could be deplasticized and accommodated a broader range of histochemical stains such as H and E, Movat, Azan, Masson, Orcein/Resorcin Fuchsin, and Verhoeff's Van Gieson (Figure 2).
Immunohistochemical Labeling
The range of immunocytochemical stains applicable to SG sections was limited to SMA for reasons previously described. TC sections were deplasticized and stained for specific markers such as SMA, vWF, and HAM56 (Figure 3). Moreover, we devised a novel method of combining fluorescent immunolabeling and histochemical staining on the same slide. For example, we immu-nolabeled specimens with a fluorescently tagged antibody to PCNA, obtained a fluorescent photomicrograph, and then performed a Masson's trichrome stain on the same section before repeating light photomicrographs of the same tissue section (Figure 4). This combined approach allowed us to specifically localize proliferating cells in relation to various components of the extracellular matrix of these lesions.
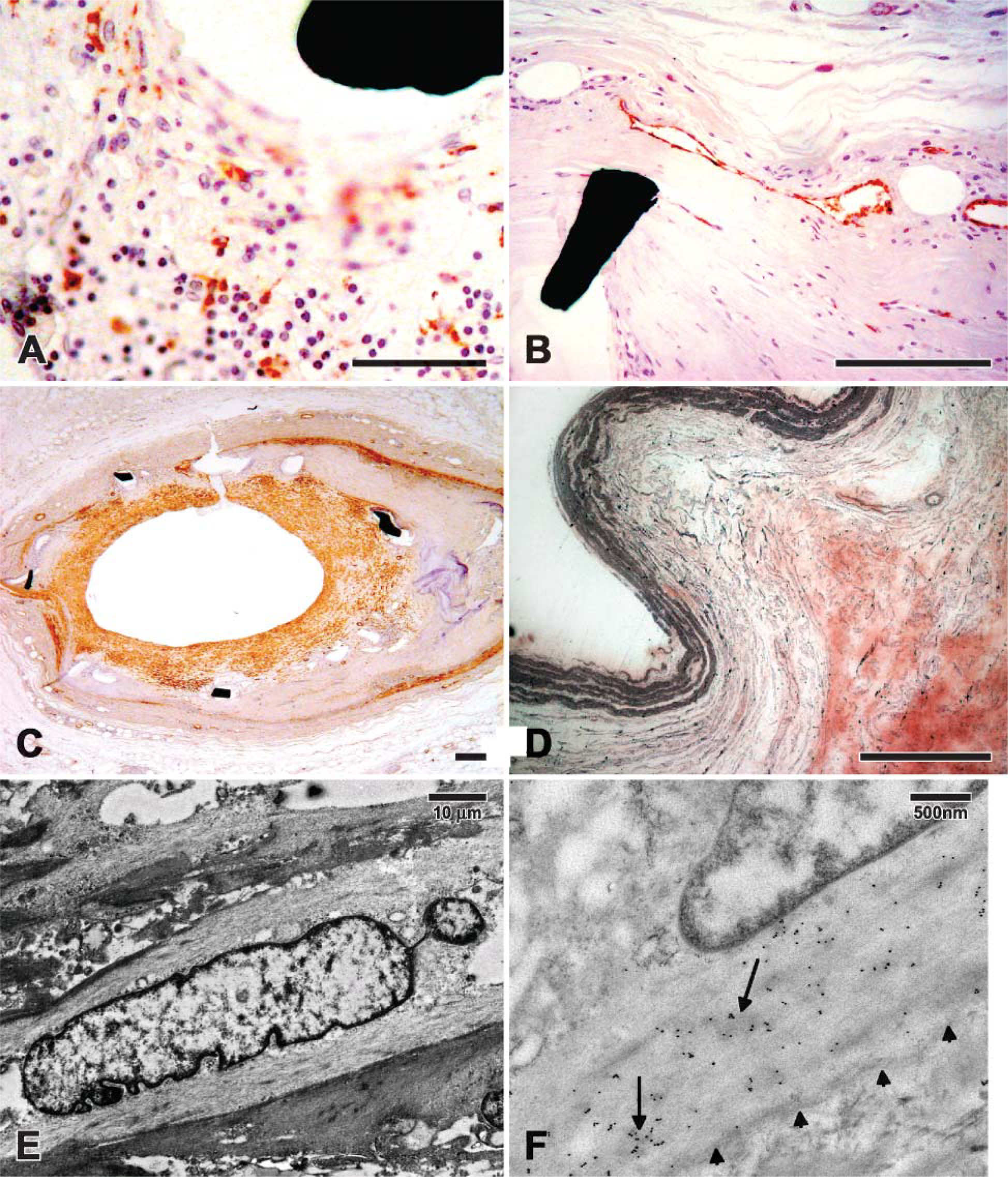
Immunohistochemistry was performed by applying various antibodies onto deplasticized TC sections and undeplasticized SG sections. (
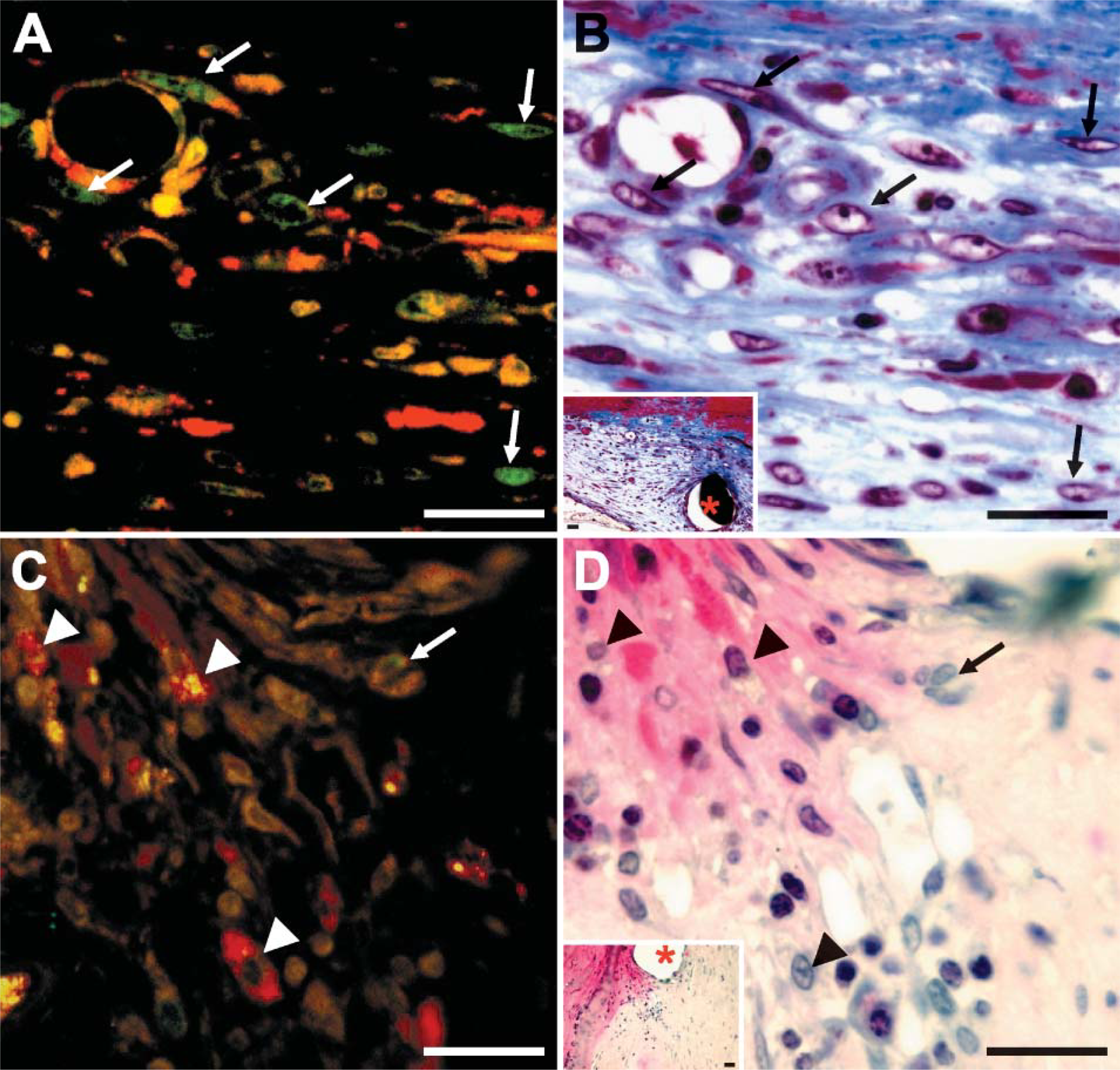
Combination of immunofluorescence and histochemistry. The double immunofluorescence was performed with rabbit antiproliferating cell nuclear antigen (PCNA) (green) and mouse anti-SMA (red) antibodies (
TEM and Gold Labeling
The MMA resin proved conducive for the performance of both routine TEM and immunogold analyses. For example, 15 nm gold-labeled anti-SMA antibody was successfully used to immunolabel smooth muscle cells (Figure 3).
Discussion
With the escalating global use of stents to treat patients with obstructive coronary artery disease, the problem of ISR is of paramount importance. Although drug-coated stents may reduce the frequency of ISR, the overall incidence of ISR is still estimated to be ∼9%, and cost may prevent the widespread use of these bio-prostheses (Waksman 2004). Currently, there is much interest in better understanding the vascular response to stent implantation, as well as identifying bona fide therapeutic targets to prevent ISR. Unfortunately, not all laboratories are adequately equipped or have sufficient experience to engage in the intricacies of sectioning, staining, and immunolabeling vascular tissues that contain metallic bioprostheses. In this manuscript, we review methodologies and outcomes of two sectioning methods for resin-embedded arteries that contain metallic stents.
The SG method consistently produces intact tissue sections without displacement of stent struts or scoring or folding of tissue. Moreover, it is superior to the TC method when stents made of more durable alloys, such as nitinol, are encountered or when longitudinal sections of stented arteries are required. Nonetheless, there are important limitations to note about the SG method. First, there is significant tissue depletion (kerf loss) with the SG technique, because only one or two sections can be produced per block. Second, because the minimum thickness of sections produced with SG is 8-10 μm, the quality of morphological studies may be inferior to that of the TC approach. Third, to produce multiple SG sections is laborious and certainly more technically challenging than producing TC sections. Fourth, the initial equipment setup for the SG method is considerably more expensive than that necessary for performing the TC method (refer to Supplemental Table 2).
The TC method consistently yields thin serial sections that are suitable for both histochemical and immunohistochemical staining of deplasticized sections (Figure 2). Although occasional scoring and folding, together with stent strut displacement, do occur in TC sectioning, in our experience, this method clearly provides superior morphological and immunolabeling detail at the tissue-stent interface. For example, using this technique, we have carefully examined stented vessels from animal arteries and determined the efficacy of novel anti-inflammatory therapies that reduce stent neointima formation (Chen et al. 2004; Ma and O'Brien 2004). This method has also provided accurate information concerning the status of human coronary stents for both forensic and hospital autopsies performed over the past 8 years at The Ottawa Hospital. Of note, others have used the SG technique utilizing Technovit 4000 glue (Heraeus-Kulzer; Friedrichsdorf, Germany) to adhere tissue sections onto slides and have obtained seemingly very acceptable immunolabeling results (Brasen et al. 2001; Rammelt et al. 2004) This has so far proven unsuccessful in our hands.
In summary, the described MMA embedding protocol adequately accommodates both the TC and SG methods of sectioning. Although either method of sectioning can be effectively utilized to evaluate the histopathological features of resin-embedded vascular stents, the TC method allows thinner sections to be used and a wider range of histochemical staining and immunolabeling procedures to be readily performed. Future studies of the vascular response to a variety of current and newer stent materials (e.g., magnesium biodegradable stent) will provide important clues to the pathogenesis of ISR and may allow the formulation of a durable scaffolding device for the growing number of patients that require percutaneous revascularization.
Footnotes
Acknowledgements
These methods were initially developed during P.R.'s former affiliation with The Ottawa Hospital Department of Pathology and Laboratory Medicine in collaboration with M.K.B. and under the supervision of J.P.V. The project was supported by an operating grant and research chair (UOP 36383, URC 57093) held by E.R.O. and jointly funded by the Canadian Institutes of Health Research and Medtronic. The Heart and Stroke Foundation of Ontario program grant no. 5275 funded some of the laboratory equipment used in this study. The method of combination of immunofluorescence and histochemistry was developed by Y-X.C., postdoctoral fellow, under the supervision of E.R.O.
The authors are indebted to Jordana Laporte for administrative support and preparation of this manuscript.