
Abstract
We demonstrate that high-frequency and high-intensity ultrasound (US) can be applied to both tissue fixation and tissue processing to complete the conventional overnight formalin-fixation and paraffin-embedding (FFPE) procedures within 1 hr. US-facilitated FFPE retains superior tissue morphology and long-term room temperature storage stability than conventional FFPE. There is less alteration of protein antigenicity after US-FFPE preservation so that rapid immunohistochemical reactions occur with higher sensitivity and intensity, reducing the need for antigen retrieval pretreatment. US-FFPE tissues present storage stability so that room temperature storage up to 7 years does not significantly affect tissue morphology, protein antigenic properties, RNA distribution, localization, and quantitation. In addition, during fixation, tissue displays physical changes that can be monitored and reflected as changes in transmission US signals. As far as we know, this is the first effort to monitor tissue physical changes during fixation. Further study of this phenomenon may provide a method to control and to monitor the level of fixation for quality controls. The mechanism of less alteration of protein antigenicity by US-FFPE was discussed. (J Histochem Cytochem 54:503-513, 2006)
Keywords
C
Cross-linking causes proteins and nucleic acids to be less effectively extracted from FFPE tissues. Though methods are available to extract macromolecules from FFPE tissues (Chu et al. 2005b), traditionally, fresh and flash-frozen tissues are used for molecular testing (Emmert-Buck et al. 2000). However, frozen sections have relatively poor morphology inadequate for definitive pathologic diagnosis. Obtaining fresh/frozen tissues requires the cooperation of at least the OR nurse, surgeon, cell culture and pathology grossing technicians, clinician, and patient. Several physicians and technicians must work coordinately for an hour or more to ensure that a tissue specimen is handled and processed properly, compared with the two to three people needed to process several hundred FFPE tissue specimens daily. In short, the high cost, labor intensiveness, and low efficiency of handling and storage all limit the use of frozen tissues in current hospital practices.
Clearly, a rapid tissue preservation technique that provides superior tissue morphology and improved macromolecular integrity with easy handling and long-term storage stability would benefit medical research and practices. Microwave-enhanced tissue fixation and processing represents one such improved breakthrough. As another physical force, ultrasound (US) has been widely used in diverse medical fields for over 50 years. Low-frequency (20-100 kHz) and high-intensity US is used for medical equipment cleaning, tissue and cell homogenization, and stone ablation. High-frequency (>0.5 MHz) and low-intensity (∼3W/cm2) US is employed for physical therapy and medical imaging. US has been successfully applied to transdermal drug delivery (Mitragotri et al. 1995), transdermal monitoring of glucose (Kost et al. 2000), and gene transfer (Danialou et al. 2002). We have previously demonstrated that high-frequency and high-intensity US can greatly enhance the rate of tissue formalin fixation to achieve superior tissue morphology and improved macromolecule preservation (Chu et al. 2005a). Here we further demonstrated that US could be applied to formalin fixation as well as to processing procedures to achieve completion of the overall FFPE procedures within an hour. Details of the US apparatus were described, US-induced temperature increase in tissue was monitored, tissue physical changes (such as hardening) during the fixation process with and without US implementation were compared, the effect of US intensity and frequency on tissue preservation was addressed, and the preservation of tissue morphology, protein antigenicity, and RNA in US-FFPE tissues after 7 years storage was evaluated.
Materials and Methods
Ultrasonic Apparatus for Tissue Fixation and Processing
A prototypic US apparatus (Bio-Quick Inc.; Silver Spring, MD) to facilitate both fixation and processing is shown in Figure 1A. Unfocused plane transducers (EDO Corporation; Salt Lake City, UT) were used to provide large acoustic fields comparable to the size of a typical tissue sample (∼25 mm in diameter). A sliced tissue sample placed in a conventional tissue cassette (Figure 1A) was positioned 3-5 cm away from transducers in 10% neutral-buffered formalin (NBF) and immediately sonicated by 1.6-1.7 MHz US for 3-15 min at an intensity of 3-20 W/cm2 acoustical power. After fixation, the tissue was immersed for 5 min each in graded ethanol and 15 min each in xylene and paraffin (preheated and kept at 58C in a water bath). US was continuously applied to the solution in each step.
Monitoring System for Temperature and Transmission US Determination
To study the effect of US on tissue fixation, tissue sample fixed to a stand was placed in a small chamber filled with fixative or processing solutions (Figures 1B and 1C). Tissue and solution temperatures were measured by a dual-channel thermometer (Fisher Scientific; Pittsburgh, PA) with needle probes for surface and internal temperature detection. At least three US transducers were used: a functional transducer for tissue fixation and processing and a pulsed signal transducer paired with a receiver transducer (King et al. 2003). Constructed in house, the signal transducer (1.385 MHz, 50 mm in diameter) was powered with a 60-dB radiofrequency power amplifier (A-500; ENI, Rochester, NY) and connected to a signal synthesizer/function generator (3325A; Hewlett-Packard, Palo Alto, CA). The receptor transducer was 1.65 MHz, 20 mm in diameter (#50-1045; APC Products, Mackeyville, PA). Pulsed US signal generated by the generator transducer was received by the receiver, captured on an oscilloscope (Tektronix, TDS 2014; Beaverton, OR), and registered by an interfacing PC for processing. Transmission signal was the US from the generator that passed through the tissue and was received by the receiver placed on the other side of the tissue. To register the transmission signals, the fixation US was turned off for 7-10 sec while the pulsed detection signal from the generator was activated and captured by the receptor. Unless otherwise stated, the acoustic power for fixation was measured by a UW-3 wattmeter (Bio-Tek; Winooski, VT).
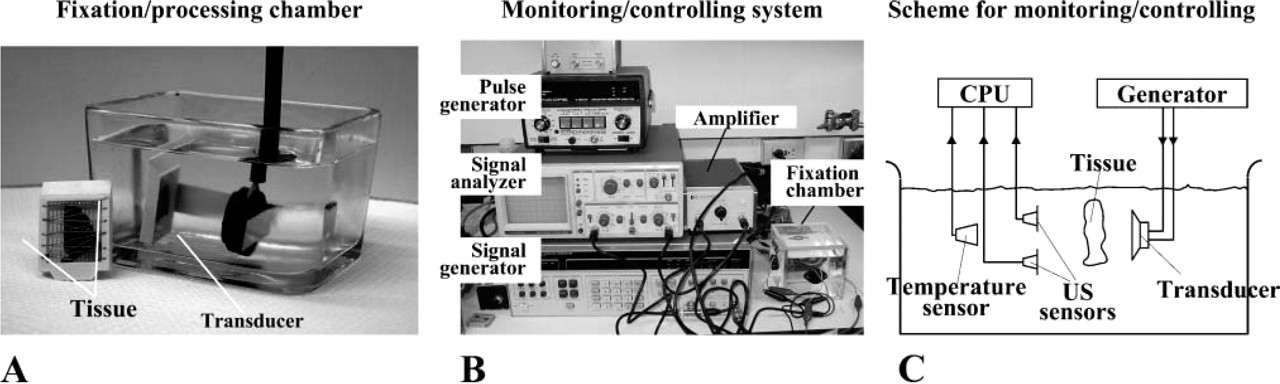
Ultrasound (US)-facilitated formalin fixation and paraffin embedding (FFPE). (
Routine FFPE and US-facilitated FFPE Procedures
A total of 86 (34 biopsy and 52 autopsy) tissues were fixed and processed with and without US. They were from 14 tissue types including arteries, brain, breast, colon, heart, kidney, liver, lung, lymph node, pancreas, prostate, skin, spleen, and uterus. All tissue specimens were obtained from Walter Reed Army Medical Center. Biopsy specimens were obtained 1-2 hr after excision, and autopsy specimens were obtained 15-32 hr postmortem. Samples were cut into 3- to 5-mm slices and stored either at −80C or processed by standard FFPE or US-facilitated FFPE procedures. Reagents and the working temperature (assuming the room temperature unless specifically stated) used for the two procedures were kept the same. Tissues preserved by standard FFPE were fixed in 10% NBF for 18-24 hr at room temperature, followed by 16-18 hr of automated processing (Tissue Tek; Miles Scientific, Naperville, IL) consisting of dehydration in graded ethanol, clearing in xylene, and infiltration in paraffin. Although apparatus for multi-sample US-FFPE was available and under further improvement, due to the availability of specimens and for better control of conditions, all tissues were preserved by the two methods, one at a time. Tissues preserved by US-FFPE were fixed in 10% NBF for 5 to 15 min, followed by dehydration, clearing, and paraffin infiltration under US for 10-15 min. US was continuously applied to the solution in each step. Fixation and processing were completed within 1 hr. For ethanol preservation, 10% formalin was replaced with 75% ethanol with all other steps unchanged. Blocks of fixed and processed tissues were either stored at room temperature for later use or cut into 4- to 5-μm sections and mounted onto glass slides. Tissue sections were deparaffinized in xylene and rehydrated in downgraded ethanol. Some were stained with hematoxylin and eosin (H and E), and others underwent immunohistochemical (IHC) procedures.
Immunohistochemistry
Rehydrolyzed tissue sections were pretreated with or without antigen retrieval by microwave (Shi et al. 1991) or pepsin digestion, as required for each antibody. The sections were then blocked with 10% horse and goat serum for 15 min to reduce nonspecific background. Primary antibodies (DAKO; Carpinteria, CA) were added onto slides at the appropriate concentrations (CD3 1:500, CD5 1:100, cytokeratin AE1/AE3 1:400) for specified time periods. Slides were washed three times in PBS and applied with biotinylated secondary antibodies for 5-30 min at concentrations specified by the providers. ABC kit (Vector Laboratories; Burlingame, CA) and chromagens diaminobenzidine (DAB) or aminoethyl carbazole (AEC) were used to develop color as previously described (Chu et al. 1999). Hematoxylin was used as the final counterstain.
mRNA In Situ Hybridization
Tissue sections were deparaffinized as described for IHC and subjected to enzymatic or MW-AR pretreatment prior to hybridization. ISH performed 7 years ago on the newly fixed/processed tissue sections was by manual protocols. Fluorescein isothiocyanate (FITC)-tagged synthetic oligonucleotide probes specific for mRNA of kappa and lambda immunoglobulin were from BioGenex (San Ramon, CA). FITC-labeled probe applied to the section was secured by a coverslip and denatured at 100C for 5 min in a steamer. Slide sections were allowed to cool and hybridize with the probe at room temperature for 1 hr. Sections were given two 3-min washes in 2X SSC, incubated for 30 min with monoclonal mouse anti-FITC, and followed by two 3-min washes in PBS. The slide section was incubated with biotinylated secondary antibody for 30 min at room temperature, followed by two 3-min washes in PBS, and then incubated with streptavidin-biotinylated peroxidase for 30 min. After PBS washing, the slide was treated with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium reagents for color development. Appropriate positive and negative controls provided by Bio-Genex were used with each reaction. ISH performed on 7-year stored tissue sections used INFORM kappa and lambda probes on a BenchMark Staining System (Ventana; Tucson, AZ) following the manufacturer's instructions.
Nucleic Acid and Protein Extraction from Tissue Sections
Four- to eight-5-μm sections per block were cut. Sections were placed in a 1.5-ml microcentrifuge tube and 800 μl of HemoDe (Fisher Scientific) was added to dissolve the paraffin. After centrifugation, the supernatant was removed, the pellet was washed three times with 400 μl absolute ethanol, and then resuspended in 0.5 ml of buffer containing 20 mM Tris (pH 7.4), 20 mM EDTA, 1% SDS, and 1 mg/ml proteinase K (Sigma; St Louis, MO). After incubating 6 hr at 55C to solubilize cellular proteins, the tissue lysate was treated twice with TRIzol LS (Invitrogen; Carlsbad, CA) to extract DNA and proteins, according to the manufacturer's instructions. RNA was extracted with the Optimum FFPE RNA isolation kit (Ambion Diagnostics; Austin, TX) and DNase I treated. Extracted DNA or RNA was resuspended in 25 μl of molecular-grade water or appropriate buffer until PCR or RT-PCR reaction.
SDS-PAGE and Western Blot
Proteins extracted from paraffin blocks by TRIzol LS method (Invitrogen) as described above were first denatured at 70C for 10 min in a non-reducing loading buffer from NuPAGE kit (Invitrogen) and then separated by electrophoresis with a 4% to 15% gradient PAGE gel. For Western blot, the protein was transferred onto a polyvinylidene fluoride membrane (PVDF; BioRad, Hercules, CA). After washing in TBS and blocking overnight with 3% dry milk-TBS buffer, the membrane was incubated in TBS-diluted anti-cytokeratin monoclonal antibody (1:5000) for 1 hr, followed by 1 hr of incubation with 1:50,000 diluted secondary antibody, alkaline phosphataseconjugated goat anti-mouse IgG. The signal was developed using the AP Color Development System (Promega; Madison, WI).
Polymerase Chain Reaction (PCR)
RNA extracted from FFPE and US-FFPE tissues was reverse transcribed into cDNA with SuperScript III First-Strand Synthesis System (Invitrogen), following manufacturer's instructions. Primers for β-actin were synthesized as two forward primers: AF1 (5′-GAAGGATTCCTATGTGGGCG-3′) and AF2 (5′-GCCAGCTCACCAT GGATGAT-3′) and four reverse primers: AR1 (5′-GCGAGAAGATGA CCCAGATC-3′), AR2 (5′-CGGGAAATCGTGCGTGACAT-3′), AR3 (5′-TGCAGAAGGAGATCACTGCC-3′), and AR4 (5′-A CAAGATGAGATTGGCATGGC-3′). PCR reaction using AF1 paired with one of the reverse primers generated amplicons of 220 bp, 489 bp, 811 bp, and 1062 bp, respectively. PCR reaction using AF2 and AR2 generated amplicons of 647 bp. PCR reaction was run for 30 to 40 cycles with 15 pmol primers, 1/10 total volume of DNA template, and 1 U Platinum
Real-time PCR
Real-time PCR was performed in a Roche LightCycler and the data analyzed by the accompanying software (version 1.1). The double-strand DNA probe SYBR Green I (Molecular Probes; Eugene, OR) was used to quantitate amplicons. All assays were carried out in 15 μl of PCR buffer (50 mM Tris, pH 8.3, 25 μg/ml BSA), 0.2 mM dNTP, 5 mM MgCl2, 1 U Platinum
Results
Technological Aspects of US Apparatus
The high-frequency, high-intensity US apparatus used in this study is shown in Figure 1 [any generator of high-frequency, high-intensity US, as specified here and in previous publications (Chu et al. 2005a), could be adapted to achieve similar results]. The apparatus generated US with a frequency of 1.6-1.7 MHz, and the intensity was adjustable in the range of 3 to 20 W/cm2 acoustical power. Sonication time was in inverse proportion to the intensity of the US and the delicacy of the tissue being processed. Tissue fixation in formalin or 75% ethanol could be finished in 5-15 min, depending on US intensity. After fixation, the tissue was immersed 5 min each in graded alcohol and 15 min in xylene at room temperature and then in paraffin at 58C for 15 min, with US at 10-15 W/cm2 acoustic power. Total fixation and processing time was ∼1 hr.
Limited Temperature Increase during US Implementation
A thermometer with dual needle probes registered no measurable difference between solution temperature and tissue temperature during US implementation. There was a slow increase in tissue/solution temperature along with US application, as shown in Figure 2A. Typically, tissues submerged in 500 ml of solution showed a temperature increase of <12C after 40 min of sonication. In general, the temperature increase gradually slowed down after solution temperature reached 32C, possibly due to normal environmental heat loss. Because no single step of fixation and processing exceeded 15 min, the temperature increase in any given tissue was <10C. Continued fixation and processing for multiple tissues would require a temperature regulation system.
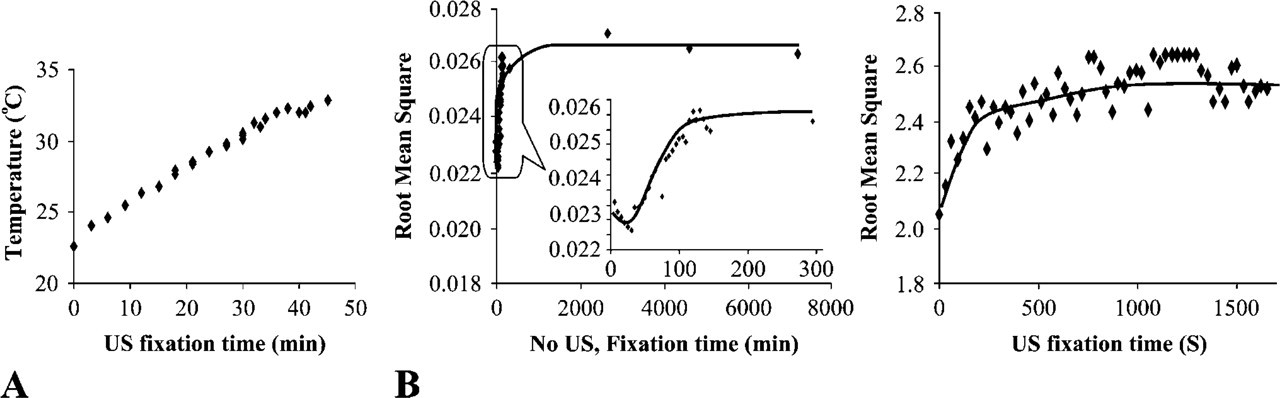
Physical properties of tissues during fixation. (
A 100-Fold Increase in Tissue Physical Change during Fixation with US
In formalin-fixed tissues, gross physical changes such as hardening, shrinkage, and color change took place as a result of water loss and tissue fixation. Other physical properties of the tissue, such as its density and its US conductivity, were also changed and could affect the transmission or backscatter (reflection) US signal accordingly. We registered the transmission US signals during tissue fixation process and presented the signals vs fixation time in Figure 2B. US receivers (or US sensors in Figure 1C) were assigned to monitor transmission US signal and the root mean square (RMS) of the registered waveform was used to characterize the signal at each point. RMS is defined as the square root of the mean of the squares of the voltage values of a waveform. RMS is not an instantaneous measurement but a statistic measure of the magnitude of a varying power output. It is used to calculate the mean power dissipation delivered into a specific load. For conventional fixation, low intensity (∼1/100 of that used for US fixation) of pulsed US was applied for 30 sec in every 5 min in the first 150 min of fixation to get the transmission signal. A slice (3- to 5-mm thick) of fresh human kidney was fixed in formalin with and without US implementation. In both cases, the RMS increased gradually at the very beginning of the fixation (Figure 2B). The low RMS at initial of fixation reflects loss of US energy due to tissue absorption and reflection, which was related to tissue physical properties (water contents, hardness, shrinkage). Within ∼7 min (or 400 sec) for US fixation and 1000 min (or 16 hr) for conventional fixation, the increase of RMS reached a plateau. The time to reach plateau was well correlated with the time to get good tissue fixation as indicated by good morphological preservation. We believed that as the RMS of transmission signal became constant, there were no substantial physical changes in tissues, indicating the completion of tissue fixation. The profile of the RMS vs time was affected by the intensity and frequency of the US and the type of tissue being fixed.
US-FFPE Preserves Good Tissue Morphology with Storage Stability
So far, 86 tissues of 14 tissue types were subjected to H and E staining and one to three IHC staining using various antibodies, as represented in Figure 3. Based on over 200 pairs parallel comparison, we concluded that compared with FFPE tissues, US-FFPE tissues constantly preserved similar superior morphology as evaluated on H and E staining, and provided improved protein antigenicity as evaluated on IHC staining. Under light microscopy, there were essentially no visible morphological differences in H and E staining between the FFPE and US-FFPE tissues in terms of evenness of stain, color balance, and visual sharpness of cellular membrane, subcellular organelles, and nuclei (Figures 3A and 3B). Improved antigen preservation resulted in increased IHC reaction and reduced the concentration of primary antibodies. The time for IHC reaction against CD3 was shortened from 1-2 hr to 10 min on US-FFPE preserved lung (brown DAB staining in Figure 3D). In contrast, under the same IHC conditions, no brown signal was detected in the FFPE tissue (Figure 3C). A 400X dilution of primary antibody against cytokeratin yielded greater staining intensity in US-FFPE tissue (brown staining in Figure 3J) than in FFPE tissues (Figure 3I), even after 7 years of storage at room temperature (Figure 3K vs Figure 3L). As another result of improved antigen preservation, antigen retrieval procedures were not necessary for CD5 (a membrane antigen) and p63 (a nuclear antigen of the p53 gene family) on US-FFPE tissue (brown in Figure 3F, red-purple AEC in Figure 3H), whereas routine FFPE tissue without antigen retrieval displayed no such signals (Figures 3E and 3G).
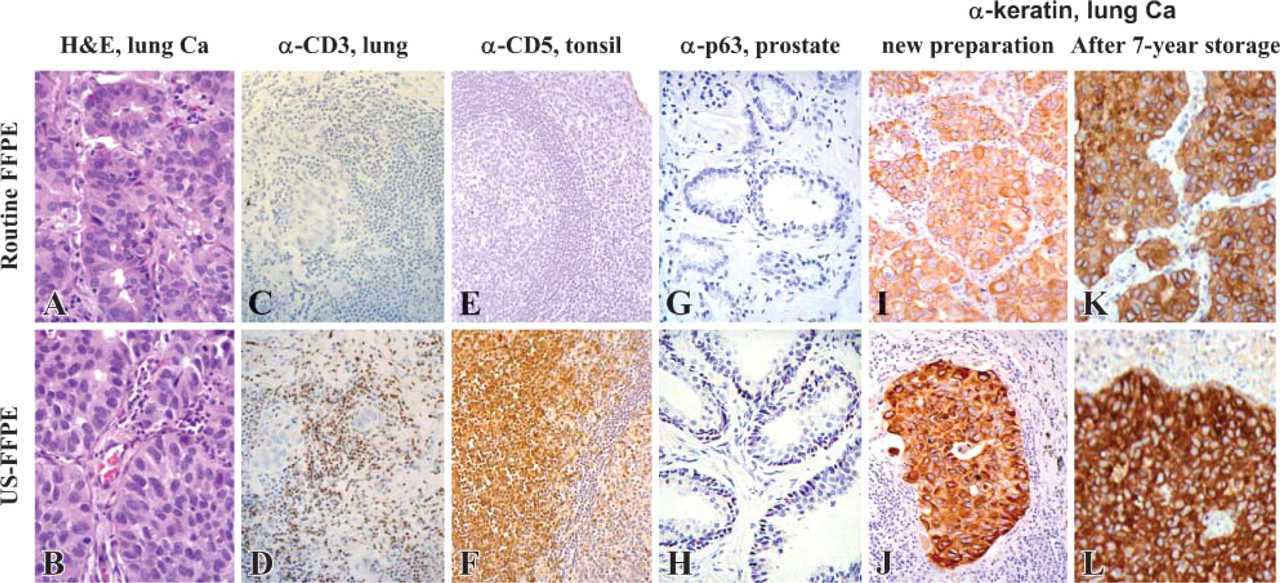
Comparison of general histology and IHC of tissues preserved by 36-48 hr of routine FFPE and 1 hr of US-FFPE. (
Under-fixation Does Not Count for Better Protein Preservation
Someone may question that better preservation of antigen properties and more extractable proteins in US-FFPE than that in FFPE tissues were simply because US-FFPE tissue was underfixed due to shortened fixation time. The following experiment refuted this reasoning. For most tissues, sonicating formalin fixative for 5-10 min at 8-10 W/cm2 acoustic power is sufficient to completely fix the tissue. Lower intensity of US resulted in longer fixation time. For kidney tissue, 30 min of US sonication at 5 W/cm2 acoustic power generated brighter H and E staining than routine FFPE (Figure 4A). When kidney tissue was sonicated for 30 min at 1.5 W/cm2, the peripheral portion of the tissue displayed good H and E staining, but the center was underfixed and showed faint staining and red blood cell lysis (Figure 4A). The amount of total extractable proteins and specific protein such as keratin extracted from this underfixed tissue was lower than that in the well-fixed US-FFPE tissues (Figure 4B). Multiple bands were detected in all three extracts because anti-keratin AE1/AE3 recognizes 56.5-, 50-, 48-, and 40-kDa keratins of the acidic subfamily plus all members of the basic subfamily.
Preservation of Tissue Nucleic Acid and Its Long-term Storage Stability
Without fixation, cellular RNA in tissue specimens degrades rapidly due to abundant cellular RNase. Thus, the integrity and the amount of preserved RNA are sensitive indicators of nucleic acid preservation. Synthetic oligonucleotide specific to kappa light chain immunoglobulin mRNA was hybridized to tonsil sections when newly preserved by conventional FFPE and US-FFPE and after 7 years storage. Compared with ISH on the newly preserved tissue sections, long-term storage did not affect mRNA distribution and localization. Whether freshly preserved or under prolonged storage, US-FFPE tissues exhibited uniform staining in both the peripheral and central portions of the tissue and greater density of total mRNA signals. Routine 24-hr formalin-fixed tissue section showed only sporadic mRNA staining signals in plasma cells along the peripheral margin, not in the central portion (Figure 5A). This edge staining has been reported previously (Nuovo and Richart 1989; Harper et al. 1992; Chu et al. 2005a). In contrast, 15-min US-formalin-fixed tonsil demonstrated intensive, specific mRNA staining of plasma cells in both central germinal centers and at the peripheral edge. Under high magnification (Figure 5B), ISH signals were well defined in cytoplasm in plasma cells but not in nuclei.
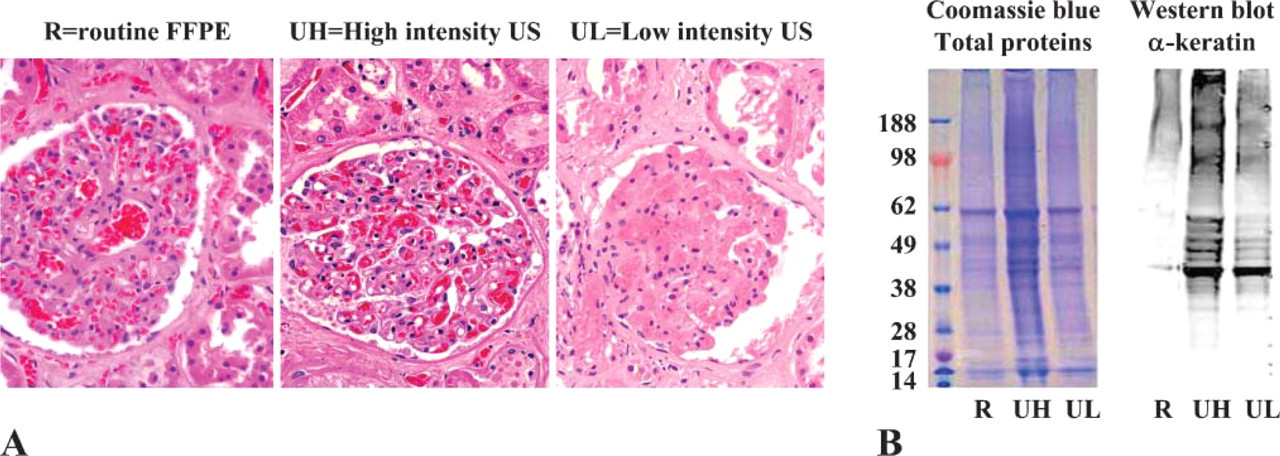
Comparison of tissue morphology and protein extracts of well-preserved and underfixed kidney tissues. (
Quantitative Studies of Extracted DNA and RNA
Applying US to fixation and processing achieved better DNA and RNA preservation. From frozen, routine FFPE, and US-FFPE tissues, DNA was extracted by the TRIzol (Invitrogen) method (which was not optimal for fixed tissues). RNA was extracted by a commercially available RNA isolation kit (Ambion Diagnostics) designated for FFPE tissues. Primer sets for β-actin were chosen to generate amplicons of 220, 489, 647, 811, and 1062 bp. As shown in Figure 6A, the amount of DNA extracted from formalin-fixed tissue was in inverse proportion to the length of the DNA fragments. DNA of 489 bp started to appear after 32 PCR cycles, whereas DNA of 811 bp needed 38-40 cycles. DNA fragments up to 800 bp were readily amplified from both routine FFPE and US-FFPE tissues, whereas a DNA fragment over 1000 bp was amplified only from frozen tissue. In Figure 6B, applying US to formalin fixation or 75% ethanol fixation preserved more DNA than routine fixation without US.
To better compare extracted RNA from tissues fixed and processed by routine FFPE and US-FFPE, real-time RT-PCR was used to determine the Ct value. RNA extracted from various tissues was reverse transcribed into the first-strand cDNA and then underwent realtime PCR. Ct was defined as the PCR cycle number at which the fluorescence (thus the amount of amplicon) was above background. It reflected the relative amount of RNA in the extracts: the higher the Ct value, the lower the cDNA template copy number to begin with. As shown in Figure 7B, the amount of extracted RNA, in proportion to the amount of cDNA, was always in inverse proportion to amplicon length. After 1.5 years storage, the RNA extracted from frozen lymph node tissues had a Ct value two cycles less (or a four-times higher amount of RNA) than that from fixed tissues. US-FFPE tissue had a lower Ct (or higher RNA amount) than the corresponding routine FFPE tissue. The difference between the Ct of FFPE and the Ct of US-FFPE increased with amplicon size: from <1 (or 2 × difference) for 220-bp fragments to >4 (or 16 × difference) for 600-bp fragments. Routine FFPE and US-FFPE lung tissues after 7 years of storage at room temperature generated actin mRNA to similar levels.
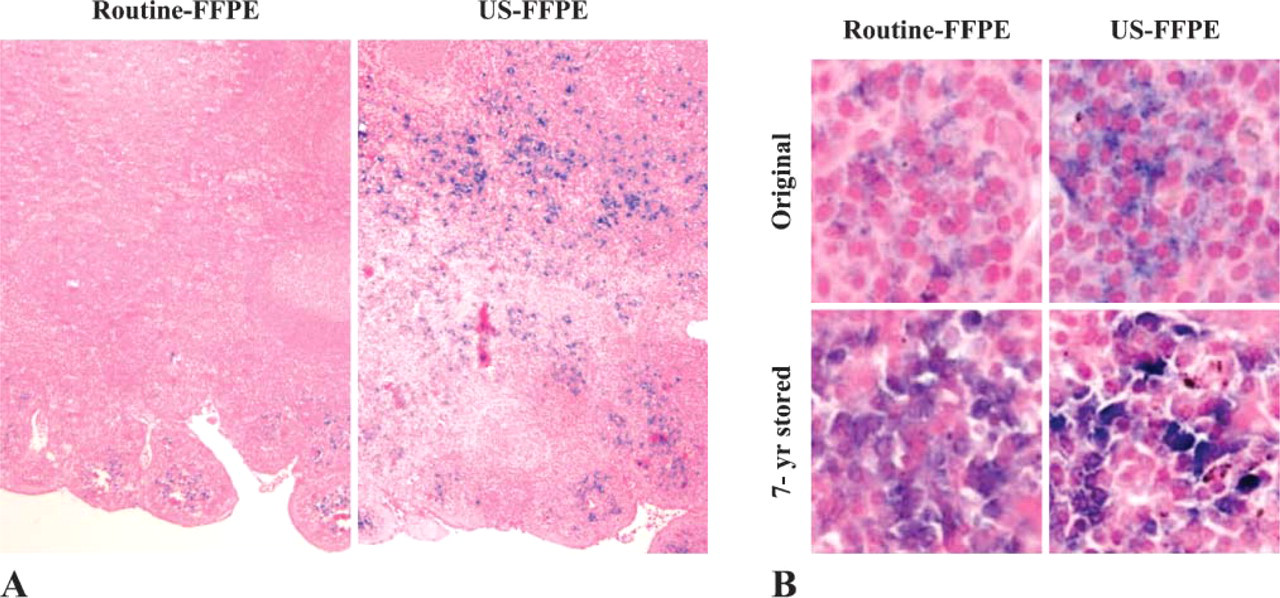
Comparison of ISH signals on routine FFPE and US-FFPE tonsil tissues when newly preserved and after 7-year storage. (
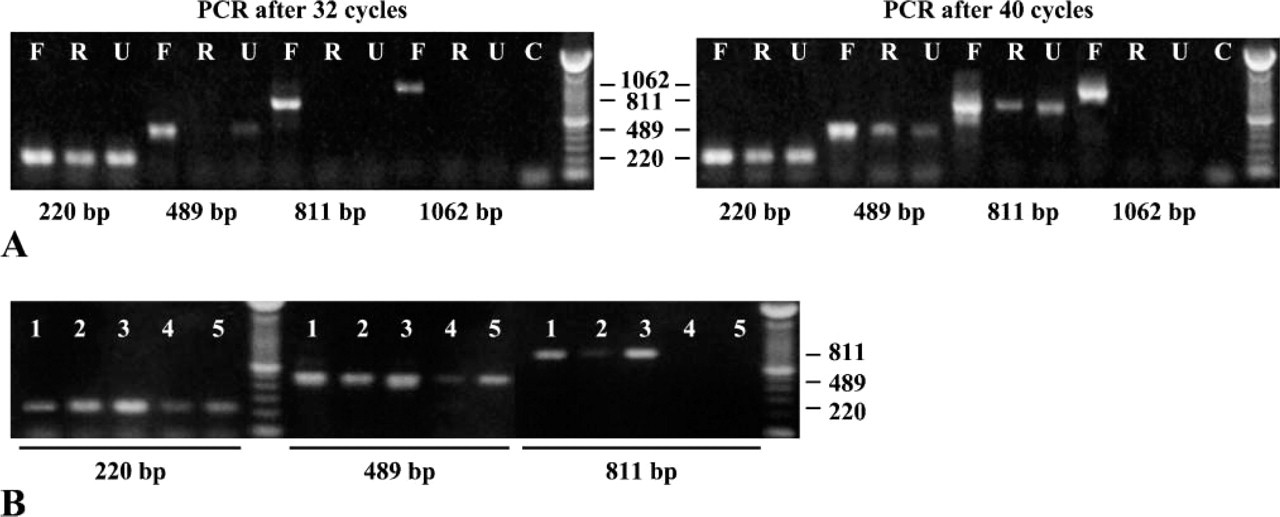
Comparison of PCR products using several primer sets for β-actin DNA, corresponding to amplicons of 220, 489, 811, and 1062 bp, using extracts of lymph nodes preserved by different methods. (
Discussion
We have previously published that applying high-intensity, high-frequency US to formalin for <15 min results in an overall improvement in morphology, antigen, and mRNA preservation (Chu et al. 2005a). Here we demonstrate that US can be applied not only to tissue fixation but also to tissue processing, allowing conventional FFPE to be completed within 1 hr. Consistent with our previous observations, US-facilitated FFPE retains superior and uniform tissue morphology. There is less alteration in protein antigenicity after US-FFPE, so that rapid IHC reactions occur with higher sensitivity and intensity, reducing the need for antigen retrieval pretreatment. Long-term storage did not affect this improved protein antigenic sensitivity or mRNA distribution. Quantitative real-time RT-PCR demonstrates that 10 times more RNA longer than 600 bp is preserved in US-FFPE tissue after 1.5 years of storage than that in routine FFPE tissues. In another case, a similar amount of RNA was extracted from 7-year stored lung tissues preserved by both methods. We do not have the data of extractable RNA from these tissue blocks when they were newly preserved 7 years ago to compare RNA degradation during the 7-year storage. In general, the long-term room temperature stability of US-FFPE tissue blocks and sections is similar to that of routine FFPE tissues. There is no substantial difference in histology, IHC, and extractable macro-molecules between newly prepared and 7-year-old specimens stored at room temperature.
We have demonstrated that, during fixation, tissue displays physical changes that can be reflected by changes in transmission US signals. As far as we know, this is the first effort to monitor tissue physical changes during fixation. Further study of this phenomenon may provide a method to control and to monitor the level of fixation for quality controls, which might be critical for standardized IHC studies (O'Leary 2001; von Wasielewski et al. 2002; Boenisch 2004). The use of transmission US signal to monitor tissue fixation is a new and useful concept. Until now, there is no means to verify and to standardize the fixation level during fixation. Because tissue preservation by routine FFPE or other fixatives is greatly affected by the type and size of samples, it is useful to control the degree of fixation to prevent over- or underfixation. We noticed significant changes in tissue color, size, and hardness upon fixation. These changes in tissue density and water content might affect the speed of sound waves passing through the tissue, which was reflected in transmission (and/or backscatter) US signal from the tissue. We found a promising correlation between transmission US signal and the level of tissue fixation (Figure 2). It is worth mentioning that this monitoring system is applicable to other tissue preservation methods using other fixatives, with or without implemented US for fixation. Further study is underway to confirm and to delineate the observed transmission US signal in correspondence to tissue fixation levels.
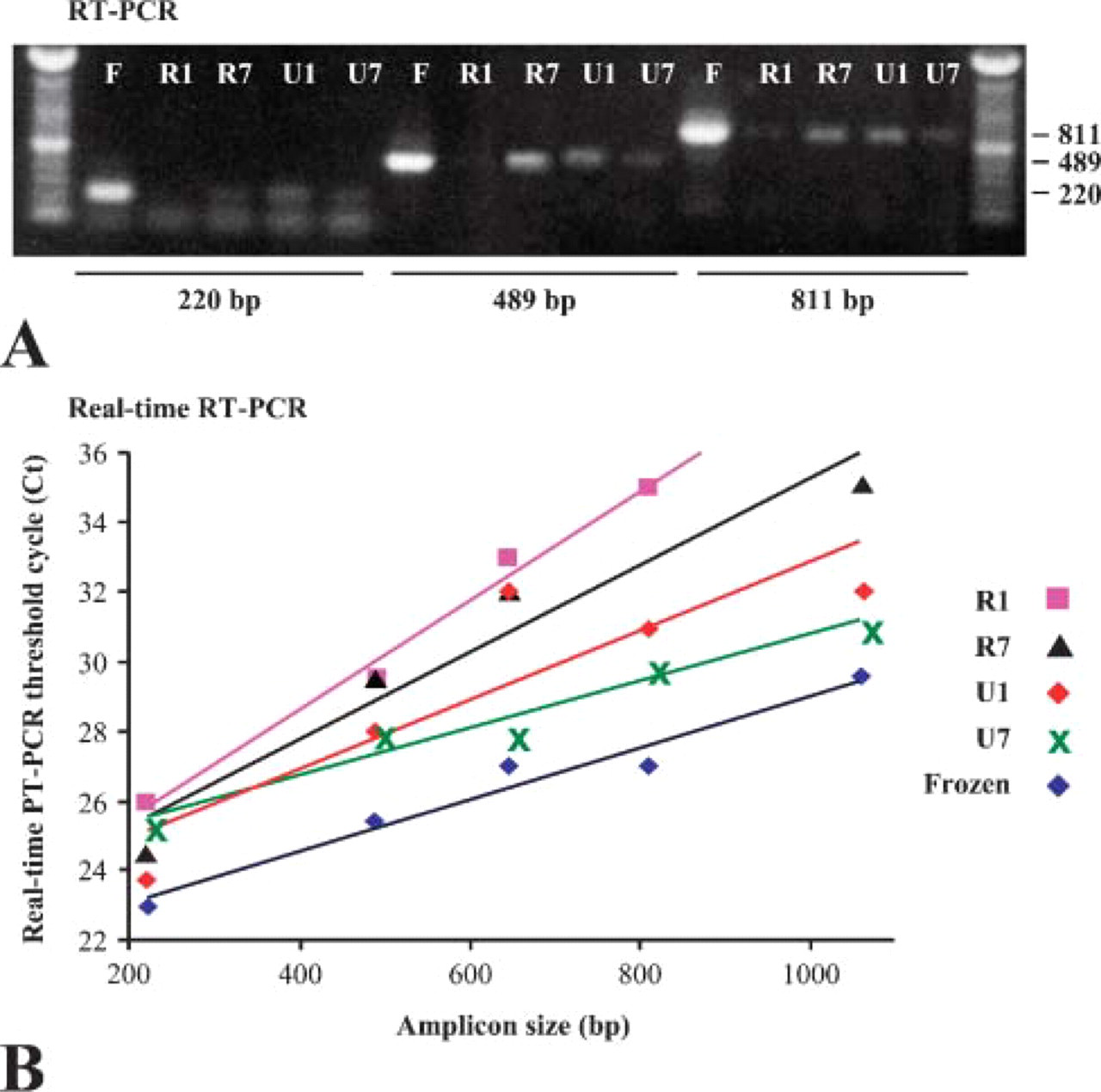
Comparison of RT-PCR products using RNA extracted from tissues preserved by conventional FFPE and US-FFPE methods after long-term storage. (
Compared with routine FFPE tissues, US-FFPE tissues seem to be less modified by formalin, so that protein antigenic properties and protein profiles are less altered, and more proteins and nucleic acids are extracted (Figure 3—Figure 7). It could be argued that US-FFPE tissues were simply underfixed compared with routine FFPE tissues. However, cellular morphologic disintegration was not observed in US-FFPE tissues, even after enzymatic or microwave treatment (data available). When we purposely underfixed a tissue by applying lower intensity US, we observed obvious tissue shrinkage, inferior staining, and lysis of erythrocytes (Figure 4A). Furthermore, this underfixed tissue generated fewer extractable proteins than the well-preserved US-FFPE tissue (Figure 4B). Underfixation causes both structural distortion and decreased amounts of extractable proteins and nucleic acids due to the possible loss of soluble components during processing.
Microwave irradiation can also speed tissue fixation and processing to within an hour (Leong and Duncis 1986; Morales et al. 2002). The intense heat generated by microwaves facilitates tissue fixation and aggregation (Leong and Duncis 1986; Login et al. 1987; Azumi et al. 1990; Morales et al. 2002) and necessitates using PBS as a fixative instead of formalin. For US-FFPE method, internal tissue temperature did increase with US in proportion to length and intensity of sonication but did not exceed 12C above room temperature during a continuous 40-min application to a 500-ml solution (Figure 2A). Because each step of fixation and processing could be completed within 15 min, the temperature increase is not significant. Thus, we believe that temperature may not be the most prominent factor in US facilitated by fixation and processing.
Though bulk temperature increase is not very remarkable, US may generate microscopically localized high temperature, high pressure, and cavitation so that a high-energy interface exists for unusual biochemical reactions such as free radical formation from water and organic solvent decomposition to occur (Henglein and Kormann 1985; Riesz and Kondo 1992). Intense US waves traveling through solvents and tissue generate cavitation and cyclic stresses that may concentrate energy and power, accelerating alcohol dehydration, xylene clearing, and paraffin impregnation reactions. As mentioned previously (Chu et al. 2005a), applying low frequency US of 20 kHz for 5 min resulted in severe tissue damage as demonstrated by cavitations throughout the tissue (data available). In contrast, high-frequency (1.6 MHz) US would not cause tissue cavitation even after 30 min of continued application. Chemical infiltration (Mitragotri et al. 1995) and permeability of tissues (Kost et al. 2000) are facilitated by US waves disrupting membrane cohesiveness and mutually exclusive forces along immiscible liquid surfaces. US-mediated emulsification into microscopic droplets favors both solvent penetration and crossover of paraffin molecules from the liquid phase into the tissue.
The main effect of US may be to induce more rapid cross-linking reactions among macromolecules, increase cell membrane permeability, and accelerate the exchange rate of water and reagents in subcellular compartments. Mitragotri et al. (1995) reported that US accelerated transdermal drug penetration and diffusion by 1000-fold. However, we believe that increased penetration is not the most prominent effect of US just as slow penetration is not the main reason for slow fixation. Using a thin layer of tissue or even using dissolved protein solution did not substantially shorten the fixation time. mRNA in a 5-μm tissue section after 5-min formalin fixation was not preserved but degraded (Fend et al. 1999). Without any need for penetration, it still required between 6 and 16 hr for complete formalin fixation of a molecular layer of peptides on the glass microscope slide (Sompuram et al. 2004). Rait found that inhibition of RNase A in formalin solution requires 15 hr or more (unpublished data), as measured by enzymatic activity and indicated by structural and thermal property changes (Rait et al. 2004a,b). As presented here and published previously, we observed that 5-15 min formalin fixation with US preserved RNA better than 16-24 hr of routine formalin fixation (Figure 5). We believe the mechanism of much accelerated fixation by US is primarily due to a similar sonochemical reaction as described by Stephanis et al. (1998) that US treatment accelerated the cross-linking between formaldehyde and macromolecules.
How to explain less modification of protein antigenic properties? Our hypothesis is that US-facilitated formaldehyde fixation greatly accelerates cross-linking reaction similar to snap-frozen macromolecules and their conformation. As summarized by Shi et al. (2000), formaldehyde is capable of forming cross-linking to lysine, arginine, tyrosine, asparagines, histidine, glutamine, and serine. In general, free amine (as in lysine and arginine) is more active than primary amides, even more so than secondary amides (Riesz and Kondo 1992; Stephanis et al. 1998; Masuda et al. 1999; Shi et al. 2000). In routine formalin fixation, cross-linking formation is a slow process, allowing methyl (from formaldehyde) to form between optimal reaction sites. For this kind of cross-linkage to occur, proteins may have to alter their local conformation (Mason and O'Leary 1991; Rait et al. 2004a,b), thus altering protein antigenicity as a result. In contrast, when formalin solution is exposed to high-frequency, high-intensity US, deprotonation of formaldehyde, amine, and amide may be much accelerated. These intermediates have high energy, so that covalent bonds between some of the less-optimal sites that are most adjacent may also be formed. Cross-links between these less-optimal sites will be easier to reverse and have less effect on conformation than that between optimal sites. Thus, US-accelerated formalin fixation is somewhat similar to the snap-frozen method, as it tends to preserve tissues close to their original conformation. Furthermore, less energy is needed to disrupt chemical bonds that form between non-optimal sites, facilitating the process of protein extraction. A study of the mechanisms of US-accelerated formalin fixation is currently underway in our laboratory.
In summary, US-accelerated FFPE is a powerful tool for the preservation of biological specimens. Treating fixatives and other processing solvents with US is simple and safe and can be easily integrated into hospital procedures. This technology combines the superior morphological preservation provided by traditional FFPE with the rapid, superior preservation of macromolecules provided by flash freezing. We believe that US-FFPE has great potential to facilitate intraoperative diagnosis and to be broadly applicable to molecular diagnostic techniques. With this technology, pathologists can provide rapid and accurate diagnosis at the morphological and molecular levels, while supporting molecular research with time and cost efficiencies.
Footnotes
Acknowledgments
This work was supported by a National Institutes of Health/National Cancer Institute Grant 1R21 CA-091166-01A1 and an American Registry of Pathology/Armed Forces Institute of Pathology Grant (UBKL) 300-1012-3029-0, both to W-SC. The opinions expressed in this article are the personal views of the authors and are not to be construed as representing the views of the Department of the Army or the Department of Defense.
Special thanks to Bonnie L. Casey for excellent editorial assistance and to Drs. Christopher R. Owner, Douglas J. Wear, Susan L. Abbondanzo, and Stephen I. Fisher for professional expertise and technical support in the preparation of this manuscript.