
Abstract
Cellular prion protein (PrPc) is a glycoprotein expressed at low to moderate levels within the nervous system. Recent studies suggest that PrPc may possess neuroprotective functions and that its expression is upregulated in certain neurodegenerative disorders. We investigated whether PrPc expression is altered in the frontal and occipital cortex in two well-characterized neurodegenerative disorders—Alzheimer's disease (AD) and diffuse Lewy body disease (DLBD)—compared with that in normal human brain using immunohistochemistry and computerized image analysis. The distribution of PrPc was further tested for correlation with glial reactivity. We found that PrPc was localized mainly in the gray matter (predominantly in neurons) and expressed at higher levels within the occipital cortex in the normal human brain. Image analysis revealed no significant variability in PrPc expression between DLBD and control cases. However, blood vessels within the white matter of DLBD cases showed immunoreactivity to PrPc. By contrast, this protein was differentially expressed in the frontal and occipital cortex of AD cases; it was markedly overexpressed in the former and significantly reduced in the latter. Epitope specificity of antibodies appeared important when detecting PrPc. The distribution of PrPc did not correlate with glial immunoreactivity. In conclusion, this study supports the proposal that regional changes in expression of PrPc may occur in certain neurodegenerative disorders such as AD, but not in other disorders such as DLBD.
P
PrPc can undergo posttranslational modification to form the disease-associated or “scrapie” isoform (PrPSc) (Harris 1999). After the production of the abnormal isoform, PrPSc is deposited extracellularly as plaques or intracellulary as fibrils or prion rods. This conformational change of the protein causes PrPSc to become partially resistant to proteases and represents the key event in the pathogenesis of a group of infectious neurodegenerative disorders termed transmissible spongiform encephalopathies or prion diseases, the most common being Creutzfeldt-Jakob disease (CJD) in humans (Budka 2003). Clinically, the human disorders present with dementia, cerebellar dysfunction, and extrapyramidal signs. Neuropathological hallmarks consist of the deposition of PrPSc, neuronal loss, spongiform vacuolation, astrocytosis, and microglial activation. It is thought that prion diseases are a consequence of either a loss of normal function or a gain in neurotoxic function of the host PrP (Jansen et al. 2001; Martins et al. 2001).
Although much is known of the conversion of PrPc to PrPSc, the normal functions of PrPc are still unclear (Martins et al. 2001; Lasmezas 2003). Structurally, PrPc contains several distinct domains: an N-terminal signal peptide, a series of five proline- and glycine-rich octapeptide repeats, a highly conserved central hydrophobic segment, and a C-terminal hydrophobic region that acts as a signal for the glycophospholipid anchor (Harris 1999). PrPc may exist in non-, mono-, or diglycosylated forms, and evidence suggests that the regional heterogeneity in PrPc glycoforms within the nervous system may contribute to the selective targeting of neuronal populations by PrPSc (DeArmond et al. 1999; Beringue et al. 2003). The prevalence of PrPc at synapses (Kitamoto et al. 1992b) has led to the idea of a possible function in modulating synaptic transmission (Collinge et al. 1994). Other putative roles include an involvement in ligand uptake, as a chelator or carrier of metal ions; in signal transduction and regulation of intracellular calcium; in cell adhesion and interaction with extracellular matrix components such as laminin; in the promotion of neurite outgrowth and neuronal differentiation; in the modulation of neurotransmitter metabolism; in the long-term survival of cerebellar Purkinje cells; and in sleep. Although mice devoid of PrP develop normally and show minimal deficits (Cohen et al. 1994), they appear to be more susceptible to seizures induced by convulsant agents (Walz et al. 1999). The absence of PrPc also confers susceptibility to oxidative stress or apoptosis (Kuwahara et al. 1999; White et al. 1999; Wong et al. 2001; Roucou et al. 2004). These findings have fueled the notion that PrPc exerts a neuroprotective function against apoptotic or oxidative stress mechanisms and may regulate the survival of neurons (Roucou et al. 2004).
Very few studies have addressed the expression of PrPc and its involvement in human neurodegenerative diseases other than the prion diseases (Hansen et al. 1989; Hainfellner et al. 1998; Esiri et al. 2000; Ferrer et al. 2001; Voigtländer et al. 2001; Kovacs et al. 2002b). Some studies have suggested that the expression of PrPc may be upregulated in certain neurodegenerative disorders (Esiri et al. 2000; Voigtländer et al. 2001). This would be consistent with a possible neuroprotective role for this protein (Roucou et al. 2004). However, experimental overexpression of PrP may itself produce pathological alterations (including focal vacuolation of the central nervous system, skeletal muscles, and peripheral nerves) in transgenic mice that harbor a high copy number of PrP transgenes (Westaway et al. 1994). Furthermore, transgenic mice harboring high copy numbers of wild-type mouse PrPc spontaneously develop neurological dysfunction in an age-dependent manner (Telling et al. 1996; Perrier et al. 2002), and accumulation of PrPc in the cytoplasm appears to be neurotoxic in transgenic mice overexpressing PrPc in the cytosol (Ma et al. 2002).
Moreover, the expression of certain forms of host PrPc could alter the susceptibility to prion diseases or be associated with particular neurodegenerative processes (DeArmond et al. 1999). For example, the expression of a truncated form of PrP [32–121/134], devoid of the N-terminal region in PrP knockout mice, specifically causes ataxia and death of cerebellar granular neurons (Shmerling et al. 1998). Separately, the toxicity induced by PrPSc should (according to the prion hypothesis) depend on the basal levels of PrPc expressed within the nervous system.
In the present study, we examined whether the immunohistochemical expression of PrPc was altered in the frontal and occipital lobe in two well-characterized neurodegenerative disorders that share certain clinico-pathological characteristics with CJD: Alzheimer's disease (AD), and diffuse Lewy body disease (DLBD) (also referred to as “dementia with Lewy bodies”), and in normal human control cases. DLBD in particular, is considered the second most common form of dementia in the elderly, after AD (McKeith et al. 1996; Rezaie et al. 1996; Hansen 1997; Förstl 1999; Luis et al. 1999; McKeith and O'Brien 1999). We further assessed whether the distribution of PrPc correlated with glial reactivity in these cases.
Materials and Methods
Tissue Samples
Frozen brain tissues were obtained from the MRC London Neurodegenerative Diseases Brain Bank, (King's College London, London, UK). All materials were obtained with informed consent and approval of the local ethical committees (Table 1). Thirty cases were selected for this study. Of these cases, 10 were neuropathologically diagnosed with “pure” AD without Lewy bodies according to the Consortium to Establish a Registry for Alzheimer's Disease criteria (Mirra et al. 1991) (mean age 70.2 years, range 58–92 years), 10 were diagnosed with DLBD (mean age 75.2 years, range 58–92 years), and 10 were neuropathologically diagnosed as normal, with no history of neurological or psychiatric disorders (mean age 79.3 years, range 66–89 years). The DLBD cases (Table 1) were further classified as “pure” forms if there was no Alzheimer-type pathology (plaques or tangles) or if they fulfilled the criteria for a Lewy body variant of AD in which plaques and tangles were also present (McKeith et al. 1996). Further clinical data (e.g., concerning the cognitive status of these cases) were not available. Frozen tissues from the frontal and occipital lobe were available from all cases for this study. Frontal blocks included Brodmann areas 6/8 (agranular-intermediate frontal cortex) and 24/32 (cingulate cortex). Occipital blocks included the calcarine sulcus (Brodmann area 17/striate cortex). Serial sections were cut from each block on a cryostat at ∼20 μm thickness and stored at −70C before immunohistochemistry. Sections from the cerebellum of a case of variant CJD (vCJD) (embedded in paraffin wax) were used as positive controls.
Immunohistochemistry
Optimal working dilutions for each of the antibodies targeting PrP were predetermined on positive control vCJD sections as well as test sections to obtain minimal background reactivity. Immunohistochemistry using antibodies to PrP: 3f4 [1:100 dilution; Senetek, Napa, CA (Kascsak et al. 1987)], sp40 [1:500 (Lantos et al. 1992)], 12f10 [1:200 (Krasemann et al. 1996)], and F89/160.1.5 [Ab3, 1:1300 dilution; CN Biosciences, Nottingham, UK (O'Rourke et al. 1998; Van Everbroeck et al. 1999)] was performed according to a standard three-step immunoperoxidase method (ABC-HRP protocol; Dako, Ely, Cambridgeshire, UK), with 3,-3'diaminobenzidine (DAB) as chromogen. The putative binding regions of these antibodies to human PrP are shown in Figure 1. Immunohistochemistry was performed on duplicate sections (processed simultaneously in entire batches) per antibody reagent (i.e., all frontal or occipital cortical samples assayed on the same occasion) to minimize interassay variability.
Control, DLBD, and AD cases investigated in this study
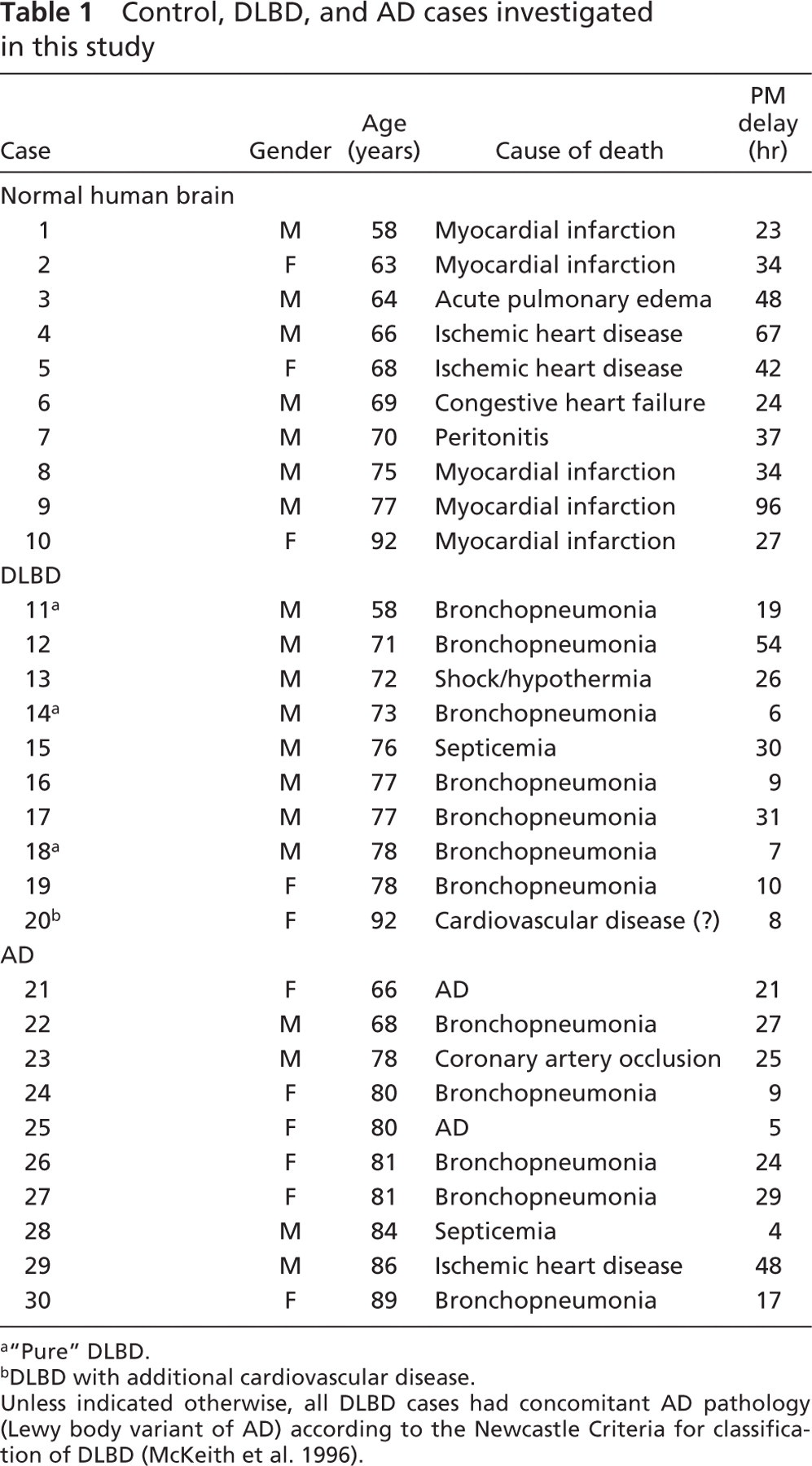
“Pure” DLBD.
DLBD with additional cardiovascular disease.
Unless indicated otherwise, all DLBD cases had concomitant AD pathology (Lewy body variant of AD) according to the Newcastle Criteria for classification of DLBD (McKeith et al. 1996).
Briefly, cryostat sections were mounted onto silane-coated slides and allowed to dry for 1 hr at room temperature before being transferred to a 37C drying oven for a further 1 hr. Sections were then immersed in a solution of methanol containing 2.5% of a 30% hydrogen peroxide solution for 1 hr. Following this, they were briefly rinsed with water, placed in phosphate-buffered saline (PBS) solution (pH 7.6) and incubated for 1 hr with lysis buffer solution consisting of Hank's balanced salt solution supplemented with 1% of each of the following: 1 M magnesium chloride, 1 M calcium chloride, bovine serum albumin, and Tween 20. Nonspecific binding was blocked by incubating sections for 1 hr with normal serum diluted 1:10 in PBS [normal rabbit serum for monoclonal mouse antibodies Ab3, 3f4, 12f10; normal swine serum (Dako) for polyclonal antibody sp40]. Primary antibody solution (diluted in a 1:200 solution of normal serum) was applied to slides and incubated overnight at room temperature in a humidity chamber. Sections were washed in PBS (two changes, 5 min each wash) and incubated for 90 min with biotinylated secondary antibody [rabbit anti-mouse IgG (Dako) for monoclonals, swine anti-rabbit IgG (Dako) for polyclonals] diluted 1:200 in PBS. After this, the secondary antibody solution was discarded, and sections were washed a further two times with PBS and incubated for 90 min with ABC-HRP complex (Dako) prepared 1 hr before use. After two more washes in PBS, sections were time-reacted with DAB (the positive control sections were used as a reference for intensity of staining), counterstained lightly with Harris's hematoxylin solution or methyl green solution, dehydrated through graded alcohols, cleared in xylene, and mounted with glass coverslips using DPX mountant (Merck; Lutterworth, UK). Slides were processed as entire batches to limit variability in immunoreactivity for subsequent image analysis. Additional immunodetection of astrocytes [glial fibrillary acidic protein (GFAP), 1:1000 dilution (Dako)] and microglia [CD68/clone PG-M1, 1:200 dilution (Dako); major histocompatiblility complex (MHC) class II/clone CR3–43, 1:100 dilution (Dako)] were also assessed qualitatively following the standard immunohistochemical procedure already described [overnight incubation of sections with the primary antibody at room temperature, ABC method (Dako)].
Controls
Sections from the cerebellum of a case of vCJD served as positive controls for detection of PrP. Negative controls included the following: preincubating test frozen sections with 5 and 10 μg/ml proteinase K, 4 M guanidinium thiocyanate (30 and 60 min), or formic acid (5 and 10 s)—treatments known to abolish detection of host cellular PrP (Bell et al. 1997). Additional controls included preincubation of test and vCJD sections with normal blocking serum, or nonspecific mouse IgG alone (omitting incubation with primary antibody).
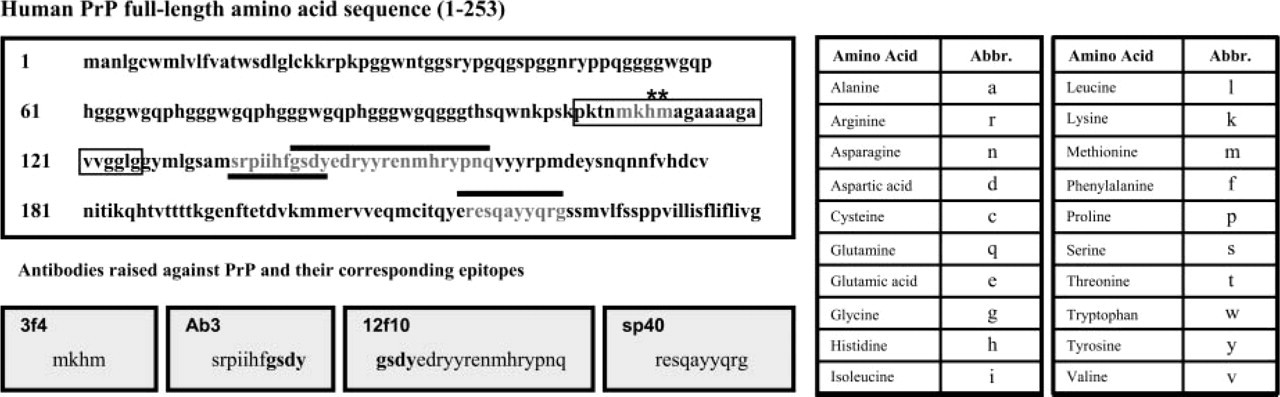
Binding sites of the antibodies used in this study to the human prion protein (PrP). The putative binding regions of antibodies to the human PrP (amino acids 1–253) are indicated. The full-length amino acid sequence of human PrP comprises a signal peptide sequence (AA1–23), octapeptide repeat sequences (AA51–95) for five Cu2+ binding sites, a highly conserved hydrophobic central region in cellular PrP (PrPc) that is neurotoxic in “scrapie” PrP (PrPSc) (AA106–126), proteolytic cleavage sites (AA111–112,∗∗), an α-helical region (AA144–154), two glycosylation sites (AA181 and AA197), and a glycophospholipid anchor (AA229–253) that can be enzymatically cleaved. Of the antibodies tested, F89/160.1.5 (Ab3) was raised against synthetic peptide residues 146–159 of bovine PrP (O'Rourke et al. 1998), and recognizes residues 135–145 in humans (Van Everbroeck et al. 1999). 12f10 binds to helix region 2 of the human PrP, residues 142–160 (Krasemann et al. 1996). 3f4 was raised against synthetic peptide Met-Lys-Hist-Met of human PrP (Kascsak et al. 1987). The polyclonal antibody sp40 was raised against sheep PrP residues 219–232, and recognizes residues 220–229 in man (Lantos et al. 1992). From the figure it can be seen that F89/160.1.5 (Ab3) and 12f10 map at either end of a sequence within the core of the protein [overlapping region indicated in bold (gsdy)]. The sp40 binding region is near the C terminus, whereas the 3f4 binding region is near the N terminus.
Quantitative Image Analysis of PrPc Reactivity
Image analysis was conducted to assess the relative levels of expression of PrPc in the following brain areas from all cases: frontal gray matter, to include the agranular/intermediate frontal and cingulate cortex (Bodmann areas 6/8, 24/32); frontal white matter; occipital striate cortical gray matter (Brodmann area 17); and corresponding occipital white matter. Quantitative image analysis has been previously employed to assess PrP immunoreactivity and associated pathology in studies on CJD (MacDonald et al. 1996; von Eitzen et al. 1998). The profile of immunoreactivity for each antibody was assessed quantitatively (mean percentage of immunoreactive product per defined area) according to established protocols (Al-Sarraj et al. 2004; Pontikis et al. 2004). The analysis was performed blind with respect to neuropathological details of the cases investigated on duplicate sections in the brain regions specified above. Field selection was randomly determined by the image analysis program controlling the movement of the microscope stage (Image-Pro Version 4.2 and Optimas version 6.2; Media Cybernetics, Silver Spring, MD).
A survey of the immunoreacted tissue sections was performed by two independent operators (PR, CP) to verify specific immunoreactivity in duplicate sections subsequently processed to quantitative image analysis. Briefly, nonoverlapping red-green-blue (RGB) images were digitally captured at random within the defined brain areas, providing a systematic survey throughout each region for each case. Images were captured via a live color video camera (JVC, 3CCD, KYF55B) mounted onto a Zeiss Axioplan microscope with a X10 objective and neutralizing gray filter (Zeiss; Welwyn Garden City, Hertfordshire, UK). All parameters including the lamp intensity, video camera setup, and microscope calibration were held constant. The optimal segmentation of immunoreactive profiles was analyzed with the Optimas image analysis program (Media Cybernetics), using a previously described semi-automated thresholding method based on the optical density of the immunoreactive product (Al-Sarraj et al. 2004; Pontikis et al. 2004). Each RGB image was processed using a blue-band filter to assess DAB reactivity (in brown), and the threshold was selected to define foreground immunostaining. Foreground immunostaining was accurately defined according to averaging the highest and lowest immunoreactivities within the sample population for each antibody [measured on a scale of 0 (100% transmitted light) to 255 (0% transmitted light) for each pixel]. A semi-automated histogram-based protocol and a manual RGB color cube-based protocol (specified in the image analysis programs) were employed to determine the optimal segmentation (threshold setting) for each antibody, rated by two independent investigators (PR, CP). Once this optimal segmentation was selected, the chosen threshold setting was then applied as a constant to all subsequent images analyzed for this antibody. Furthermore, the specificity of the detection method was also verified manually by monitoring the analysis as it progressed, per region, per case. The immunoreactive profiles were discriminated according to their intensity (a property that reflects the intensity of the antigen-antibody immunoreacted complex), to determine the specific immunoreactive area (the mean pixel gray value was obtained by subtracting the total mean gray value from the nonimmunoreacted value per defined field). Each field measured 475 μm wide, with a height of 320 μm, the total area assessed for each region corresponding to 19 × 12.8 mm. Macros were subsequently recorded to transfer the data to a spreadsheet. The data were analyzed using the Statistical Package for Social Sciences (SPSS v.11) program (SSPS Inc.; Chicago, IL). Data were plotted as the mean percentage area of immunoreactivity per field ± SEM for each brain region (Figure 3). Statistical significance was assessed using Mann-Whitney U and Wilcoxon Z tests. Linear regression and Spearman correlation tests were used to assess the relationship between PrPc expression, postmortem delay, and age. A p value of 0.05 or less was considered significant.
Qualitative Analysis of Glial Reactivity
A simple graded scoring system was adopted for visual interpretation of PG-M1, MHC class II, and GFAP immunoreactivities, using a standard light microscope with a X20 objective. The scoring was as follows: 0, no reactivity; 1, weak/mild reactivity on a few scattered cells; 2, moderate reactivity; 3, intense reactivity on numerous cells. For cases of AD, plaque-associated glial reactivity was further assessed according to the following graded scores: 0, no reactivity; 1, weak/mild reactivity on one or two plaque-associated cells; 2, moderate reactivity surrounding plaques; 3, intense reactivity on many cells associated with plaques. The results are presented in Table 2.
Results
Microscopic Analysis
The four antibodies demonstrated variability in their detection of host PrPc. F89/160.1.5 (Ab3) and 12f10 showed similar and consistent immunoreactivity profiles for PrPc. By contrast, 3f4 showed variable and inconsistent reactivity, whereas sp40 was least effective at detecting PrPc in all samples (data not shown). Nevertheless, all four antibodies proved to be reliable when detecting PrPSc on positive control sections of vCJD cerebellum. In our study, pretreatment with 10 μg/ml proteinase K (Figure 2G), 4 M guanidinium thiocyanate (60 min), and formic acid (10 s)—treatments known to denature PrPc—abolished the detection of PrPc, thus confirming specificity of the antibodies. In most of the cases examined, the expression of PrPc was confined to the deeper cortical layers and was particularly notable as a distinctly visible diffuse band at the boundary between the gray and white matter (Figures 2A–2C). This pattern of immunoreactivity was suggestive of an association with projections of thalamocortical fibers within these regions. Microscopically, PrPc expression was predominantly cellular (it was mainly present in neurons) (Figure 2F) but was also diffuse within the neuropil of the gray matter (Figures 2D, 2H, and 2L). By contrast, and with the exception of DLBD cases, immunoreactivity was very weak and diffuse when present within the white matter (Figures 2E and 2I). We further noted that PrPc was weakly associated with blood vessels of control and AD cases (Figures 2J and 2K). However, this was markedly upregulated on blood vessels, particularly within the white matter in the majority of DLBD cases (Figures 2M–2O).
Qualitative analysis of PG-M1 (CD68) and MHC class II immunoreactivity in the frontal and occipital cortex in cases of DLBD and AD and in normal controls
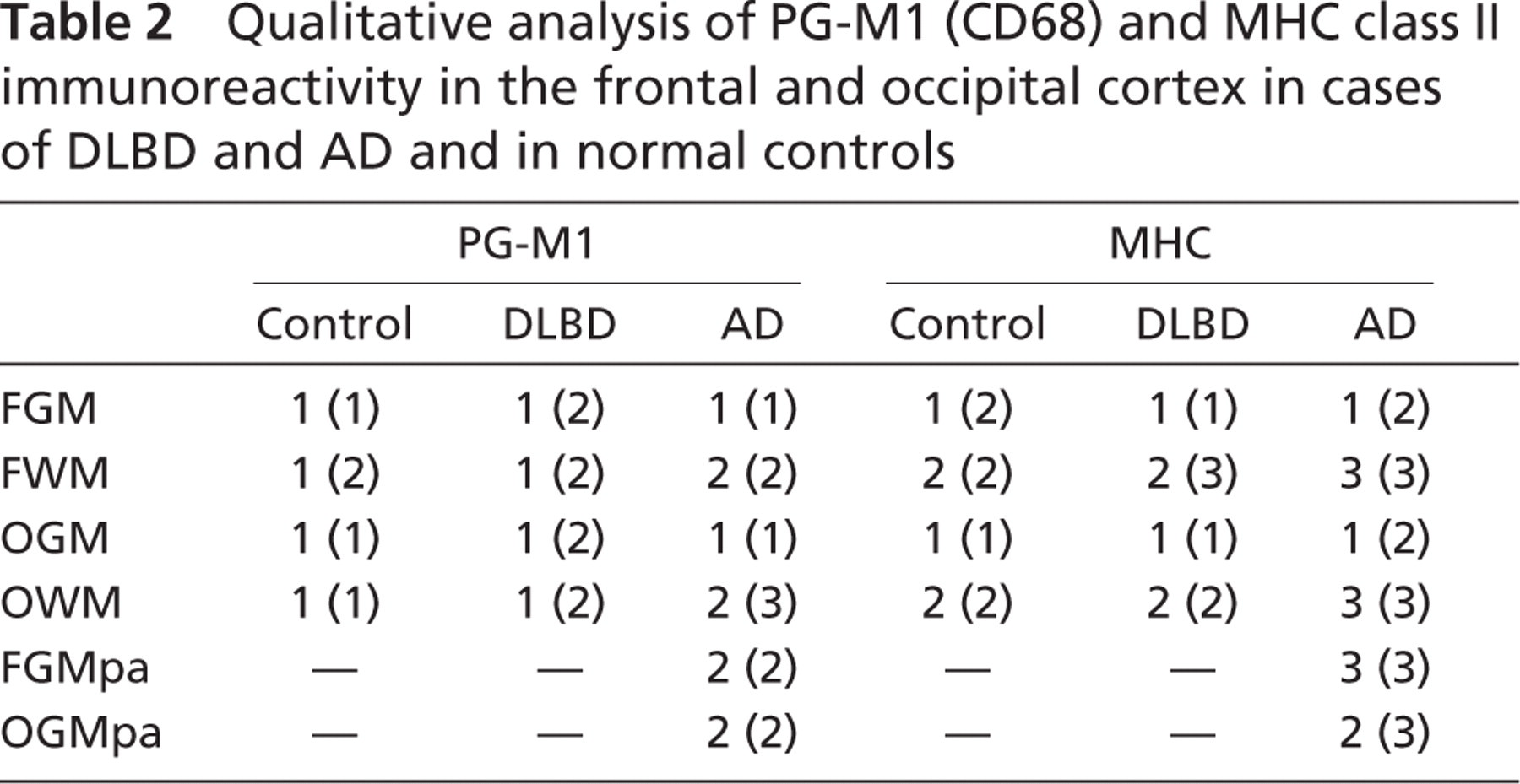
The following graded scoring system was used for qualitative assessment of immunoreactivity: 0, no reactivity; 1, weak/mild reactivity on a few scattered cells; 2, moderate reactivity; 3, intense reactivity on numerous cells. For cases of Alzheimer's disease, plaque-associated glial reactivity in the frontal and occipital cortical gray matter was further assessed according to the following graded scores: 0, no reactivity; 1, weak/mild reactivity on one or two plaque-associated cells; 2, moderate reactivity surrounding plaques; 3, intense reactivity on many cells associated with plaques. Data are presented as median scores, with maximum scores per region indicated in brackets (n=10 cases for each patient group). Examination of PG-M1 and MHC class II immunoreactivities revealed no marked differences between control and DLBD brains. By contrast, immunoreactivity for both markers was higher in AD cases: particularly in association with perivascular reactivity within the white matter and spatially distributed focal clusters of reactivity within the gray matter, consistent with plaques. Both PG-M1 and MHC class II revealed the presence of numerous activated microglia and perivascular cells. FGM, frontal cortical gray matter; FWM, frontal white matter; OGM, occipital gray matter; OWM, occipital white matter; FGMpa, plaque-associated cellular immunoreactivity in the cortical gray matter of the frontal lobe; OGMpa, plaque-associated cellular immunoreactivity in the cortical gray matter of the occipital lobe.
Quantitative Image Analysis of PrPc Expression
The quantitative analysis of PrPc immunoreactivity detected with Ab3 (F89/160.1.5), 12f10, and sp40 is shown graphically in Figure 3. There were significant differences in the expression of PrPc (p≤0.05) between gray and white matter and between occipital and frontal cortex in cases of AD and DLBD and in controls. Immunoreactivity with 3f4 antibody was variable and inconsistent within the frontal and occipital cortex of the cases examined (data not shown). Of the four antibodies, sp40 was least effective at detecting PrPc in all samples analyzed (Figure 3C). By contrast, F89/160.1.5 (Ab3) and 12f10 demonstrated consistency in detecting PrPc in all samples (Figures 3A and 3B). The similarity in immunoreactivity profiles demonstrated with F89/160.1.5 (Ab3) and 12f10 may be associated with an overlap in the binding region within the core of the human PrP recognized by these two antibodies (Figure 1). This was not shared by 3f4, which recognizes four amino acid residues (containing the cleavage site for PrP) closer to the N-terminal of the PrP. Similarly, sp40, which recognizes a sequence near the C-terminal, was far less effective at immunohistochemical detection of PrPc at optimal dilutions. The fact that all four antibodies proved to be highly effective at detecting PrPSc on positive control sections suggests that the differential detection was more likely to be related to the epitope specificity of these antibodies, associated with regional variations in the tertiary structure (i.e., glycosylation) of PrPc, although this requires further investigation.
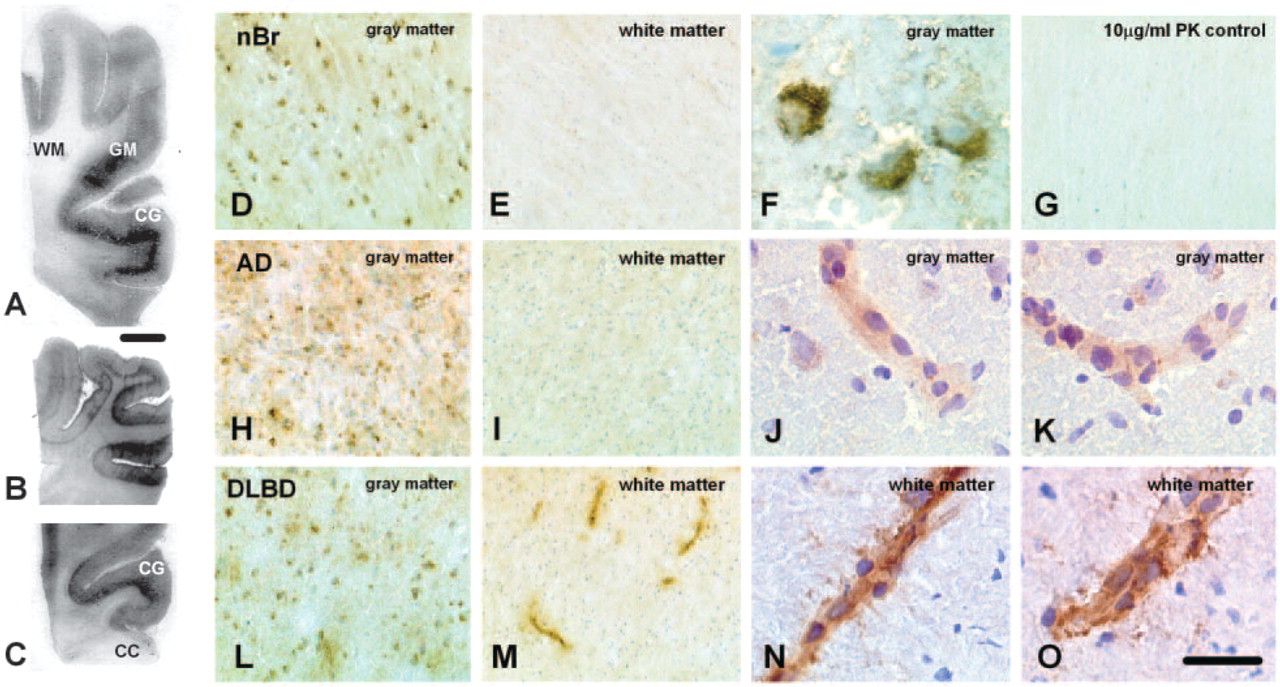
Patterns of PrPc immunoreactivity in the frontal and occipital cortex, and immunohistochemical expression of cellular prion protein (PrPc) in normal aged brain and in Alzheimer's disease (AD) and diffuse Lewy body disease (DLBD) 12f10 (
Quantitative image analysis of the immunoreactive profiles detected using F89/160.1.5 (Ab3) and 12f10 confirmed that the expression of PrPc was predominantly confined to the gray matter, with greater levels (3- to 8-fold) detected in the occipital than frontal cortex of normal control and DLBD cases (Figure 3A,B). Expression of PrPc did not differ significantly between DLBD and control cases in any of the areas examined. By contrast, both F89/160.1.5 (Ab3) and 12f10 detected markedly higher levels of PrPc (p≤0.05) within frontal cortical gray matter in AD and lower levels within the occipital cortex compared with DLBD and control cases. A slightly higher level of expression was also detected within the frontal cortical gray matter of AD cases with sp40 (Figure 3C), reflecting to a much lesser degree the results obtained using F89/160.1.5 (Ab3) and 12f10 in this region. Although linear regression and correlation analyses showed no statistically significant relationship between PrPc expression, postmortem delay or age, a general reduction in the expression of PrPc (detected using 12f10 and F89/160.1.5) was noted with advancing postmortem delay (data not shown). There was no significant correlation of PrPc levels with increasing age. However, the number of cases may have been limiting, and a greater cohort of cases is required to verify these observations.
Qualitative Analysis of Glial Markers
No significant qualitative differences in the expression of PG-M1 and MHC class II (markers that detect microglia) were noted between cases with DLBD and controls (Table 2). Immunoreactivity for GFAP (a marker for astrocytes) was very weak, diffuse, and inconsistent on frozen sections, and was therefore not assessed qualitatively. Although there was an upregulated microglial response in AD (mainly perivascular reactivity within the white matter and plaque-associated clusters of cells within the gray matter) (Table 2), the expression of PrPc did not visibly correlate with PG-M1 or MHC class II immunoreactivity. In particular, PrPc expression was not obviously associated with plaques in AD (as determined on the basis of morphological characteristics), which were clearly invested with microglia. However, further assessments of the colocalization of PrPc with β-amyloid protein are required to clarify this relationship.
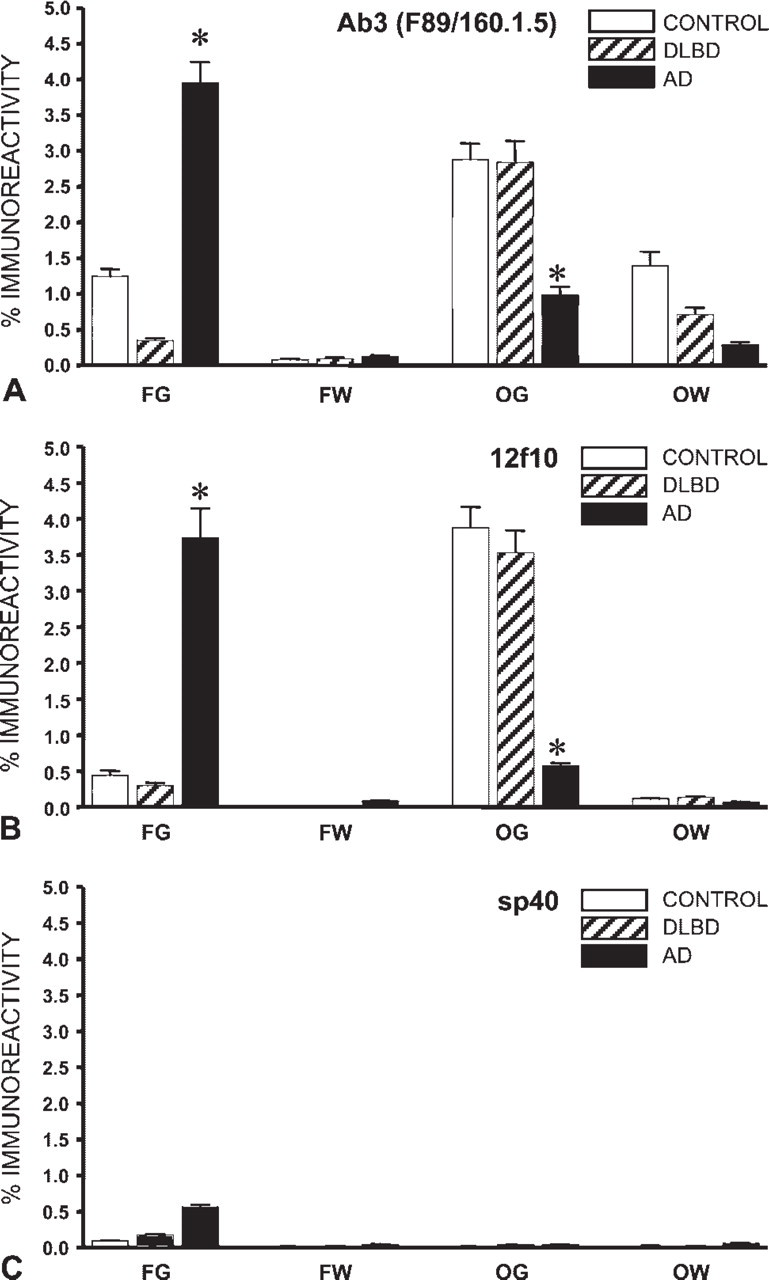
Quantitative analysis of PrPc expression in the frontal and occipital cortex in cases of AD and DLBD and in normal brain. Data are presented as the mean percentage immunoreactivity ± SEM (based on analysis of 400 fields on duplicate sections from 10 cases per group). Expression of PrPc was significantly higher within the cortical gray matter (p≤0.05) than in the white matter in all cases examined. Expression in DLBD did not differ significantly from controls in all brain regions examined. However, there were apparent differences in the detection of PrPc between the antibodies used. F89/160.1.5 (Ab3) and 12f10 displayed similar and consistent profiles of immunoreactivity in AD and DLBD and in control brain, demonstrating higher reactivity within the occipital cortex of DLBD and control cases than within the frontal cortex. By contrast, both F89/160.1.5 (Ab3) and 12f10 detected significantly higher levels of PrPc (p≤0.05) expression within frontal cortical gray matter in AD and lower levels within the occipital cortex compared with DLBD and control cases∗∗∗. sp40 was least effective at detecting PrPc in all samples analyzed. Even so, there was a slightly higher expression detected within the frontal cortical gray matter compared with DLBD and control cases, reflecting the results obtained using F89/160.1.5 (Ab3) and 12f10. Differences in the levels of detection and regional distribution of PrPc may either reflect variation in the glycosylation patterns of this protein and/or epitope specificity of the antibodies. FG, frontal gray matter; FW, frontal white matter; OG, occipital gray matter; OW, occipital white matter.
Discussion
In view of the recent reports suggesting that PrPc is upregulated in certain neuropathological conditions (Esiri et al. 2000), including in AD (Ferrer et al. 2001; Voigtländer et al. 2001), we examined the expression of PrPc in frozen sections from the frontal and occipital lobe in AD and DLBD—two well-characterized neurodegenerative disorders that share certain clinical and pathological characteristics with CJD (DeArmond 1993; Budka et al. 1995; Hsiao 1997; Haïk et al. 2000; Tschampa et al. 2001; Castellani et al. 2004), and in normal control cases. This is significant because unlike routine paraffin-embedded materials, the tissues in our study had not been previously subjected to long-term fixation in aldehyde-based fixatives (notably formalin) and antigenicity was therefore well preserved by comparison. Our results indicate that the epitope specificity of antibodies directed at PrPc clearly has an impact not only on the detection of this protein in the human nervous system but also on the subsequent interpretation of the data obtained in both normal and pathological conditions. It is possible that the use of frozen versus paraffin-embedded material and fixation of tissues (as well as a host of other factors such as postmortem delay, age, and duration of illness, where applicable) will also have some bearing on the results obtained in different laboratories using an immunohistochemical approach.
In our study, we found that of the four epitope-specific antibodies used to detect PrPc, the ones that produced consistent labeling were targeted toward the central region of the protein [F89/160.1.5 (Ab3) and 12f10] (Figure 2). By contrast, both 3f4 (which binds to an epitope closer to the N terminus) and sp40 (which binds to an epitope close to the C terminus) were far less useful in detecting PrPc in the cases examined, even though all four antibodies proved to be reliable when detecting PrPSc on positive control sections from the cerebellum of a case with variant CJD. Pretreatment with proteinase K, 4 M guanidinium thiocyanate or formic acid abolished PrP immunoreactivity on all sections apart from the positive vCJD control, demonstrating that the staining on sections represented cellular PrP. The variability in staining and poor immunoreactivity that has been obtained previously using 3f4 (Esiri et al. 2000; Salès et al. 1998), as well as in our study, may either be due to fixation and processing of samples, masking of the antibody binding region that depends on the folding of the PrP, or possibly the normal proteolytic cleavage of PrP by lysosomotropic amines at amino acid residues 111 (histidine) and 112 (methionine) (Figure 1) (Chen et al. 1995; Lehmann et al. 1999). It is recognized that excessive cross-linking of proteins which occurs following prolonged exposure to aldehydes, as well as conventional paraffin wax histological preparations, markedly affect the antigenicity of proteins. Therefore, detection will depend not only on the antibody employed, but also on the extent of protein cross-linking within specimens. Because the use of frozen sections should, in theory, prove more reliable, our findings point to enzymatic cleavage of the protein as a determinant for the variability in detection with the 3f4 antibody.
PrPc occurs in several conformational states in situ within the human brain (Salès et al. 1998). However, the regional variability in topological forms of PrPc (i.e., secreted cleavage products, transmembrane or cytosolic compartmentalized fragments) have not been investigated. Use of epitope-specific antibodies in combination with other techniques such as Western blot and electrophoretic analysis would aid in identifying these forms. Due to the limited source of frozen material, these studies could not be pursued in the present investigation. Moreover, considering that Western blot examination would be reliant on the number of freeze/thaw cycles to which a tissue is exposed and that PrPc is readily degraded, the use of this technique would be restricted in this respect. Furthermore, we were limited in our investigation to examining the frontal and occipital cortices, because these were available for all cases. Although these selected brain regions are known to be targeted in neurodegenerative diseases (including prion diseases), it would be important to examine more fully the topographical distribution of PrPc expression in cases of AD and DLBD and in normal controls.
In our samples, cellular and diffuse PrPc immunoreactivity were detected within the cortical gray matter (predominantly associated with neurons), and in a distinct band along the border between gray and white matter in the majority of cases investigated. Because the neocortex is a major target for CJD pathology (Jansen et al. 2001), and the primary structure of the host PrP influences the distribution of abnormal PrP in the nervous system (Kitamoto et al. 1992a), this pattern of PrPc expression could in theory influence the synthesis of PrPSc regionally within the brain. To support this, we previously noted that some cases diagnosed with sporadic CJD showed PrPSc deposits within the deeper cortical layers (IV–VI), which correlated with microglial reactivity in the same region (Rezaie and Lantos, unpublished observations). Similar findings have been reported by other groups (von Eitzen et al. 1998; Verghese-Nikolakaki et al. 1999), and it is possible that the deposition of PrPSc in these areas precedes glial activation (Rezaie and Lantos 2001).
Our findings of PrPc expression confined largely to neurons within the cerebral cortex in normal brains generally support recent reports (Salès et al. 1998; Esiri et al. 2000; Moya et al. 2000; McLennan et al. 2001; Voigtländer et al. 2001). However, there is some discrepancy between our results and reports of the distribution of PrPc message within the neocortex of the normal human brain in paraffin-sectioned materials. In situ hybridization studies by Jansen and colleagues (Jansen et al. 2001) found PrPc mRNA mainly in upper cortical neurons as well as in cerebellar Purkinje cells, and McLennan and colleagues (2001) demonstrated higher levels of PrP mRNA in fixed paraffin human tissue in layers II (external granular layer) and III (pyramidal cell layer) compared with layers V (ganglionic layer) and VI (fusiform layer). However, these authors could not detect PrP mRNA within the subcortical white matter; likewise, Salès et al. (1998) found very limited PrPc staining within the white matter of the human brain. Other studies have found no specific laminar distribution of the PrP message or protein in the cortex (McLennan et al. 2001; Kovacs et al. 2002b). These differences could possibly be explained by differences between the transcription and translation of PrPc or by the state of tissues analyzed (frozen as in this study versus paraffin-embedded tissues in the other studies). Our immunohistochemical findings of minimal PrPc expression within the white matter of the normal brain are consistent with these studies. We also noted minimal expression in the white matter of AD cases in our study. By comparison, immunoreactivity confined to blood vessels in the white matter was a finding specific to cases of DLBD.
PrPc has been detected on the surface of human endothelial cells (human umbilical vein endothelium, Simak et al. 2002; Starke et al. 2002; human microvascular endothelium, Starke et al. 2002,2003b) maintained in culture, using immunohistochemical and flow cytometric methods. Endothelial cells of both macrovascular and microvascular origin maintained in vitro are also capable of releasing PrPc slowly and constitutively into the tissue culture medium (Simak et al. 2002; Starke et al. 2002), particularly following proapoptotic stimulation (Simak et al. 2002). RT-PCR analysis has further confirmed the presence of PrPc mRNA in these cultured endothelial cells. Furthermore, Starke et al. (2002,2003a) have proposed that endothelial cells could be a likely source for the soluble plasma pool of PrPc. However, it has been questioned whether endothelial cells in situ also express detectable levels of PrPc under normal conditions (Sivakumaran 2003; Sivakumaran et al. 2003). Specifically, Sivakumaran and colleagues (2003) could not detect PrPc in paraffin sections of human umbilical cord vessels or adult saphenous vein and aorta using the 3f4 antibody. Although these authors noted the expression of PrPc by neurons in the human cerebral cortex (used as a positive control in their study), they presented no further data regarding PrPc expression on blood vessels in the brain. In this study, we found weak and minimal PrPc expression on blood vessels in the brain of control and AD cases. A separate study in rodents attributed blood vessel-associated PrP immunoreactivity to astrocytic foot processes terminating on these vessels (Verghese-Nikolakaki et al. 1999). However, due to the inconsistent staining patterns for GFAP on our frozen tissue samples, we could not determine whether vessel-associated PrP reactivity was associated with astrocytic GFAP expression in our study. For this reason, this particular issue remains inconclusive at present.
Our observation of increased PrPc expression in white matter blood vessels in DLBD was unexpected. The significance of this finding is presently unclear. Upregulation of PrPc could be associated in some way with the vascular pathology that has been noted in the white matter of patients with DLBD (particularly the frontal cortex) (Londos et al. 2000). Another possibility could be related to an upregulation of haparan sulfate proteoglycans on vascular endothelium, to which PrPc binds (Warner et al. 2002), in DLBD. However, both of these suggestions are purely speculative at present and will require further investigation. It is also important to note that inflammatory mediators do not appear to alter the expression of PrPc by endothelial cells maintained in vitro (Simak et al. 2003). Once again, this observation will require further clarification in the context of AD and DLBD.
Our findings relating to AD also differ slightly from the observations previously noted (Ferrer et al. 2001; Voigtländer et al. 2001; Kovacs et al. 2002a); we could not detect PrPc immunoreactivity specifically at the periphery of senile plaques within the cortex of AD cases. Interestingly, Kovacs and colleagues (2002a) noted that out of a panel of 10 antibodies directed against PrP, 6H4 (raised against residues 144–152) and 12f10 failed to give this type of labeling at the periphery of, or throughout, senile plaques∗∗∗. 3f4 separately appears to produce variable results when detecting PrP (Esiri et al. 2000; Moya et al. 2000). Therefore, the use of differing antibodies, fixation, or processing (frozen versus paraffin sections in particular); the brain region examined [frontal and occipital cortex (this study); hippocampus (Ferrer et al. 2001); hippocampus, subiculum, entorhinal cortex, and temporal cortex (Voigtländer et al. 2001); hippocampus, temporal cortex, cerebellum (Kovacs et al. 2002a,b)]; or detection of different forms of PrPc (Liu et al. 2001; Warner et al. 2002) may account for these differences. Nevertheless, we are in agreement with these authors that PrPc immunoreactivity is likely to be upregulated regionally in AD (in this case, the frontal cortical gray matter), depending on the use of specific antibodies (i.e., F89/160.1.5 (Ab3) and 12f10 in our study). It is unclear at present whether the accumulation of PrPc occurs as a result of increased synthesis, reduced degradation or vice versa.
Recent in vitro experiments suggest that neurotoxicity of the PrP is dependent on the presence of microglia (for a review, see Rezaie and Lantos 2001) and astrocytes (Brown 1999). Microglia represent the resident mononuclear phagocytes in the central nervous system (CNS), whose primary function is to respond to pathological insults by becoming activated to perform phagocytosis, present antigen (via MHC class II molecules), and release various inflammatory mediators (Rezaie and Lantos 2001). Their activation correlates with deposition of PrPSc and occurs before neuronal cell death in vivo; microglia are located in regions of plaque formation, vacuolation, and accumulation of PrP in the form of diffuse deposits or plaques (von Eitzen et al. 1998). Given these observations, we determined qualitatively whether glial reactivity corresponded spatially with the PrPc expression detected in DLBD or AD brains compared with controls. The results from the few studies that have investigated the microglial response in DLBD have been somewhat contradictory (Mackenzie 2000; Rozemüller et al. 2000; Shepherd et al. 2000; Mackenzie 2001). However, we found that PG-M1 (CD68) and MHC class II immunoreactivities were similar within the frontal and occipital cortex of control and DLBD cases—there was no overt evidence for a microglial response in the frontal or occipital cortex of either “pure” DLBD cases or the LB variant of AD in our study. This is in keeping with the results of a similar investigation by Rozemuller and colleagues (Rozemüller et al. 2000) and Shepherd and coworkers (2000), and may be taken together to further support the lack of an intrinsic inflammatory component to this disorder. By comparison, we found moderate to intense PG-M1 and MHC class II reactivity in AD brains, with clusters of microglia (corresponding to sites of AD plaques) evident throughout the cortical gray matter and intense, mainly perivascular reactivity within the white matter in all samples. Because PrPc was widely expressed throughout the cortical gray matter in AD and did not colocalize with these focal clusters of microglia, it is more likely that microglial aggregates corresponded to the distribution of senile plaques (identified on adjacent hematoxylin & eosin sections; data not shown) containing β-amyloid. Therefore, the expression of PrPc in our study did not correlate with glial reactivity per se but was instead predominantly associated with neurons in the neorcortical gray matter.
In conclusion, this study has shown that the expression of PrPc in neocortical gray matter does not differ significantly between DLBD and control cases. However, differential expression between the frontal and occipital cortex were noted in AD, with higher levels of PrPc detected in the frontal cortical gray matter compared with DLBD cases or controls. Significantly, the frontal cortex and particularly the cingulate gyrus, are brain regions known to be primarily affected in neurodegenerative diseases. The varying levels of PrPc noted in AD suggest that PrP is involved in some way in this neurodegenerative disorder. Changes in the levels of expression of PrPc could reflect a protective cellular response against oxidative stress (e.g., upregulation of PrPc) or associate with susceptibility to neuronal damage (e.g., lower level of expression of PrPc) (Kim et al. 2004; McLennan et al. 2004; Roucou et al. 2004). Whether these findings are directly related to the regional specificity in glycoform patterns and neuron-specific differences in PrPc glycosylation (Beringue et al. 2003) warrants further critical assessment. Separately, it is well recognized that there is an inflammatory component in AD, and this includes elevated microglial reactivity and expression of proinflammatory cytokines within the CNS (Eikelenboom et al. 2002). There are substantial differences between patients with DLBD and AD in terms of the degree of brain inflammation; cases with DLBD have a much weaker inflammatory response, if present. This observation introduces the question of whether an inflammatory response could regulate the expression of PrPc or whether the function of PrPc could in some way be associated with neuroinflammation. Future studies should therefore not only clarify the relative levels of expression of PrPc within other brain regions known to be affected in AD (such as temporal and parietal cortex, hippocampus, thalamus, and basal ganglia) and in a larger cohort of “pure” cases of DLBD vs cases of DLBD with concomitant AD pathology, but further assess the topographical expression of this protein in other neurodegenerative (e.g., Parkinson's disease, motor neuron disease, Huntington's disease) and neuroinflammatory disorders (e.g., multiple sclerosis).
Footnotes
Acknowledgements
We thank Nadeem Khan (Coordinator, MRC London Neurodegenerative Diseases Brain Bank, Department of Neuropathology, Institute of Psychiatry, King's College London, London, UK), for providing tissue materials used in this study. We are grateful to Dr. Steve Whatley and Prof. Brian Anderton (Department of Neuroscience, Institute of Psychiatry, King's College London, London, UK) for the generous gift of the sp40 antibody. 12f10 was a kind gift from Prof. G. Hunsmann, Drs. S. Krasemann and M. Groschup, and Prof. W. Bodemer (The German Primate Centre, Göttingen, Germany). This work was partially supported by the European Union Biomed-2 Grant QLK2-CT-2001-01924 (“Strategies for the Prevention and Treatment of Prion Diseases”). The work was in part presented as a poster at the 102nd meeting of the British Neuropathological Society.