
Abstract
We investigated the localization of several markers for lysosomes and aggresomes in the chromatoid bodies (CBs) by immunoelectron microscopy. We found so-called aggresomal markers such as Hsp70 and ubiquitin in the core of the CBs and vimentin and proteasome subunit around the CBs. Ubiquitin-conjugating enzyme (E2) was also found in the CBs. In tubulovesicular structures surrounding the CBs, lysosomal markers were detected but an endoplasmic reticulum retention signal (KDEL) was not. Moreover, proteins located in each subcellular compartment, including the cytosol, mitochondria, and nucleus, were detected in the CBs. Signals for cytochrome oxidase I (COXI) coded on mitochondrial DNA were also found in the CBs. Quantitative analysis of labeling density showed that all proteins examined were concentrated in the CBs to some extent. These results show that the CBs have some aggresomal features, suggesting that they are not a synthetic site as proposed previously but a degradation site where unnecessary DNA, RNA, and proteins are digested.
I
On the other hand, it has been reported that when the amount of misfolded protein in a cell exceeds the capacity for degradation, the misfolded proteins accumulate in the cytoplasm to form aggresomes (Johnston et al. 1998). Overexpression of cytosolic protein chimera also causes the formation of aggresomes (García-Mata et al. 1999). Recent studies have shown that a diverse array of human diseases, including amyloidosis and neurodegenerative disorders, are caused by the accumulation of misfolded proteins due to an impaired degradation system (Johnston et al. 2000; Junn et al. 2002; McNaught et al. 2002; Namekata et al. 2002; Riley et al. 2002; Ryan et al. 2002). Thus, during their long evolution process, cells have acquired a mechanism in which cells assemble misfolded or unnecessary proteins to small aggregates and transport to aggresomes where abnormal proteins are degraded by the ubiquitin proteasome system or eventually by autophagy (see review Kopito and Sitia 2000; Kopito 2000; Garcia-Mata et al. 2002; Wójcik and DeMartino 2003). The aggresome pathway functions as a quality control of proteins (Kopito and Sitia 2000).
As the CBs appear in the vicinity of the Golgi apparatus and are membrane-free inclusions containing many proteins, the CBs have a very similar profile to the aggresomes. To elucidate which types of proteins are contained in the CBs, in the present work we studied the localization of lactate dehydrogenase (LDH) and enolase as cytoplasmic protein, PHGPx and ATP synthase subunit α (F1α) and subunit β (F1β) as the mitochondrial proteins coded on nuclear DNA, cytochrome oxidase subunit I (COXI) as a mitochondrial protein coded on mitochondrial DNA, and histone H2B, acetylated histone H2B, and acetylated histone H3 as nuclear proteins using a quantitative immunoelectron microscopic method. Furthermore, we investigated the localization of aggresomal markers, including chaperones, ubiquitin, ubiquitin-conjugating enzyme (E2), and proteasome subunits in the CBs, and of lysosomal markers such as cathepsins, lysosome-associated membrane protein 1 (LAMP1), and leucine aminopeptidase (LAP) in vesicles surrounding the CBs. As we found that the CBs contained the proteins of all the subcellular compartments examined as well as several aggresomal markers and the vesicles surrounding the CBs included lysososmal markers, we proposed that the CBs are a site of degradation rather than synthesis.
Materials and Methods
Animals
Nine-week-old male Wistar albino rats, weighing 200-250 g, were fed the appropriate standard diets for each animal type and water ad libitum until used. The animal experiments were performed in accordance with the Guidance for Animal Experiments, University of Yamanashi.
Antibodies
Rabbit anti-ubiquitin antibody was prepared as described (Hershko et al. 1982). Briefly, 5 mg of bovine erythrocyte ubiquitin (Sigma-Aldrich; St Louis, MO) was conjugated with 8 mg of limpet hemocyanin (Sigma-Aldrich) using glutaraldehyde. The conjugates were mixed with 2% SDS and heated for 5 min. After cooling in ice water, SDS was removed by aluminum chloride. The conjugates were then emulsified with the same volume of Freund's complete adjuvant. The emulsion containing 400 μg of ubiquitin was injected four times at intervals of 2 weeks into the back of Japanese white rabbits. Two weeks after the last injection, blood was collected and the immunoreactivity was checked with ubiquitin. Ubiquitin-specific antibody was purified by affinity chromatography using ubiquitin-coupled Sepharose. Antibodies to lysosomal proteins were described previously: rabbit anti-rat cathepsin H (Yokota and Kato 1987); rabbit anti-cathepsin B, D, and L (Yokota et al. 1985, 1988; Yokota and Kato 1987); rabbit anti-PHGPx (Haraguchi et al. 2003); rabbit anti-LAMP1 (Akasaki et al. 1990); and rabbit anti-LAP antibodies (Haraguchi and Yokota 2002). Rabbit anti-LDH was a gift from Dr. Ohsumi (Department of Life Science, Himeji Institute of Technology, Hyogo, Japan). Mouse anti-Hsp70 antibody and rabbit anti-enolase antibody were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); mouse anti-F1α and F1β antibodies, mouse anti-COXI, and Alexa Fluor 546 conjugated goat anti-rabbit IgG were from Molecular Probes (Eugene, OR); mouse anti-ER (endoplasmic reticulum) retention signal (KDEL) and rabbit anti-proteasome activator PA700 and PA28α antibodies from Calbiochem-Novabiochem (San Diego, CA); rabbit anti-histone H2B polyclonal antibody was from Chemicon International (Temecula, CA); mouse anti-vimentin antibody was from Sigma-Aldrich; mouse anti-20S proteasome subunit (p52) antibody was from Progen Biotechnik Gmbh (Heidelberg, Germany); goat polyclonal antibody to ubiquitin-conjugating enzyme E2 (UBC3B) was from Abcam Limited (Cambridgeshire, UK); mouse anti-actin antibody was from ICN Pharmaceuticals (Costa Mesa, CA); rabbit anti-bovine DNase I was from Upstate Biotechnology Inc. (Lake Placid, NY); and rabbit anti-bovine RNase A was from Nordic Immunological Laboratories (Tilburg, The Netherlands).
Tissue Preparation
Testes were dissected out from rats and sliced in ice-cold fixative consisting of 4% paraformaldehyde, 0.1-0.2% glutaraldehyde, 0.01% CaCl2, and 0.2 M Hepes-KOH buffer (pH 7.4). For immunoelectron microscopy, the tissue slices were fixed in the same fixative containing 0.2% glutaraldehyde for 1 hr at 4C and then cut into small tissue blocks. The tissue blocks were dehydrated in a graded ethanol series and embedded in LR White resin at −20C. For immunofluorescence microscopy, the tissue was fixed with the same fixative containing 0.1% glutaraldehyde.
Post-embedding Immunoelectron Microscopy
Thin sections of rat testis embedded in LR White were cut with a diamond knife equipped with a Reichert Ultracut R (Leica; Vienna, Austria), mounted on nickel grids, and incubated overnight with the primary antibodies against lysosomal proteins (2 μg/ml), PHGPx (X1,000), vimentin (X200), Hsp70 (X1000), F1α and F1β (X200), COXI (2.5 μg/ml), 20S proteasome subunit (p52) (X5), KDEL (X4000), histone H2B (X500), proteasome activators PA700 and PA28α (X40), enolase (X200), LDH (X500), actin (X1000), UBC3B (2 μg/ml), RNase (X2000), and DNase (X1000) antibodies at 4C. After washing, sections were treated with rabbit anti-mouse IgG (X2000) or anti-goat IgG (X2000), depending on the primary antibody. Finally, the antigens were visualized using protein A-gold probes 15 nm in diameter. For the immunocytochemical control experiment, the incubation of sections with the primary antibody was omitted, followed by the secondary antibody or by protein A-gold probe. Sections were stained with 2% uranyl acetate for 10 min and lead citrate for 30 sec and examined with a Hitachi H7500 electron microscope (Hitachi; Tokyo, Japan) at an acceleration voltage of 80 kV.
Quantitative Analysis of the Labeling Density in the CBs and Cytoplasmic Matrix
After the immunogold staining for 22 antigens with a 15-nm protein A-gold probe, 10 electron micrographs of spermatids in the developing steps 3-6 were taken for each antigen at X15,000 magnification and enlarged fourfold. The steps of spermatids were determined according to the morphological criteria of Russell et al. (1990). For analysis of gold labeling density, the areas of the CBs (excluding their clear spaces) and cytoplasmic matrix of spermatids were estimated using a digitizer tablet and SigmaScan software (Jan-del Scientific; San Rafael, CA) attached to a computer. For lysosomal enzymes, the areas of the CBs including surrounding vesicles were estimated. Gold particles located in the estimated areas were counted. The labeling density was expressed as gold particles/μm2 for each compartment. Background labeling density was measured for section area using the immunocytochemical controls in which incubation with the primary antibodies was omitted.
Routine Electron Microscopy
Tissue slices of rat testis were fixed in 2% glutaraldehyde for 1 hr at 4C and cut into small blocks. After a brief wash in phosphate-buffered saline (PBS), the tissue blocks were post-fixed in 1% reduced osmium tetroxide for 1 hr at room temperature. The tissue blocks were dehydrated in a graded ethanol series and embedded in Epon 812. Thin sections were stained with lead citrate and examined with the same electron microscope described above.
Results
Routine Electron Microscopy
In early pachytene spermatocytes at stages IV to VI, the CBs appeared in the juxtanuclear cytoplasm and were composed of loose aggregates of dense material, being accompanied by many small vesicles (Figure 1A). Solitary dense materials were also observed in the cytoplasm (Figure 1A, large arrow). In later pachytene at stages VIII to × and in step 1 spermatids, the core of CBs became compact and homogeneous (Figure 1B). Many tubules and vesicles enclosed the CBs, and some small vesicles invaded into the core region. These tubulovesicular structures were not decorated with ribosomes. In the spermatocytes, after step 10, the CBs moved to the cytoplasm around the neck and decreased in size. In many cases, multivesicular bodies accompanied the CBs.
Immunoelectron Microscopy
Localization of ER and Lysosomal Markers in the Compartments Surrounding the CBs. The ER retention signal (KDEL) was present in the lumen of ER (Figure 2A) but not in the tubulovesicular structures. LAMP1 was detected on the membrane of tubulovesicular structures surrounding the CBs but not in the core of the CBs (Figure 2B), strongly suggesting the lysosomal nature of the tulubovesiclular structures. Lysosomal enzymes, LAP, cathepsin D, and cathepsin H were present in these structures (Figures 3A-3C). Cathepsins B and L were also detected in the tubulovesicular structures.
Localization of Aggresomal Markers in the CBs. The CBs were heavily stained for ubiquitin (Figure 4A). The gold particles were present in the core. Strong ubiquitin staining of the CBs was noted in spermatids of steps 2-6. The gold signals for 20S proteasome sub-unit p52 were closely associated with the CB (Figure 4B), and weak signals were present throughout the cytoplasm. Strong signal for ubiquitin-conjugating enzyme E2 (UBC3B) was detected in the core of the CBs (Figure 4C). Proteasome activators PA700 (data not shown) and PA28α were also localized to the surface of the CB core as well as the cytoplasm (Figure 4D). The CBs were strongly stained for Hsp70 (Figure 4E). The staining intensity tended to decrease as the differentiation progressed. The intermediate filament protein vimentin was present around the CB (Figure 4F).
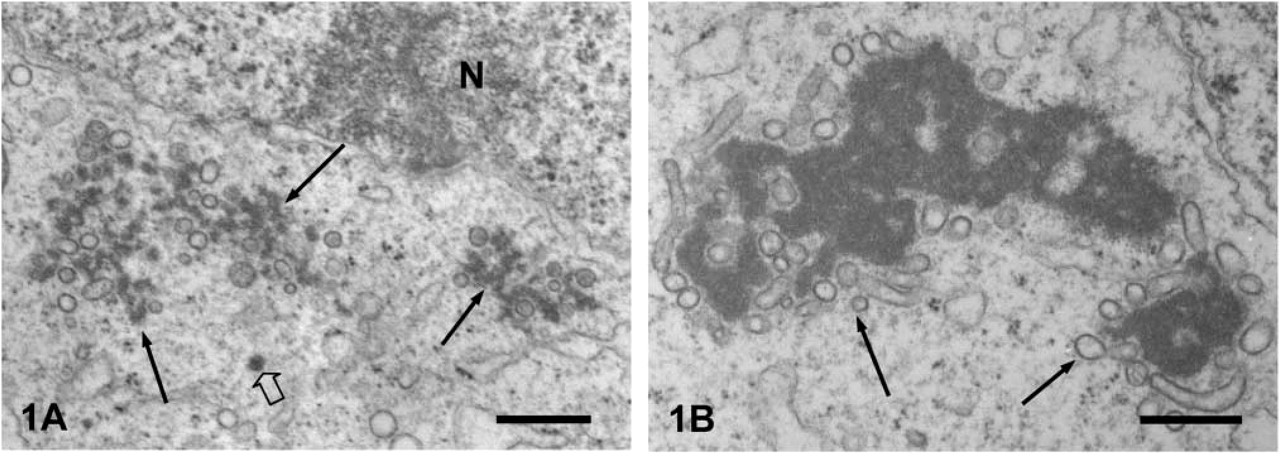
Routine EM of an early pachytene spermatocyte.
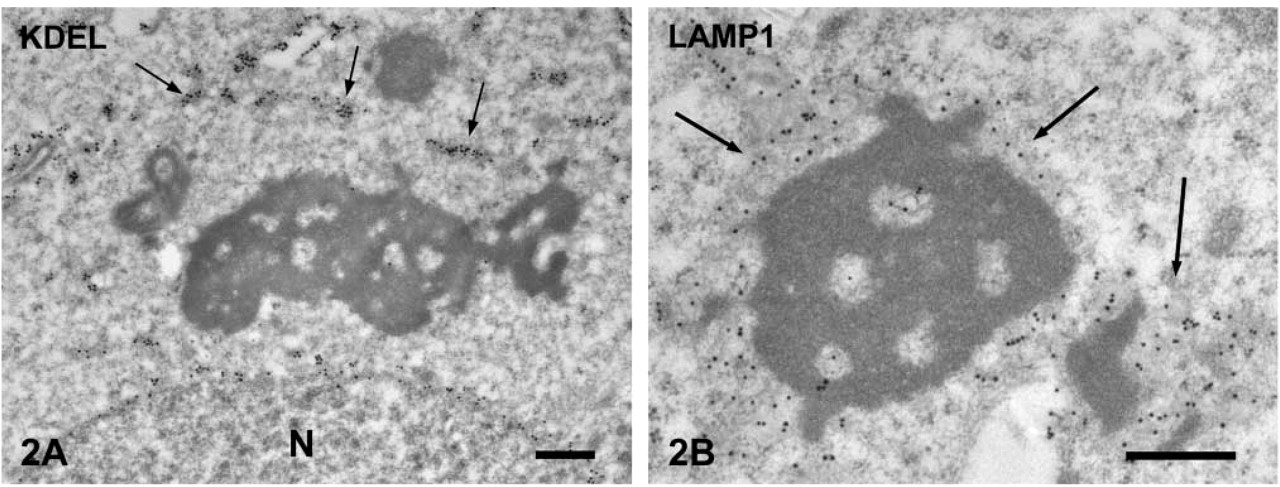
Immunogold staining of ER retention signal (KDEL) in spermatids.
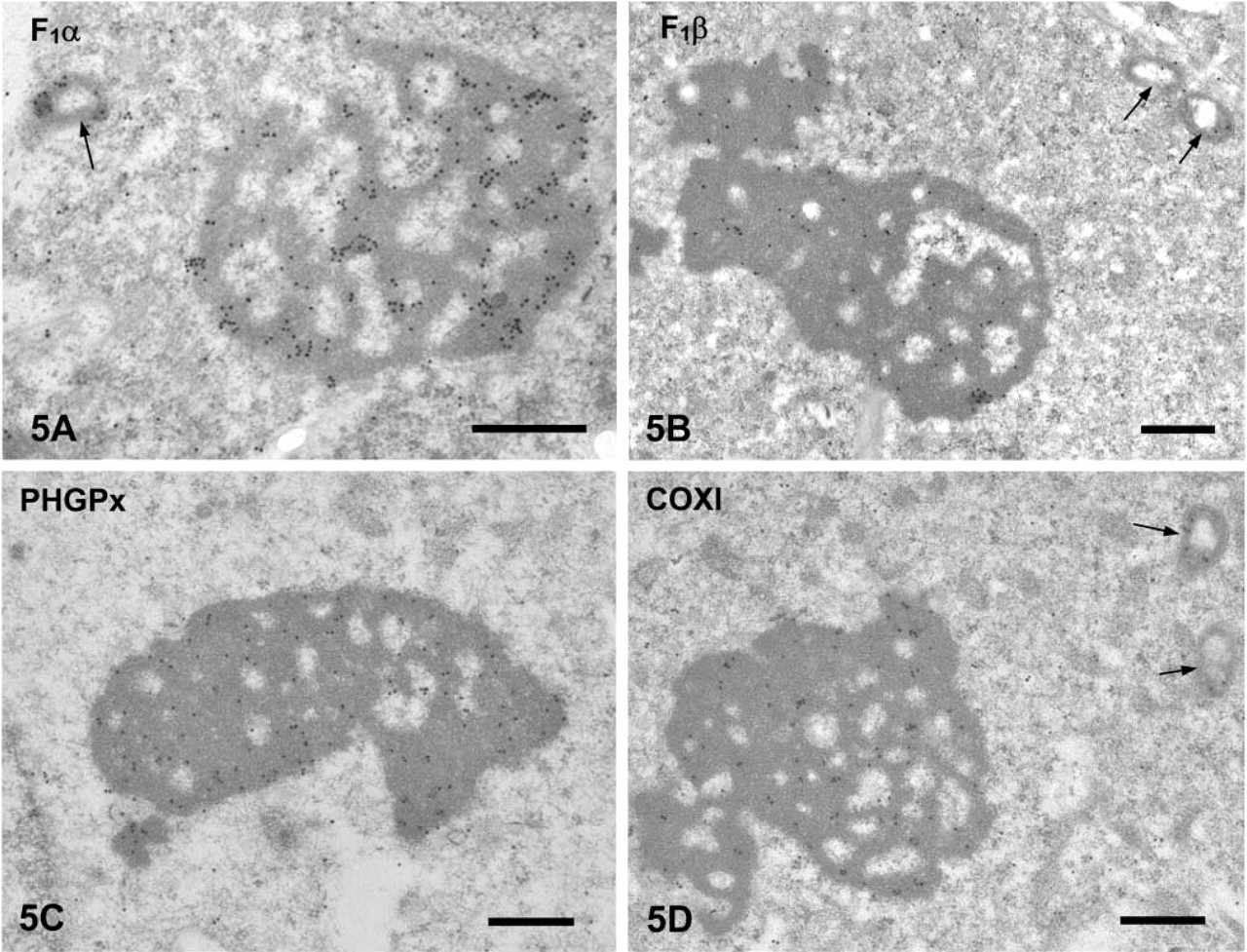
Localization of Other Proteins in the CBs. Nuclear DNA-coded Mitochondrial Proteins. ATP synthase subunits α and β were detected in the core of the CB as well as in the mitochondria (Figures 5A and 5B). As the differentiation of spermatids progressed, the labeling intensity in the CBs decreased (data not shown). Heavy signals for PHGPx were found in the CB core (Figure 5C) as described previously (Haraguchi et al. 2003). Their intensity also decreased as the differentiation of spermatids progressed.
Mitochondrial DNA-coded Protein. Cytochrome oxidase subunit I (COXI) was examined. It was detected in the CBs as well as in the intermembrane space of mitochondria (Figure 5D). The labeling density decreased after step 9.
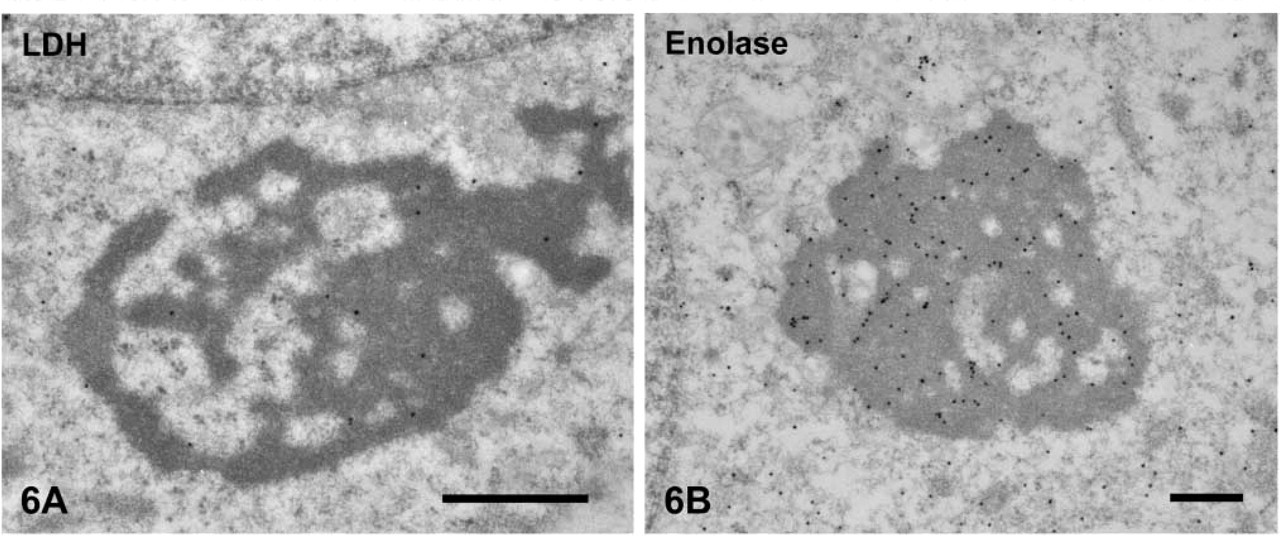
Cytosolic Protein. Lactate dehydrogenase (LDH) was stained in the CBs (Figure 6A). Enolase was detected in the core of the CBs from pachytene spermatocytes to step 9 spermatids (Figure 6B). The labeling intensity for these proteins decreased as spermatids differentiated. The CBs in spermatids after step 9 were almost all negative.
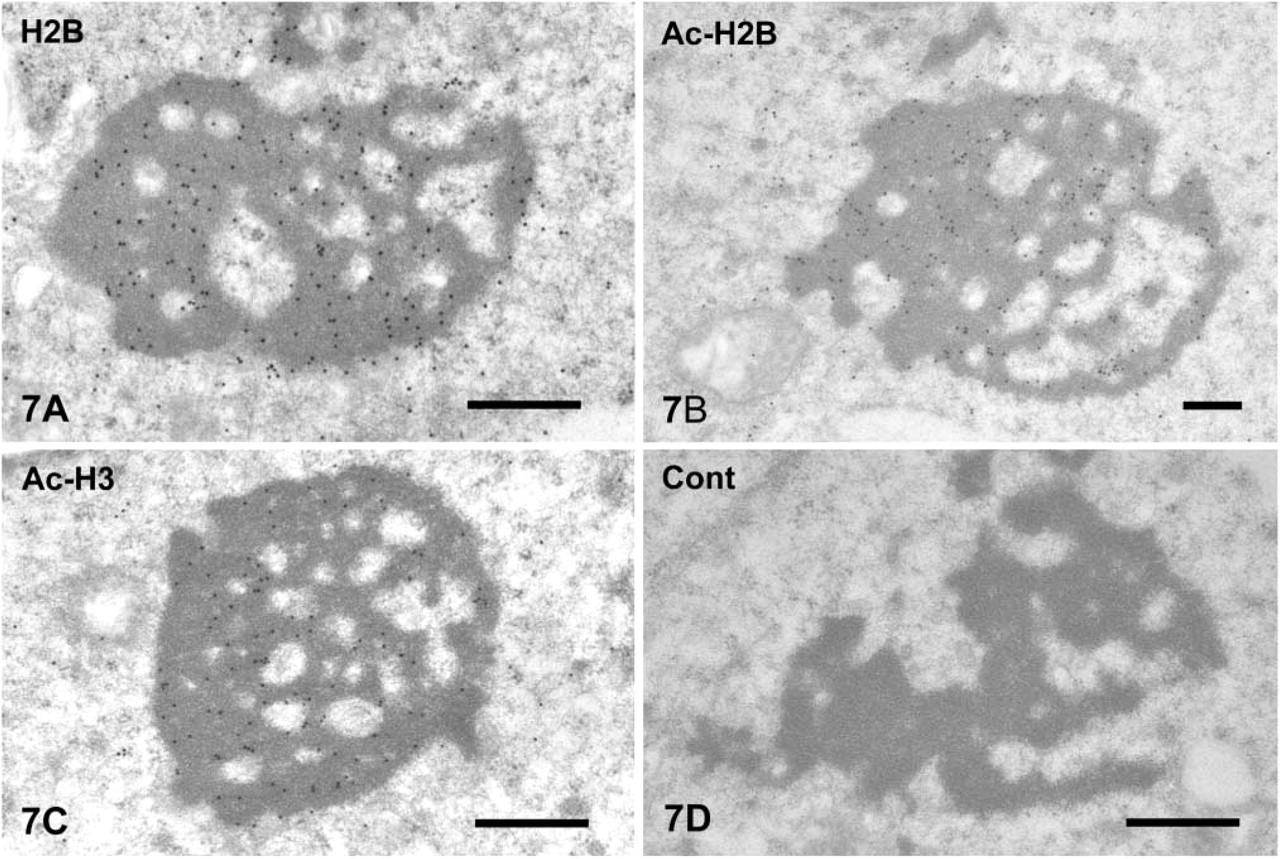
Nuclear Protein. Histone H2B, acetylated histone H2B, and acetylated histone H3 were detected in the CBs of spermatocytes from pachytene to spermatids of step 6 (Figures 7A-7C). The gold signals were mainly present in the core of the CBs. The staining intensity decreased as the spermatids differentiated. Nuclei of early spermatogenic cells, including spermatogonia and pachytene spermatocytes, were stained heavily for histone H2B and acetylated histone H2B but very weakly for acetylated histone H3 (data not shown). Immunocytochemical control sections that were not incubated with the primary antibodies, followed by secondary probes, showed very weak background labeling (Figure 7D).
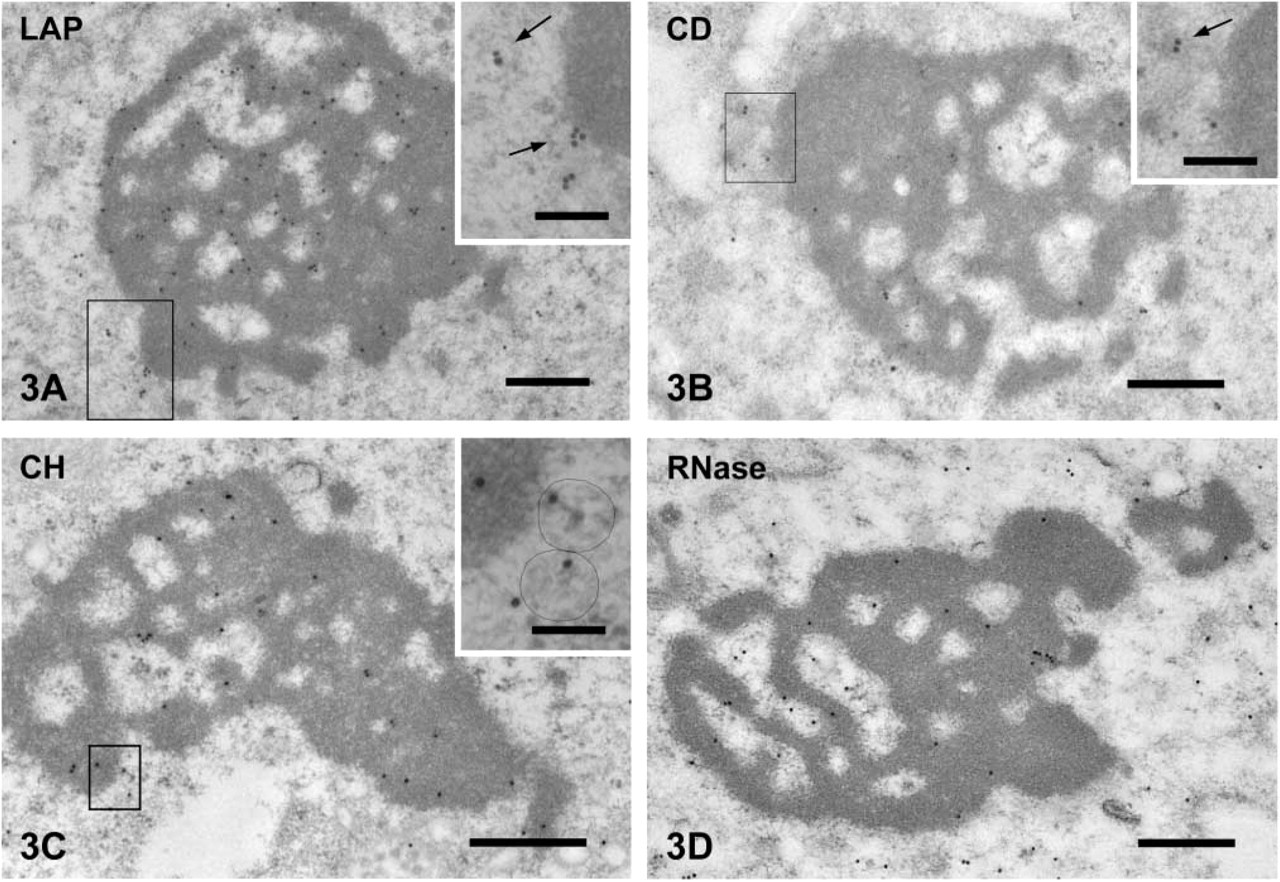
Immunogold staining of lysosomal enzymes and nucleic acids in spermatids.
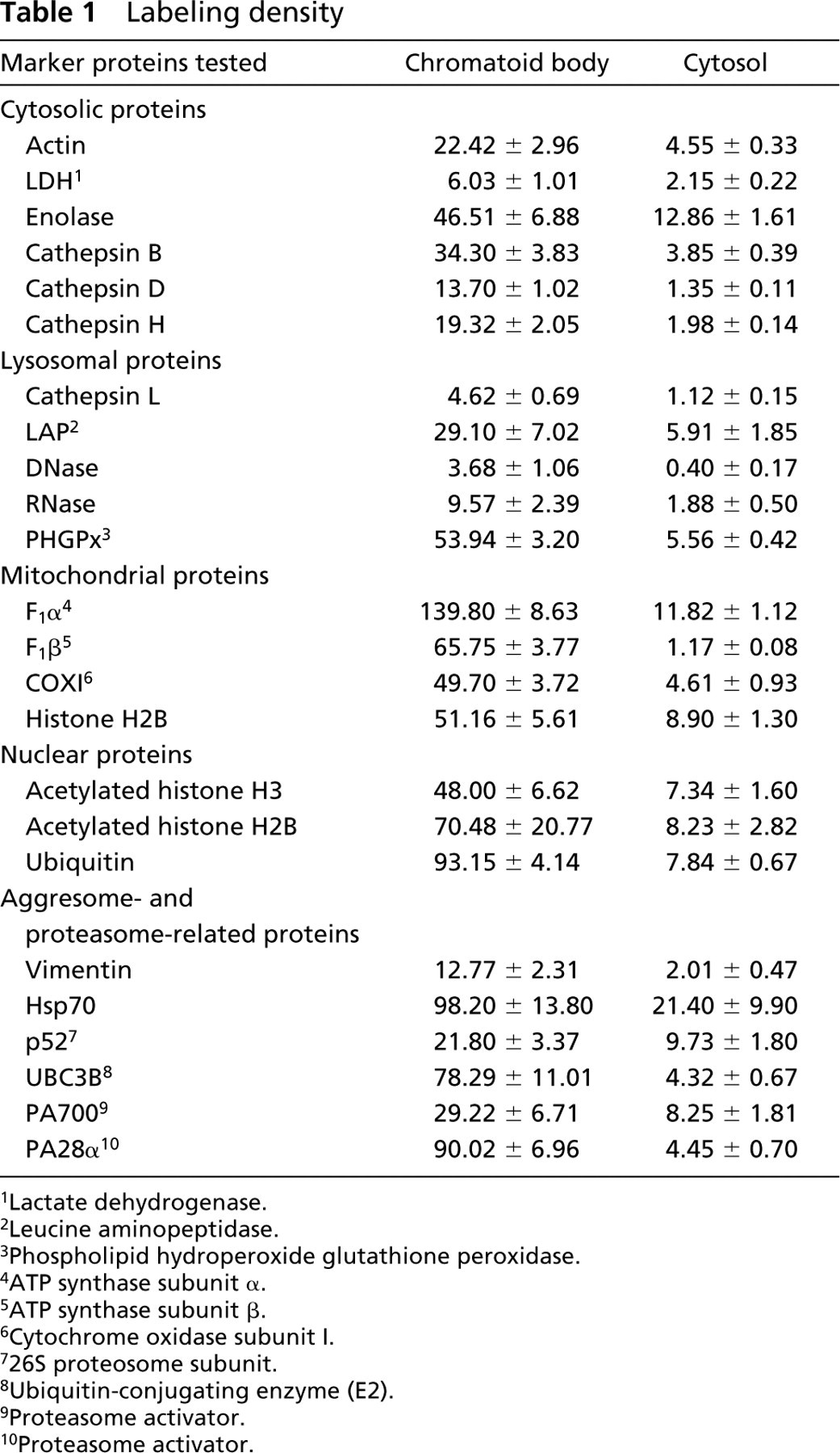
Quantitative Analysis of the Gold Labeling in the CBs
In order to know the concentration in the CBs of the examined proteins, we determined the labeling density (gold particles/μm2) for each protein in the CBs and cytoplasmic matrix of spermatids at steps 3 to 6. All the proteins examined were significantly concentrated in the CBs. The results are shown in Table 1. The values shown were subtracted by the background labeling density obtained from immunocytochemical controls. The labeling density for cytosolic proteins such as actin, LDH, and enolase in the CBs was 3.6- to 9.7-fold higher than that in the cytoplasmic matrix. The cytoplasmic labeling of lysosomal and mitochondrial proteins showing background noise of the labeling was significantly lower than that in the CBs. The labeling density for nuclear proteins, histone H2B, and acetylated histone H3 was ~6-fold higher in the CBs than in the cytoplasmic matrix. The labeling density for aggresomal marker proteins and proteasome-related proteins was also higher in the CBs than in the cytoplasmic matrix.
Discussion
In early studies it was considered that the CBs were the site where proteins necessary for the late spermiogenic cells were synthesized, because virtually all transcription activity ceased in late spermatids (Söderström and Parvinen 1976a; Söderström 1978; Parvinen and Parvinen 1979; Saunders et al. 1992). Ribonucleoproteins, mRNA, and mRNA-binding proteins (p48/52) were detected in the CBs (Biggiogera et al. 1990; Saunders et al. 1992; Oko et al. 1996). Also, polysome-like structures were found in isolated CBs (Figueroa and Burzio 1998). These findings support the idea that mRNA molecules transcribed before the occurrence of nuclear condensation should be stored in the CBs until they are required. On the other hand, it was reported that mRNAs of transition protein 1 and protamine 1 and poly (A)+ RNA were not present in the CBs but scattered in the cytoplasm (Morales et al. 1991; Morales and Hecht 1994). Recent studies suggest a potential connection between nuage and the micro-RNA and/or RNAi pathway in Drosophila and Xenopus oocytes (Findley et al. 2003; Bilinski et al. 2004). Also, a possibility was demonstrated that the CBs are involved in intercellular transport of Golgi-derived haploid gene products between early spermatids through the cytoplasmic bridge in rat (Ventelä et al. 2003). Thus, the role(s) of the CBs are still controversial.
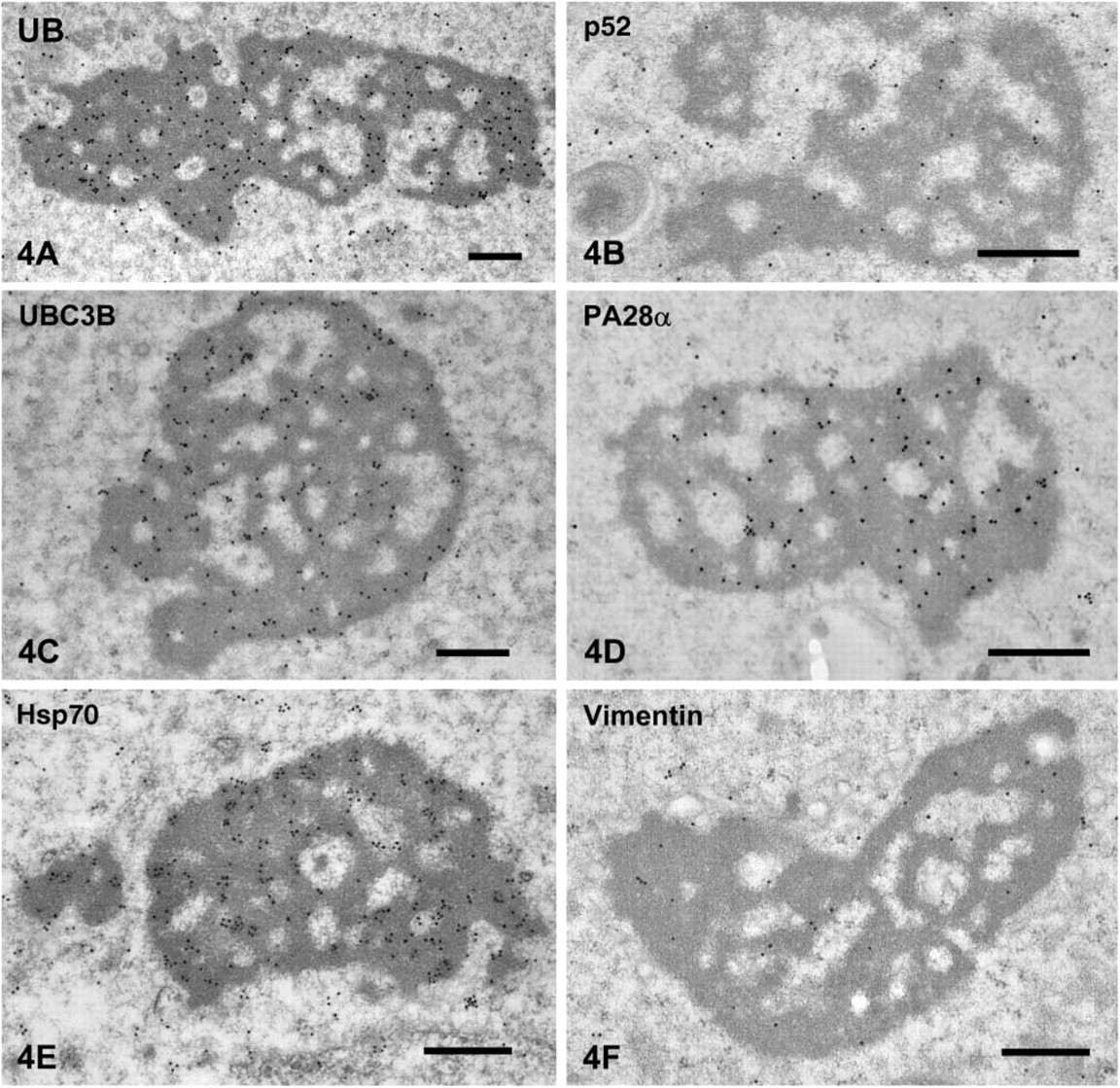
Immunogold staining of aggresomal markers in CBs of spermatids.
Immunogold staining of mitochondrial enzymes in CBs of spermatids.
Immunogold staining of cytosolic proteins.
Immunogold staining of nuclear proteins.
We reported previously that PHGPx signals were observed in the nuclear material and in the intermitochondrial cement, which were previously proposed as the origin of CBs (Fawcett et al. 1970; Russell and Frank 1978). Also, we have found by immunoelectron microscopy that PHGPx is contained in the CBs, although PHGPx was synthesized before its appearance in the CBs. These observations tempted us to hypothesize that PHGPx is degraded rather than synthesized in the CBs. In this study, we detected three other mitochondrial proteins in addition to PHGPx in the core of the CBs, of which one (COXI) was encoded on the mitochondrial DNA and two (F1α and F1β) on the nuclear DNA. Mitochondria have their own protein synthesis system within the compartment, and some of the codons used in mitochondria differ from those used in the nucleus. Thus, if proteins whose genes are on the mitochondrial DNA are synthesized in the CBs, the amino acid sequences of synthesized proteins in the CBs should be different from those in the mitochondria. The COXI gene contains 17 TGA codons used as a stop codon in the universal codon system; therefore, the COXI protein is unable to be synthesized in the CBs. This strongly suggests that the CBs are not a site for protein synthesis.
The present study also confirmed the earlier findings showing that the CBs had no limiting membrane and were surrounded by small tubules and vesicles (Sud 1961; Fawcett et al. 1970; Susi and Clermont 1970; Russell and Frank 1978; Anton 1983; Thorne-Tjomsland et al. 1988). Furthermore, in the present study we showed that these tubules and vesicles contained LAMP1 and lysosomal proteinases as well as DNase and RNase. Enzyme histochemical studies showed that acid phosphatase and NADPase activities were present in these tubulovesicular structures (Anton 1983; Thorne-Tjomsland et al. 1988). Various proteins, including histone H4, actin, cytochrome c, and basic proteins, have been detected in the core of the CBs (Krimer and Esponda 1980; Walt and Armbruster 1984; Hess et al. 1993; Werner and Werner 1995). These results suggest that the CBs have an intracellular garbage-like function.
Labeling density
Lactate dehydrogenase.
Leucine aminopeptidase.
Phospholipid hydroperoxide glutathione peroxidase.
ATP synthase subunit α.
ATP synthase subunit β.
Cytochrome oxidase subunit I.
26S proteosome subunit.
Ubiquitin-conjugating enzyme (E2).
Proteasome activator.
Proteasome activator.
Recently, it has been shown that cells have an aggresomal pathway by which aggregates of misfolded proteins or mutated proteins are transported to the area around the microtubule organizing center (MTOC) where they form large aggresomes (Johnston et al. 1998; García-Mata et al. 1999; Kopito 2000; Garcia-Mata et al. 2002). Typical features of aggresomes are as follows: they locate in the pericentriolar region, contain ubiquitinated proteins, chaperones such as Hsp70 and Hsp40, and abnormal proteins, and are surrounded by vimentin filament and proteasomes. To examine whether the CBs have these features, we tried to stain these antigens. Strong signals for ubiquitin were noted in the CBs. The ubiquitin molecules present in the CBs are thought to be conjugated to proteins and function as a targeting signal for proteasome degradation. This is supported by the presence of the ubiquitin-conjugating enzyme E2 in the core of the CBs. The CBs were heavily stained for Hsp70, suggesting their binding to proteins or the accumulation of Hsp70 to be degraded. Signals for vimentin and the 20S proteasome subunit were detected around the CBs. Moreover, in pachytene spermatocytes, the CBs were frequently observed in the vicinity of the Golgi apparatus (Tang et al. 1982; Thorne-Tjomsland et al. 1988) and later in differentiation, they moved to the caudal pole of the nucleus where the centrioles are located. These results strongly suggest that the CBs function as aggresomes. The aggresomes are essentially the site of degradation, and the cytoplasmic proteolytic apparatus, the proteasome, is thought to be the main machinery for gathering proteins to the aggresomes.
For the degradation of proteins sequestrated to the aggresome, an autophagic pathway has been proposed (Kopito 2000). In the autophagic process, macroautophagy and microautophagy have been recognized. Macroautophagy is induced by amino acid deprivation in perfused rat liver (Mortimore and Schworer 1977). In microautophagy, lysosomal compartments incorporate the cytoplasm and specific proteins (de Duve and Wattiaux 1966). The autophagic vacuoles are rarely observed in spermatogenic cells, and those containing CBs are hardly encountered, suggesting that aggregated proteins in the CBs are not degraded through the macroautophagic pathway. The present study showed that the CBs were closely surrounded by the tubulovesicular structures that had several lysosomal markers. It is likely that microautophagy occurs between the CBs and these vesicles. It is not clear whether the vesicles take up in bulk the CBs or directly incorporate proteins through a chaperone-mediated process (Dice 1990). The proportion of proteasomes associated with the centrosome is only 1% in unstressed cells (Fabunmi et al. 2000). This amount of proteasomes might be enough to degrade ubiquitinated proteins used in cell-cycle control but not enough for large amounts of aggregated proteins in the CBs. Thus, the CBs may require the autophagic process in addition to the ubiquitin-proteasome system.
Although any conclusion concerning the function of the CBs must wait until the CBs can be isolated and their composition analyzed, we would like to speculate here the possible role of CBs. During spermatogenesis, the CBs appear in the early pachytene stages, maximize their sizes in the spermatids of steps 7-8, and disappear from those of steps 16-18. In the early stages, mitochondria form a clustered structure with the intermitochondrial cement and later dramatically change to the condensed form. This structural change is thought to make mitochondria more active for ATP production. After this process, mitochondria separate, and some of them move to the flagellum at step 7 and finally wind around flagella at step 12. In the present study, immunocytochemical localization of three mitochondrial proteins was confirmed in the CBs. One of the proteins is coded on mitochondrial DNA, suggesting that at least one material contained in the CBs is derived from the mitochondria. It might originate from mitochondria dismantled by means of an unknown mechanism, and the mitochondrial proteins are possibly transported to the CBs for degradation.
On the other hand, somatic histones are replaced with transition proteins beginning at step 9 of spermatogenesis. At step 13, the transition proteins are completely replaced by protamines (Kistler et al. 1996). These replaced nucleoproteins also might be degraded in the CBs within a limited period. We observed nucleoproteins such as histone H2B, acetylated histone H2B, and acetylated histone H3 on the CBs. Thus, the CBs contain specific proteins that became unnecessary during definitive periods of spermiogenesis. Quantitative analysis of labeling showed that the labeling density of all proteins examined was higher in the CBs than in the cytoplasmic matrix, even if it contained nonspecific background labeling. It is likely that of these proteins, lysosomal enzymes and aggresome- and proteasome related proteins are concerned with the function of the CBs. However, cytosolic proteins, mitochondrial proteins, and nuclear proteins examined are gathered to the CBs to be degraded, because the concentration of the latter proteins must be higher in their original locations than in the CBs if the CBs are synthetic sites of these proteins. From these observations, we propose that the CBs might be involved in the degradation of proteins released from the mitochondria and nucleus during this period to control the quality of these organelles and to adjust the function specific for spermatozoon.
Footnotes
Acknowledgments
This work was supported in part by a grant-in-aid (14580693) and the Center of Excellence (COE) program from the Ministry of Education, Science, Culture and Sport (Japan) to SY.