
Abstract
Several types of cytotoxic insults disrupt endoplasmic reticulum (ER) homeostasis, cause ER stress, and activate the unfolded protein response (UPR). The role of ER stress and UPR activation in hypersensitivity pneumonitis (HP) has not been described. HP is an immune-mediated interstitial lung disease that develops following repeated inhalation of various antigens in susceptible and sensitized individuals. The aim of this study was to investigate the lung expression and localization of the key effectors of the UPR, BiP/GRP78, CHOP, and sXBP1 in HP patients compared with control subjects. Furthermore, we developed a mouse model of HP to determine whether ER stress and UPR pathway are induced during this pathogenesis. In human control lungs, we observed weak positive staining for BiP in some epithelial cells and macrophages, while sXBP1 and CHOP were negative. Conversely, strong BiP, sXBP1- and CHOP-positive alveolar and bronchial epithelial, and inflammatory cells were identified in HP lungs. We also found apoptosis and autophagy markers colocalization with UPR proteins in HP lungs. Similar results were obtained in lungs from an HP mouse model. Our findings suggest that the UPR pathway is associated with the pathogenesis of HP:
Keywords
Introduction
Hypersensitivity pneumonitis (HP) is an inflammatory interstitial lung disease (ILD) resulting from an exaggerated immune response after repeated inhalation of organic or inorganic environmental antigens. HP may occur in a variety of occupational, home, and recreational environments and has a variety of names based on the source of exposure (e.g., farmer’s lung, pigeon breeder’s disease, sauna-takers disease).1,2 HP clinical behavior is heterogeneous and may present as a fibrotic and non-fibrotic disease. Fibrotic HP is associated with higher morbidity and mortality. Furthermore, the clinical features and disease behavior of idiopathic pulmonary fibrosis (IPF), the most aggressive ILD, and fibrotic HP can be indistinguishable, making it challenging to provide a precise diagnosis.3,4 IPF is characterized by repeated injury to the alveolar epithelium with the emergence of aberrant types of lung epithelial cells and abnormal tissue repair. Previous studies have reported endoplasmic reticulum (ER) stress in the alveolar epithelium in lung biopsies from patients with IPF, suggesting a pathogenic role; however, the putative participation of this process in HP has not been clearly defined.5,6
The ER is a key protein quality-control organelle that orchestrates the synthesis, folding, and structural maturation of proteins, maintaining protein homeostasis (or proteostasis). Multiple factors can cause disruptions in this organelle, altering protein translation, folding, and degradation, leading to the accumulation and aggregation of misfolded or unfolded proteins, a condition known as ER stress. 7 ER stress can trigger unfolded protein response (UPR) activation to restore proteostasis. UPR signaling is mediated by three ER transmembrane receptors or sensors: activating transcription factor 6 (ATF6), inositol requiring kinase 1 (IRE1), and double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK). 8 Another element that controls the activation of UPR signaling is the chaperone BiP/GRP78, which constitutively binds to the luminal domains of IRE1α, PERK, and ATF6 receptors and prevents their activation. However, when misfolded proteins accumulate in the ER, BiP dissociates from these UPR sensors and activates downstream cascades; within them, the synthesis and nuclear translocation of the transcription factor form sXBP1.8,9
The UPR integrates information about the intensity and duration of ER stress. Irremediable and chronic ER stress turns UPR signaling toward apoptosis. ER stress-mediated apoptosis has been associated with the activation of ER stress-specific caspases 4 and 12 and the proapoptotic transcription factor C/EBP homologous protein (CHOP). 10 Considerable progress has been achieved in the understanding of the cellular and molecular mechanisms involved in the pathogenesis of HP. However, it has not been fully elucidated whether ER stress–mediated UPR activation is involved in the development of this disease. Thus, the aim of this study was to investigate the lung expression and localization of the key UPR effectors BiP, sXBP1, and CHOP in HP patients and in the Saccharopolyspora rectivirgula–induced mouse model of HP to determine whether ER stress and the UPR pathway play a role in its pathogenesis.
Materials and Methods
Patient and Control Groups
Lung biopsies from nine adults diagnosed with fibrotic HP (mean age 56 ± 5.5 years) and three controls (mean age 57 ± 6.9 years, nonsmokers) obtained from autopsies of patients who died from non–lung-related causes were included in the study (Table 1). The diagnosis was performed by a multidisciplinary team that included pulmonologists, radiologists, and pathologists and was based on combined clinical criteria (history of exposure to antigens and serum-specific IgG, BAL lymphocytosis, high-resolution computed tomography of the chest, and histopathological features compatible with fibrotic HP). The research protocol (#4623) was approved by the Ethics Committee of the National Institute of Respiratory Diseases (INER), Mexico. Formalin-fixed and paraffin-embedded (FFPE) lung biopsies from HP patients and control subjects were used to obtain 5-μm-thick serial sections. Serial sections were stained with hematoxylin-eosin (H&E) and Masson trichrome stain and also used to perform immunohistochemistry (IHC) and immunofluorescence.
Characteristics in Control Subjects and HP Patients (Age, Race/Ethnicity, and Comorbidities).
Mouse Model of HP Induced by S. Rectivirgula
C57BL/6 mice were intranasally instilled with 50 μg of S. rectivirgula (obtained from the American Type Culture Collection, Manassas, VA; strain designation ATCC 55441) in 25 μL of sterile saline for 3 weeks (three times per week) and were euthanized 3 days after the last exposure (S. rectivirgula–exposed group, n=9 mice). The control groups received the same volume of sterile saline (control group, n=6 mice). All experiments were performed with 8- to 10-week-old male mice. All mice were provided free access to food and water and maintained under specific pathogen-free conditions. For tissue harvesting, the lungs were perfused with sterile saline from right to left ventricle of the heart. Left lungs were removed for fixation overnight in 4% paraformaldehyde, and right lungs were snap frozen in liquid nitrogen followed by storage at –80C until protein extraction. Mouse lung sections were stained with H&E stain, and histological changes were analyzed by the pathologist. The experiment was approved by The Bioethics Committee of the Faculty of Sciences from Universidad Nacional Autónoma de México (protocol approval PI_2022_23_07_Cabrera) and performed in strict accordance with approved guidelines.
Immunohistochemistry
Lung biopsies from HP patients and control subjects and mouse lungs were FFPE. Tissues from all cases were sectioned in 5-μm-thick sections. The tissue sections were deparaffinized and rehydrated using graded ethanol (100%, 80%, and 50%) followed by water for 5 min. Endogenous peroxidase activity was eliminated using 3% H2O2 in methanol for 10 min and then washed in phosphate-buffered saline (PBS) 1×. Heat-induced antigen retrieval was performed in 10-mM citrate buffer (pH 6.0) for 6 min using a microwave. Lung tissues were blocked with a universal blocking solution (HK085–5K; BioGenex, Fremont, CA) for 10 min and then incubated overnight at 4C with the following primary antibodies: anti-sXBP1 (SC-7160; Santa Cruz Biotechnology, California, USA), anti-sXBP1 (83418; Cell Signaling Technology, Danvers, MA), anti-CHOP (SC-7351; Santa Cruz Biotechnology, California, USA), anti-SQSTM1/p62 (P0068; Merck, Darmstadt, Germany), and anti-LC3B (L7543; Merck). A secondary antibody cocktail followed by horseradish peroxidase–conjugated streptavidin (HK330-5K; BioGenex) was used according to the manufacturer’s instructions. The chromogenic substrate 3-Amino-9-ethyl-carbazole (HK092-5K; BioGenex) in acetate buffer containing 0.05% H2O2 was used, and sections were counterstained with hematoxylin.
Imaging Acquisition and Analysis
From each slide, 15 nonoverlapping fields for human lung tissue (HP patients n=9 and control subjects n=3) and 9 non-overlapping fields for mouse lung tissue (S. rectivirgula–treated n=9 and saline controls n=6) were captured using 40× objective and analyzed by using QuPath bioimage analysis software (https://qupath.github.io). 11 Images were taken using the Eclipse E600 Nikon microscope (Nikon, New York, USA) equipped with a DXM1200C digital camera (Nikon, New York, USA) and with the Nis Elements 3.0 software (Nikon, New York, USA). The brightfield digital image files were uploaded, a full-image annotation was created, and QuPath’s Positive Cell Detection algorithm was performed. The wand and brush tools were then used to annotate regions of tissue to define epithelium and immune cells. The measurement table was exported for statistical analysis (Percentage of positive cells = positive cells/total nuclei × 100). The mean percentage of all fields was calculated for each tissue sample and plot.
Immunofluorescence Analysis
Tissue slides were processed equally as IHC slides and incubated with the primary antibody combinations anti-sXBP1 plus anti-p62 (SC-28359, Santa Cruz Biotechnology) and anti-CHOP plus anti-Caspase 12 (PA5-20033; Thermo Fisher Scientific, Waltham, MA). After washing, tissues were incubated with secondary antibodies Alexa 546 anti-mouse (Green), Alexa 568 anti-rabbit (Green), Alexa 647 anti-mouse (Red), or Alexa 647 anti-rabbit (Red) (Thermo Fisher Scientific). Nuclei were stained with Hoechst 33342 (Blue) Staining Dye Solution (62249, Thermo Fisher Scientific). Samples were imaged with a Confocal Laser Scanning Microscope TCS SP8 using a CS2 Plan Apochromat 63× (Leica, Wetzlar, Germany). A minimum of three representative, non-overlapping fields from lungs were evaluated per tissue section. Background noise for the fluorophore emitting at the 546, 647, and Hoechst channels was reduced by subtraction of the negative noise signal using the ImageJ program and applied to the entire images.
Immunoblot
Lung tissue was homogenized in a 20-mM Tris buffer pH 7.4, containing 150-mM NaCl, 1% Triton X-100 (Sigma-Aldrich, T8787), 10-mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitor cocktail. Then, tissue homogenate was centrifuged at 15,000 × g at 4C, and supernatant fractions were collected. Protein concentration was quantified by using the bicinchoninic acid technique (Micro BCA protein assay kit 23235, Thermo Fisher Scientific). A total of 25 µg of protein was loaded on either 8% or 13% sodium dodecyl sulfate (SDS)-polyacrylamide gels. After electrophoresis, gels were electrotransferred onto polyvinylidenedifluoride membranes (PVDF, 1620177, Bio-Rad, Hercules, CA), and then membranes were blocked with 5% nonfat dried milk in PBT (PBS with 0.05% Tween 20 [P9416, Merck]) and incubated overnight at 4C with primary antibodies diluted in an antibody diluent (003118; Thermo Fisher Scientific). The same primary antibodies used for IHC were used for incubating the membranes, except for the anti-β-actin antibody (A5441; Merck). After three washes with TBS-T, the membranes were incubated with the corresponding secondary antibody at a 1:3000 dilution in 1.5% milk in TBS-T and developed with Immobilon Western Chemiluminescent HRP substrate (WBKLS0500, Merck Millipore, Darmstadt, Germany). Image capture was performed using a ChemiDoc XRS Gel Imaging System (Bio-Rad) and analyzed using the ImageJ program.
Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Data analysis was performed with The R Foundation for Statistical Computing Package (R version 4.3.2). Variables were assessed for normality using the Shapiro-Wilk test for normality. Differences between groups were assessed with independent t-tests for variables with normal distributions and with Mann–Whitney U test for variables where normality could not be assumed. Graphs were performed using Prism version 6.0 (Graph-Pad software), and p<0.05 was considered significant.
Results
Lung Localization of the UPR Proteins BiP, sXBP1, and CHOP in HP Lungs
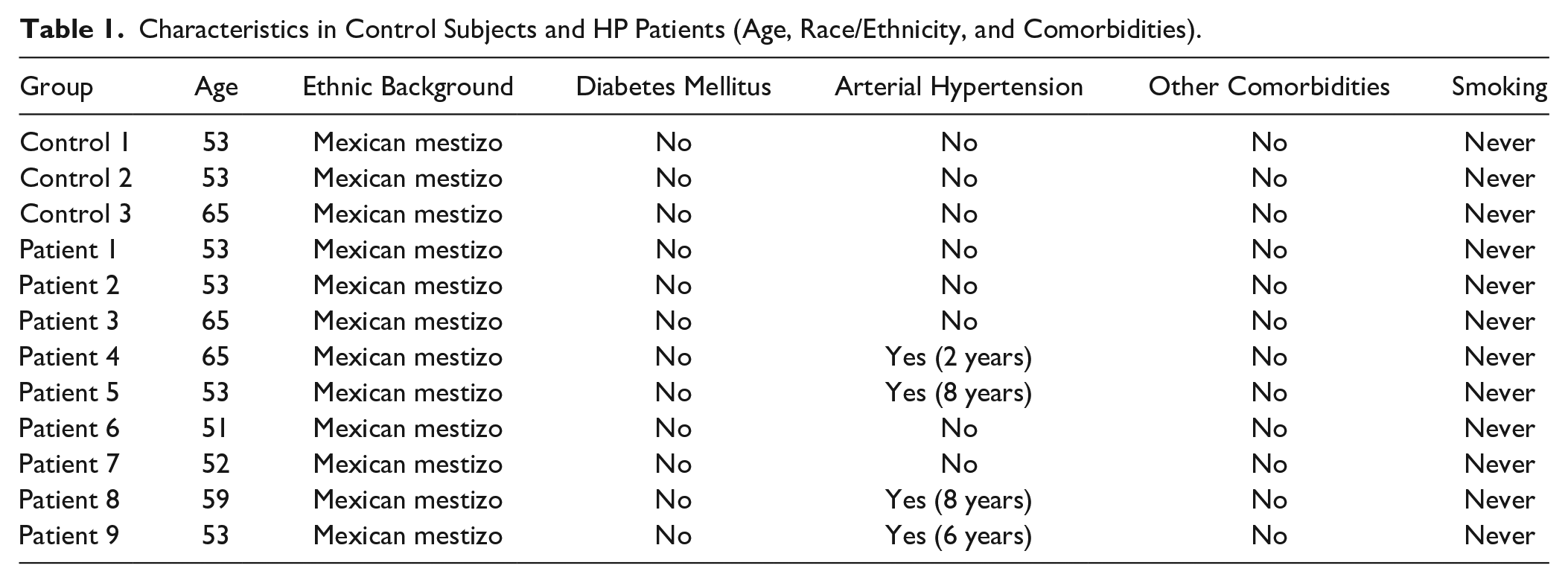
To explore whether ER stress and UPR activation were involved in the HP pathogenesis, the distribution and localization of BiP, sXBP1, and CHOP were evaluated by IHC in lung tissue from control subjects and HP patients. In lungs from control subjects, we observed very weak positive staining of BiP in the bronchial epithelium, in some free alveolar macrophages, and in some macrophages attached to the alveolar walls. In addition, some alveolar epithelial cells (AECs) were positive for BiP (Fig. 1A and Fig. A1 [arrows]). No immunoreactivity for the transcription factors sXBP1 and CHOP was observed in the control lungs (Fig. 1A and Fig. A1).
UPR proteins BiP, sXBP1, and CHOP in control and HP lungs. (A) Representative photomicrographs of BiP, sXBP1, and CHOP immunostaining in lung tissue from controls and (B) HP patients. The positive signal was observed in red. All sections were counterstained with hematoxylin (blue). Percentage of (C) BiP-positive, (D) sXBP1, and (E) CHOP epithelial and inflammatory cells in control compared to HP lungs. Data are presented as mean ± SD. Statistical significance was determined with t-test or Mann–Whitney U test (**p<0.001, ***p<0.0001). Images are at 40× magnification (Scale bars = 50 µm).
The histopathological changes in the lungs from the HP patients were characterized by interstitial infiltration of macrophages, neutrophils, and lymphocytes. We noticed aggregates of alveolar macrophages, alveolar foamy macrophages, and multinucleated giant cells. Furthermore, we observed alveolar and bronchiolar epithelial hyperplasia and centrilobular, peribronchial, and bridging fibrosis (Figs. 1B and 3A and D). Compared to control lungs, we detected significantly increased BiP-positive staining in HP lungs. Hyperplastic and cuboidal AECs close to areas of fibrosis were strongly positive for BiP. In addition, we identified intense BiP-positive staining in alveolar macrophages and in few plasma cells (Fig. 1B and C and Fig. A2).
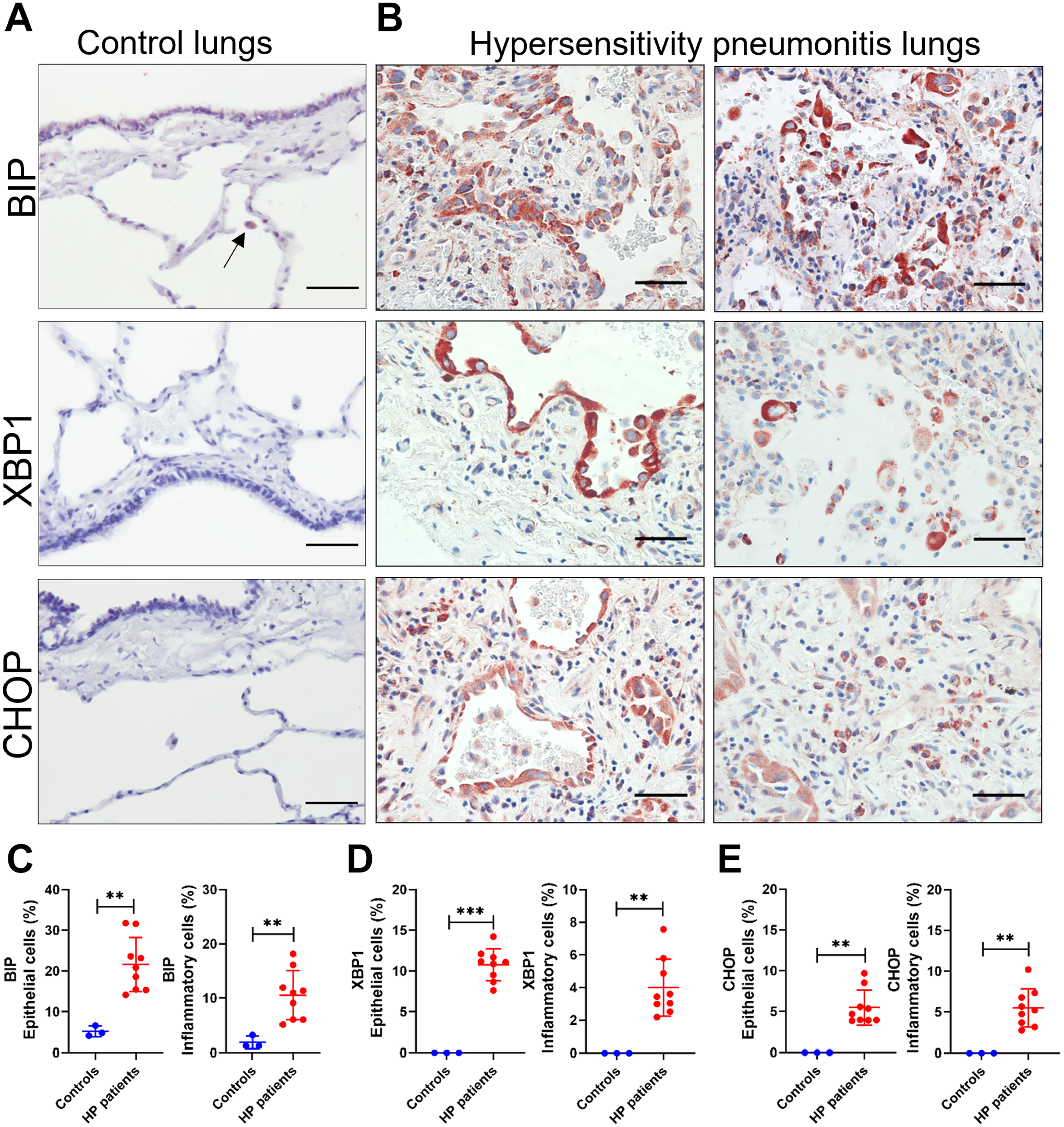
A strong sXBP1 signal was frequently observed in cuboidal AECs lining fibroblastic foci but also in bronchial epithelium in HP lungs. Macrophages showed moderate and strong immunoreactivity for sXBP1, and some neutrophils and muscle cells were also positive (Fig. 1B and D and Fig. A3). CHOP was observed in hyperplastic AECs, macrophages, neutrophils, and plasma cells around the bronchioles and in fibrotic areas centered around the small airways (Fig. 1B and E and Fig. A4). A significantly increased percentage of BiP, sXBP1, and CHOP-positive cells was found in the lungs from HP patients compared with controls, and these findings were corroborated by Western blot analysis, which revealed an increased BiP, sXBP1, and CHOP protein level in lung tissue extracts from HP patients compared to controls (Fig. 2A and B).
BiP, sXBP1, and CHOP protein level in control and HP lungs. (A) Representative BiP, sXBP1, and CHOP immunoblots using total lung tissue extracts from control and HP lungs. β-actin was used as a loading control. (B) Densitometry. Protein levels normalized to β-actin were expressed in arbitrary units. Results represent mean ± SD. Statistical significance was determined with t-test or Mann–Whitney U test (*p<0.05).
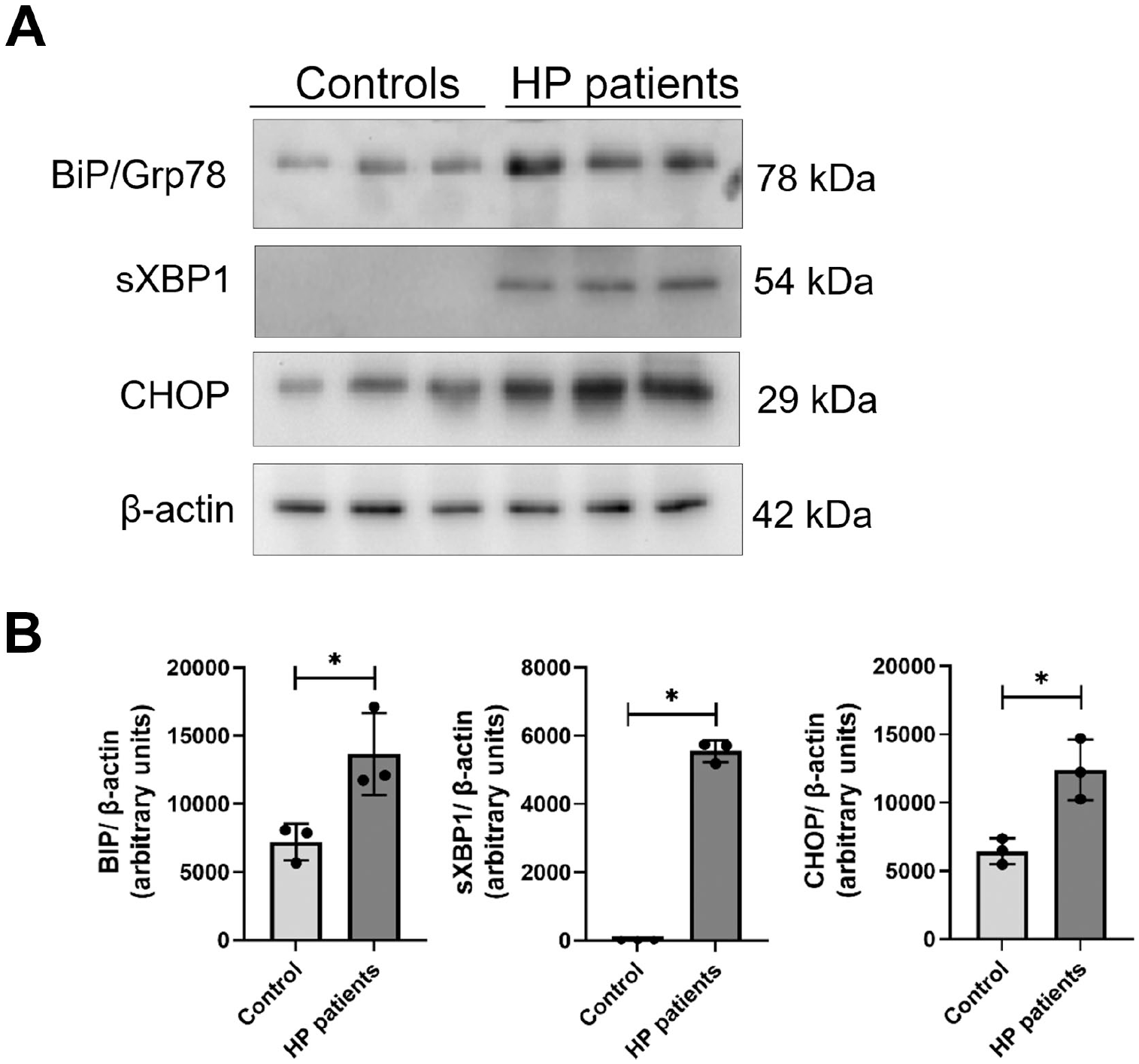
By immunofluorescence using a rabbit polyclonal anti-sXBP1 antibody combined with a mouse monoclonal anti-SPC antibody, we colocalized sXBP1 and SPC in alveolar epithelial type II cells (AECIIs) in HP lungs (Fig. 3A). Furthermore, since CHOP plays an essential role in ER stress-induced apoptosis and caspase 12 (Cas12) is localized to the ER and activated by ER stress, 10 we evaluated the localization of both proteins in HP lungs by immunofluorescence using a rabbit polyclonal anti-Cas12 antibody combined with a mouse monoclonal anti-CHOP antibody. We observed CHOP and Cas12 nuclear colocalization in bronchial epithelial cells (Fig. 3B). These results confirmed our IHC findings indicating that the UPR pathway is activated in alveolar and bronchial epithelium and suggest that CHOP- and Cas12-mediated apoptosis could occur in bronchial epithelial cells in HP lungs.
UPR protein sXBP1 colocalizes with SPC in alveolar epithelial cells and CHOP with Cas12 in bronchial epithelial and inflammatory cells in HP lungs. (A) Representative confocal fluorescence images of sXPB1 (green) and p62 (red) in HP lungs. (B) Representative confocal fluorescence images of CHOP (green) and Cas12 (red) in HP lungs. Nuclei were stained with Hoechst (magnification 63× scale bars = 50 µm).
UPR and autophagy biomarkers colocalize in the lungs of HP patients.
ER stress is also a potent trigger of autophagy, and the UPR pathway can regulate the autophagic response to alleviate stress for cell survival.7,8 We have previously shown that LC3B and SQSTM1/p62 (proteins from the autophagy machinery) are highly expressed in epithelial and inflammatory cells in the lungs of HP patients. 12 Histological changes in parenchymal tissue from HP lungs was evaluated by H&E staining (Fig. 4A), and deposition of extracellular matrix was evaluated by Masson’s trichrome staining (Fig. 4D). We use serial tissue sections to compare the localization of UPR and autophagy proteins by IHC in the same fields analyzed by H&E and Masson’s trichrome–stained sections (as indicated in Fig. 4A–F by an asterisk). We found that sXBP1, BiP, and p62 stained epithelial cells’ lining areas of dense fibrosis depicted by dotted boxes in micrographs (Fig. 4B, E, and F). Aggregates of alveolar macrophages showed positive staining for both BiP and LC3B, as depicted by dotted circles in micrographs (Fig. 4B and C).
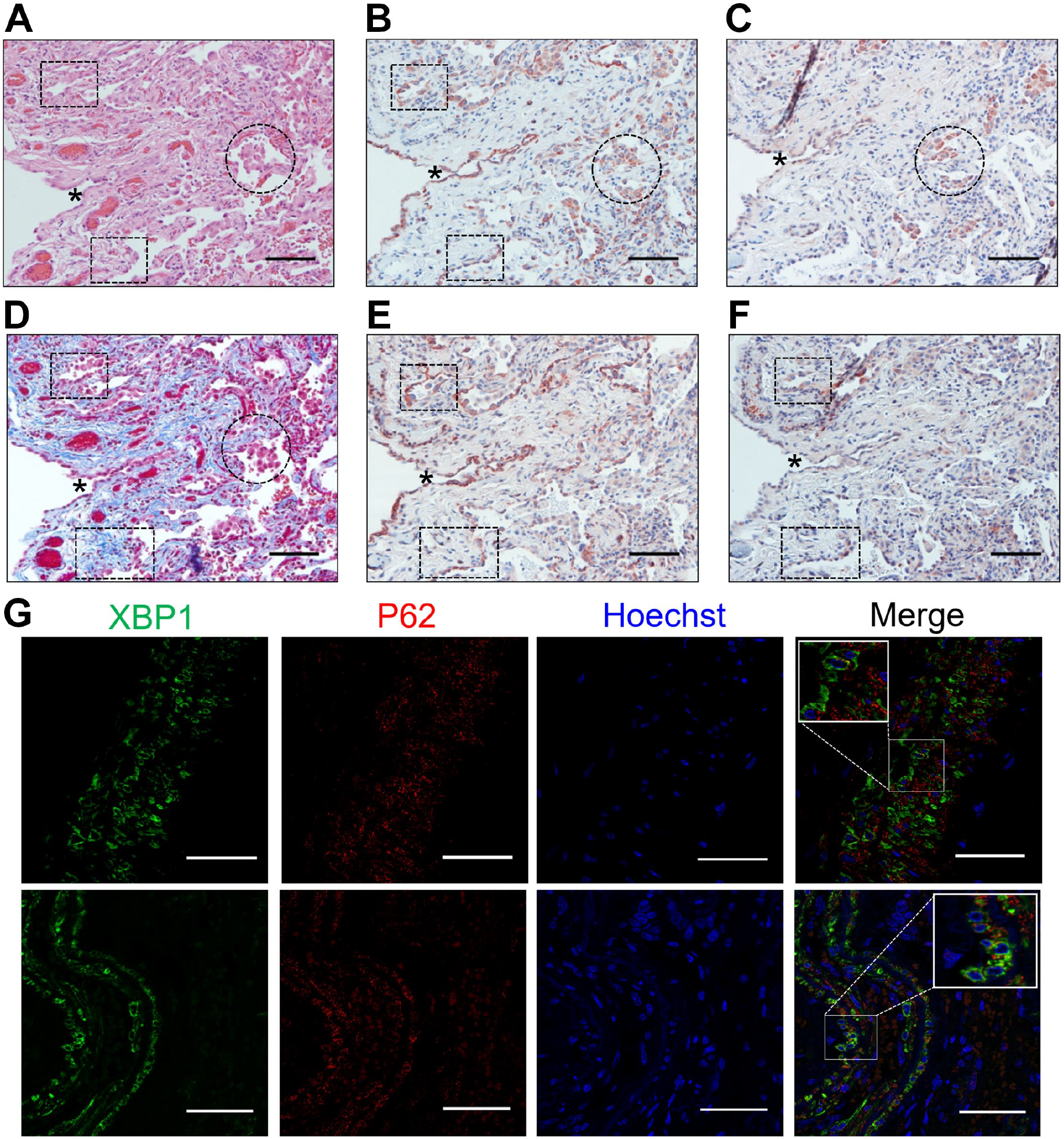
UPR and autophagy biomarkers colocalize in lungs from HP patients. (A) Representative photomicrographs of serial sections stained with hematoxylin and eosin, (B) BiP, (C) LC3B, (D) Masson’s trichrome, (E) sXBP1, and (F) p62 in lung tissue from HP patients. Asterisk and dotted circles and boxes highlight the same areas in serial sections. The positive signal was observed in red. Nuclei were counterstained with hematoxylin (blue). (G) Representative confocal fluorescence images of XPB1 (green) and p62 (red); nuclei were stained with Hoechst magnification 63× scale bars = 50 µm). Immunohistochemistry images are at 20× magnification (scale bars = 100 µm).
By immunofluorescence, using a rabbit polyclonal anti-sXBP1 antibody combined with a mouse monoclonal anti-p62 antibody, we were able to colocalize sXBP1 and p62 in epithelial cells and some endothelial and muscle cells. The sXBP1 staining pattern was diffuse in the cytoplasm, while p62 showed a granular pattern (Fig. 4G). Together, these results suggest that ER stress induction and UPR activation could regulate autophagy in macrophages, epithelial cells, and muscle cells in HP lungs.
Lung Localization of the UPR Proteins BiP, sXBP1, and CHOP in the S. rectivirgula Mouse Model of HP
Farmer’s lung disease is one of the most common types of HP, and it is caused by repeated inhalation of the gram-positive thermophile S. rectivirgula commonly found in moldy hay.1–4 We used the S. rectivirgula mouse model of HP to better elucidate whether ER stress was induced and UPR was activated in the lung after this antigen challenge. 13 H&E-stained tissues were evaluated by light microscopy to assess general morphology after S. rectivirgula exposure. Saline controls showed a normal lung morphology (Fig. 5A), while S. rectivirgula–treated mice developed severe inflammation after 3 weeks of antigen exposure. We observed intra-alveolar infiltration of neutrophils, macrophages, and lymphocytes, without evidence of giant cells (Fig. 5B and D). We also identified neutrophilic and lymphocytic infiltration of the perivascular and peribronchiolar areas and the development of bronchus-associated lymphoid tissue (BALT) in S. rectivirgula–treated mice (Fig. 5C and D). Masson’s trichrome staining was performed to analyze lung collagen deposition, and we found thickening of alveolar interstitial tissue and small fibrotic foci only in 33% of S. rectivirgula–exposed mice 3 weeks after treatment (Fig. 5E and F).
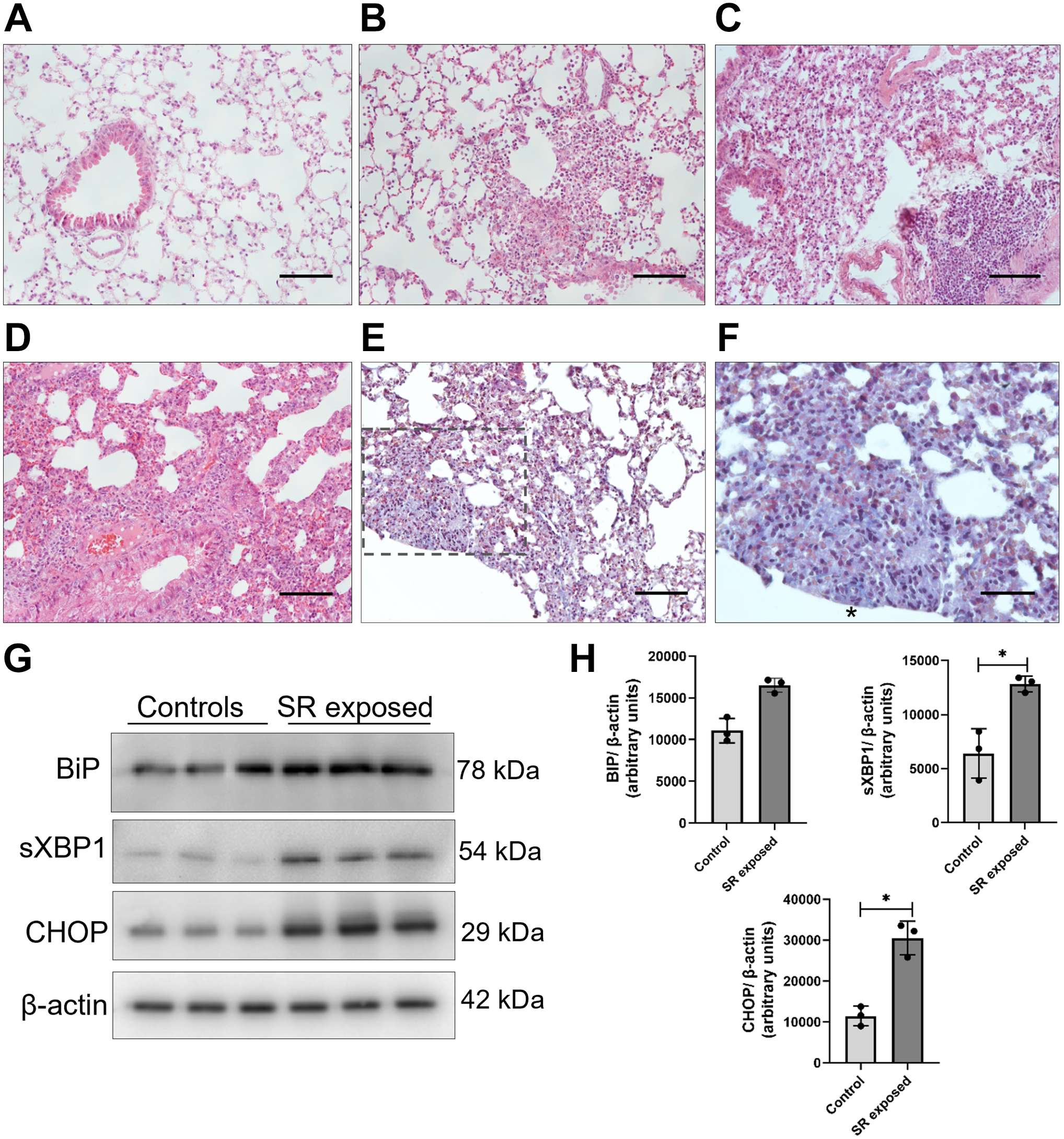
Lung histopathology and BiP, sXBP1, and CHOP protein level in Saccharopolyspora rectivirgula mouse model of HP. (A) Representative photomicrographs of control and (B, C, D) S. rectivirgula–induced HP lungs stained with hematoxylin and eosin. (E) Representative photomicrograph of Masson’s trichrome–stained S. rectivirgula–treated mouse lung. (F) Higher magnification image of the area duplicated from the black dotted box in (E) showing collagen deposition. (G) Representative BiP, sXBP1, and CHOP immunoblots using total lung tissue extracts from control and S. rectivirgula lungs. β-actin was used as a loading control. (H) Densitometry. Protein levels normalized to β-actin were expressed in arbitrary units. Results represent mean ± SD. Statistical significance was determined by t-test (*p<0.05). Photomicrographs are at 20× and 40× magnification (scale bars = 100 and 50 µm, respectively).
Then, we measured BiP, sXBP1, and CHOP protein levels in total lung tissue extracts from control and S. rectivirgula–treated mice. The sXBP1 and CHOP levels were significantly increased in S. rectivirgula lungs compared with control lungs. We did not find significant differences in the BiP protein level, possibly because it is constitutively expressed and regulated by relocalization in the ER when dissociating from the UPR transducers, although a tendency to increase was noticed (Fig. 5G and H).
In saline control lungs, BiP-positive staining in the bronchial epithelium and few positive intra-alveolar macrophages were observed (Fig. 6A). Lungs from S. rectivirgula–treated mice showed a significant increase in BiP-positive staining in epithelial cells, macrophages, and neutrophils (Fig. 6B and C). Regarding sXBP1, we found very few positive macrophages in saline control lungs but a significantly higher number in lungs from S. rectivirgula–exposed mice. We did not observe sXBP1 immunoreactivity in epithelial cells in control lungs, and in contrast, epithelial cells near inflamed areas were stained positive for sXBP1 after S. rectivirgula treatment (Fig. 6A, B, and D). CHOP showed a less-intense signal than BiP and sXBP1, but we identified positive staining in epithelial cells, macrophages, and some neutrophils. No immunoreactivity for CHOP was found in saline controls (Fig. 6A, B, and E).
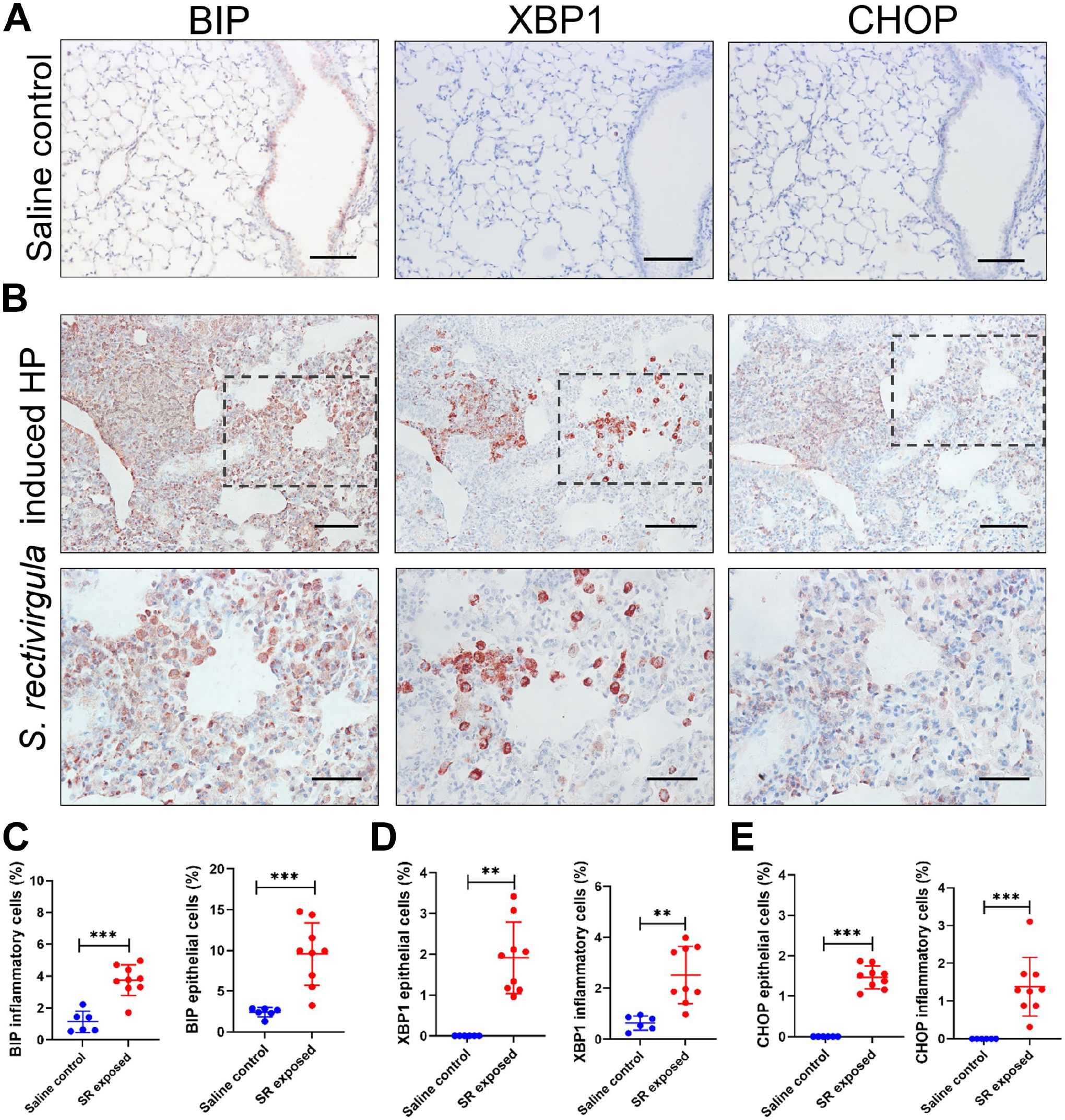
Lung localization of UPR proteins BiP, sXBP1, and CHOP in Saccharopolyspora rectivirgula mouse model of HP. (A) Representative photomicrographs of BiP, sXBP1, and CHOP immunostaining in lung tissue from control and (B) S. rectivirgula–exposed mice. The lower-row images are enlargements of the black dotted boxes in the middle row to show image details at higher magnification. The positive signal was observed in red. All sections were counterstained with hematoxylin (blue). (C) Percentage of different BiP-, (D) sXBP1-, and (E) CHOP-positive cell types in control and S. rectivirgula lungs. Data are presented as mean ± SD. Statistical significance was determined with t-test or Mann–Whitney U test (**p<0.001, ***p<0.0001). Images are at 20× and 40× magnification (Scale bars = 100 and 50 µm, respectively).
Moreover, by immunofluorescence and paralleling the results in human HP, we colocalized sXPB1 and SPC in some AECIIs in S. rectivirgula–treated lungs, supporting the notion that S. rectivirgula HP antigen induces ER stress and UPR activation in AECIIs (Fig. 7A). We also identified CHOP and Cas12 coexpression in S. rectivirgula–challenged lungs, mainly in alveolar and interstitial macrophages (Fig. 7B). Our HP mouse model corroborates that there is an activation of the UPR pathway in lungs after antigen exposure and is similar to what we observe in the human disease.
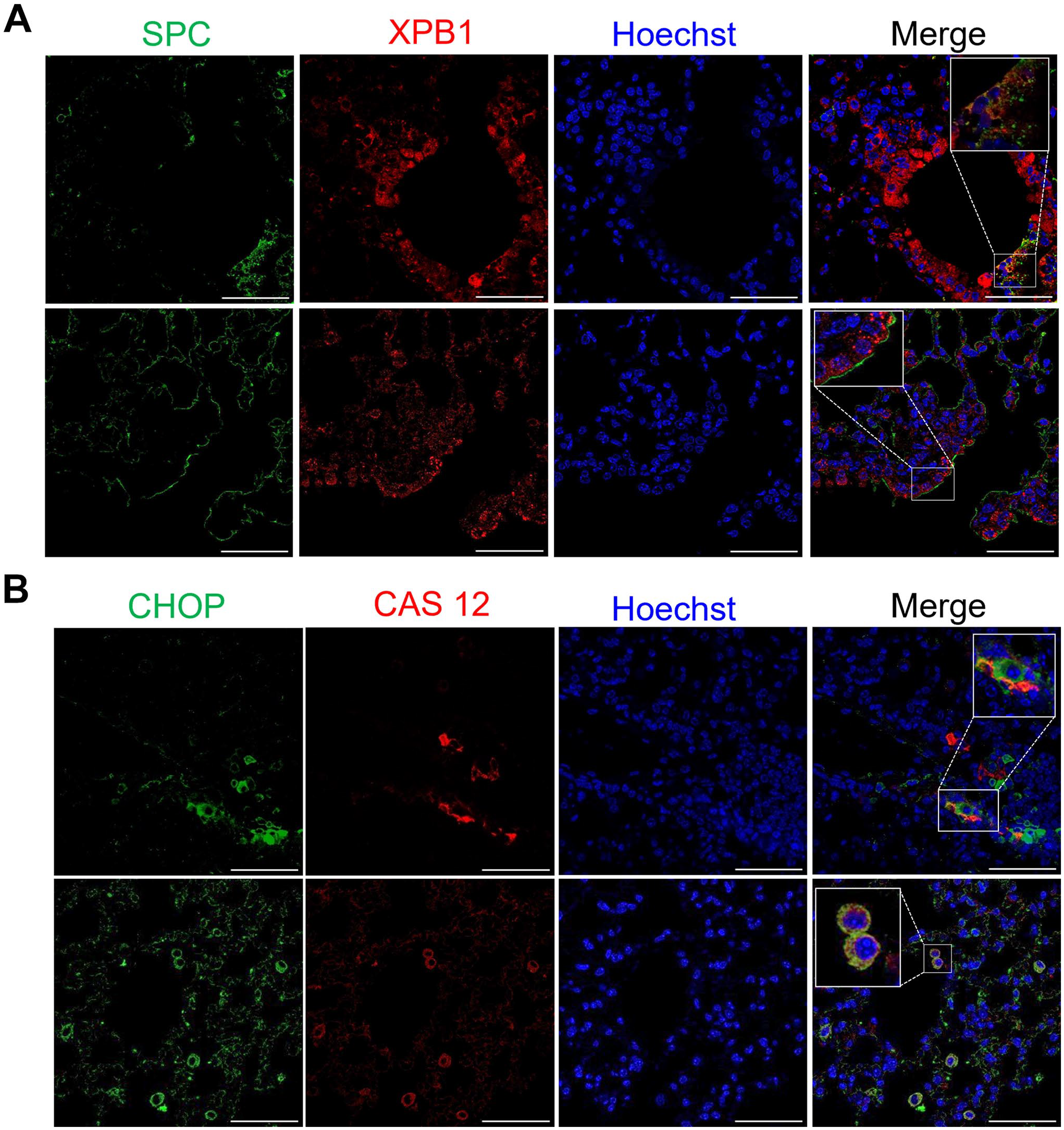
UPR protein sXBP1 colocalizes with SPC in alveolar epithelial cells and CHOP with Cas12 in macrophages in lungs from S. rectivirgula–exposed mice. (A) Representative confocal fluorescence images of SPC (green) and sXPB1 (red) in S. rectivirgula lungs. (B) Representative confocal fluorescence images of CHOP (green) and Cas12 (red) in S. rectivirgula lungs. Nuclei were stained with Hoechst (magnification 63× scale bars = 50 µm).
Overall, our findings show increased BiP, sXBP1, and CHOP protein levels in human and mouse HP lungs, suggesting that ER stress induction and UPR pathway activation are induced in HP and may play a role in the pathogenesis of this disease.
Discussion
Recent evidence links ER stress and UPR signaling to the pathogenesis of several diseases in many organs. Importantly, the proapoptotic and proinflammatory effects of ER stress have been associated with fibrosis.5,6,14 The lungs of patients with IPF showed increased ER stress, characterized by elevated levels of BiP/GRP78. In addition, it has been proposed that BiP expression in the lungs may serve as a marker of lung injury due to smoke since smokers and chronic obstructive pulmonary disease (COPD) patients express elevated BiP levels.14,15
The respiratory epithelium is often exposed to organic and inorganic agents, which could lead to dysregulation of the epithelial barrier and result in lung injury.1–4 In this context, we speculate that oxidative stress and epithelial damage associated with lung inflammation could induce ER stress and UPR activation in the lungs of HP patients. UPR activation in HP lungs has not yet been delineated. Thus, we aimed to evaluate whether the UPR effectors BiP, sXBP1, and CHOP were induced and which cells expressed these proteins in HP lungs. In the present study, we found basal expression of BiP in some epithelial cells and macrophages, since BiP is a constitutively expressed resident protein of the ER of all eukaryotic cells. However, BiP-positive cells were significantly increased in HP lungs, mainly bronchial epithelium, hyperplastic and cuboidal AECs in areas close to fibrosis, and in macrophages and plasma cells. Specific loss of BiP in AECIIs using an inducible knockout mouse model led to increased apoptosis and senescence and impaired progenitor capacity of AECIIs, resulting in spontaneous lung fibrosis. 16 This evidence indicates that BiP epithelial expression is crucial for lung epithelium integrity. We also described intense positive staining of sXBP1 and CHOP proteins in bronchial and AECs of HP lung tissues, while no staining was found in control lungs.
Healthy aging of the lung involves not only structural changes but also a decline in central mechanisms of cellular quality control such as proteostasis. An imbalance between increased ER stress and decreased UPR results in loss of proteostasis, and this is a hallmark of aging.17,18 Although HP can affect anyone of any age, it is considered an adulthood illness since it is most often diagnosed in people of ages 50–55 years.1,2 In the current study, we compare controls and patients diagnosed with HP of the same age range (51–65 years). In this context, we cannot rule out the possibility that aged lung experiences changes that could influence ER stress induction and changes in UPR activation. However, absent immunoreactivity for XBP1 and CHOP and weak staining of BiP were found in controls, while a strong signal for the three UPR biomarkers and significantly increased number of positive cells were observed in the HP lungs. These results were confirmed by Western blot analysis, where significantly increased BiP, sXBP1, and CHOP protein levels were found in HP compared with control lung tissue extracts, suggesting that activation of this pathway is associated with pathogenesis rather than aging. However, age-matched cohort studies including a larger sample size and HP animal models comparing young versus aged mice are needed.
We suggest that the expression of BiP, sXBP1, and CHOP increases in epithelial cells from HP lungs as a reparative mechanism after the exposure to inciting agents such as microbes, proteins, or inorganic matter but also after the exposure to chemokines and other mediators secreted by inflammatory cells. Another possibility is that changes in the biochemical and mechanical properties of the extracellular matrix could induce sustained epithelial ER stress and phenotypic alterations creating a profibrotic positive feedback loop that could drive to the fibrotic form of HP. Our current findings show that the hyperplastic and cuboidal epithelial cells that line thickened fibrotic alveolar septa and flattened epithelial cells overlying fibroblastic foci were strongly positive for BiP, sXBP1, and CHOP.
Previous evidence has shown that ER stress is induced in AECIIs exposed to hypoxia and that the activation of UPR pathway contributes to epithelial-mesenchymal transition (EMT), a pathogenic event linked to the development of lung fibrosis. 19 Another important finding in our study was the colocalization of the UPR biomarkers with autophagy proteins mainly in epithelial and inflammatory cells from HP lungs. ER stress and autophagy are closely related to each other, 20 and UPR activation begins the ER-associated degradation (ERAD) response, inducing autophagy to mitigate ER stress. Autophagy stabilizes the ER function via the degradation of unfolded, misfolded, or aggregated proteins or the damaged ER organelle itself, but it can also induce cell death under extreme conditions.21,22 For example, Caspase 3 and CHOP-positive AECIIs near dense fibrotic zones have been detected in IPF lungs, indicating that ER stress and apoptosis occur in AECIIs in this disease. 23 Therefore, the UPR pathway could determine whether epithelial cells survive, undergo phenotypic changes, or die in response to sustained or prolonged ER stress.
Evidence from animal models indicates that asbestos, cigarette smoke, and environmental particulates induce ER stress in AECs. 24 Next, we used the S. rectivirgula mouse model of HP and described here for the first time that S. rectivirgula treatment significantly increased UPR proteins compared to control lungs. Basal expression of BiP was identified in bronchial epithelial cells and very few macrophages, but after S. rectivirgula exposure, a significant increase in BiP staining was observed in epithelial cells and macrophages. We found that sXBP1 colocalizes with some SPC-positive AECs, confirming that S. rectivirgula–induced injury causes ER stress in AECIIs in our HP mouse model. Similar upregulation of BiP and sXBP1 was found in AECIIs in L188Q SFTPC transgenic mice, which was associated with lung fibrosis. 24 In addition, aged mice exposed to MHV68 infection showed XBP1 and SPC colocalization in AECII cells. 25
Regarding the UPR activation in inflammatory cells, sXBP1-positive staining was significantly increased in alveolar and interstitial macrophages in both human fibrotic HP and mouse lungs after S. rectivirgula treatment. From Western blot analysis, we corroborate that the sXBP1 protein level significantly increased in both human fibrotic HP and mouse lungs after S. rectivirgula treatment compared with their respective controls. Macrophages play an essential role as antigen-presenting cells during the development of HP, and it has been described that Toll-like receptors (TLRs) trigger sXBP1 activation in macrophages, and this process regulates cytokine production. 26 Further studies are required to evaluate the role of sXBP1 in regulating macrophage responses in HP pathogenesis.
Finally, we reported here that no CHOP signal was observed in control lungs, while strong staining was found in macrophages, neutrophils, and plasma cells in HP lungs. It has been shown that CHOP activation in plasma cells is necessary to produce immunoglobulins since Chop knockout mice have abnormal plasma cells with a lower immunoglobulin secretion rate. 23 Increased immunoglobulin production and the formation of immune complexes also play a role in the pathogenesis of HP. In this context, it will be interesting to define with future experiments whether CHOP regulates immunoglobulin secretion by plasma cells in HP lungs.
In addition, high levels of CHOP have been detected in IPF lungs and were mainly found in M2 macrophages. In addition, Chop deficiency significantly reduced M2 macrophage polarization and attenuated bleomycin-induced lung fibrosis. 27 In our mouse model of HP induced by S. rectivirgula, we observed CHOP- positive macrophages and epithelial cells, while control lungs were CHOP negative. Interestingly, we observed Cas12 and CHOP colocalization mainly in alveolar and interstitial macrophages. CHOP-mediated macrophage apoptosis was also found in bleomycin-induced fibrosis. 28 Together, this evidence indicates that CHOP could regulate macrophage apoptosis after lung injury in HP pathogenesis.
It should be noted that our HP mouse model only recapitulates the inflammatory stage of the disease that replicates nonfibrotic HP, but not the fibrotic HP. The most common HP animal model involves repeated exposure to S. rectivirgula for 3 weeks, and we performed this protocol and found minimal fibrotic foci formation not comparable with the HP fibrotic lung. We must not lose sight of the fact that fibrotic HP is frequently caused by a very long-term exposure to the antigen (or combination of antigens), and the genetic background of patients could also determine if HP progresses to the chronic fibrotic form.2–4 Some HP animal models have extended the antigen exposure period to 14 weeks, but, although collagen deposition was observed, the fibrotic response did not recapitulate fibrotic HP. 29 Using another HP protocol where mice received intranasal instillations of pigeon serum on three consecutive days per week for either 3 weeks (acute) or 12 weeks (chronic), interstitial inflammation was observed with the presence of perivascular infiltration and only some giant cells; however, no peribronchiolar or interstitial fibrosis was found in the chronic exposed group.30,31 Therefore, further studies using long-term exposure to S. rectivirgula or another antigen or combinations of antigens are needed to obtain a fibrotic response and acquire a better understanding of how UPR activation regulates the dynamics of the fibrotic stage of HP. Moreover, future research in HP could include studies with knockout animal models (i.e., Chop-, Xbp1-, Bip-deficient mouse) to better define the specific role of ER stress and UPR activation in HP pathogenesis.
The UPR pathway exerts diverse effects mainly via protein-protein interactions, changes in the subcellular localization of the effector proteins, phosphorylation, and proteolytic cleavage.7–10 In this context, we choose as a first approach to evaluate the localization and protein levels of BiP, sXBP1, and CHOP effector proteins in the lungs from controls and HP patients. However, although the immunohistochemical staining and Western blot analysis are valuable techniques, one limitation of our study is that our findings are supported exclusively by antibody approaches, and complementary gene expression analysis are needed to demonstrate upregulation in UPR biomarkers and to establish specific conclusions about their role in the HP pathogenesis. Another limitation of our study is the small sample size, primarily attributed to the restricted availability of human tissue. While the sample size is small for drawing definitive conclusions, this exploratory and qualitative study could provide initial insights into the involvement of ER stress and UPR activation in the HP pathogenesis.
There are a number of several UPR issues linked with HP pathogenesis that have yet to be resolved, such as the specific role of BiP, XBP1, and CHOP in every cell type (i.e. bronchial epithelial, alveolar epithelial, inflammatory, and so on); UPR-mediated aberrant phenotypes; UPR-cytoprotective or UPR-proapoptotic outputs; and age-related changes in proteostasis. By addressing these unresolved questions and leveraging experimental animal model development, we can enhance our understanding of HP pathophysiology and identify novel therapeutic targets.
Footnotes
Appendix
Acknowledgements
We thank LANSBIODYT UNICUA-UNAM (CONACYT) and Dr. Edgar Jiménez-Díaz for the technical support. We thank Dr. Luis Jiménez Álvarez and Christian Alan Cabello Hernández for their support with the mouse model development.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have contributed to this article as follows: funding (SC), study design (SC), experiments (SC, CR-B, AG-V, PG, AS), animal mouse model (SC, AS), data analyses (MG, PG, SC), writing the initial draft of the manuscript (SC), provided clinical insights relevant to the study (MG, MS), reviewed and edited the manuscript (MS, AP, PG), and all authors read and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT IN202221), DGAPA-UNAM.