
Abstract
Hepatic stellate cells (HSCs) are the major site of retinoid storage, and their activation is a key process in liver fibrogenesis. We have previously shown that expression of the retinoic acid receptor alpha (RARα) is upregulated in activated rat HSCs at a post-transcriptional level and that these RARα proteins showed a speckled distribution in the cytosol, despite their possession of a nuclear localization signal (NLS). In this report, we further characterize these cytosolic RARα proteins by using exogenously expressed RARα protein fragments or mutants tagged with a green fluorescent protein. Substitution of four amino acids, 161–164 from lysine to alanine, abolished the NLS. Exogenously expressed RARα protein fragments containing an NLS were localized exclusively in the nuclei of activated rat HSCs and never colocalized with the endogenous RARα proteins in the cytosol, suggesting that the NLS of endogenous RARα proteins is masked. Biochemical analysis showed that 65% of RARα proteins in activated HSCs were insoluble in a mixture of detergents. The insolubility of RARα proteins makes it difficult to identify RARα proteins in activated HSCs. Therefore, we propose that insoluble, speckled cytosolic distribution of RARα proteins represents a new marker of HSC activation. (
Keywords
Vitamin A and its active metabolites (retinoids) exert most of their physiological activities by transcriptional regulation (Mangelsdorf et al. 1995; Chambon 1996). All-
The fact that HSC activation is accompanied by a loss of vitamin A has led to the idea that retinoid signaling is diminished during liver fibrosis (Ohata et al. 1997; Milliano and Luxon 2005). This opinion is supported by the findings that mRNAs for RARα, −β, and −γ were decreased during HSC activation (Weiner et al. 1992; Ohata et al. 1997). However, we have recently found that retinoid signaling is upregulated in activated HSCs by posttranscriptional upregulation of RARα gene expression (Mezaki et al. 2007). RARα protein was never detected in quiescent HSCs and increased gradually during activation. We also reported that the upregulated RARα proteins are distributed in the cytosol in a speckled fashion. There are several reports describing the cytosolic distributions of RARα (Akmal et al. 1996; Mey et al. 2007), RARβ (Sommer et al. 1999), and RXRα (Ghose et al. 2004) proteins in specific cell types. However, to the best of our knowledge, there is no report describing a speckled distribution of RARα proteins in the cytosol. Therefore, we investigated the cytochemical and biochemical features of this curious cytosolic distribution of RARα proteins in activated HSCs. We demonstrate in this report that over-expression of RARα protein fused with a green fluorescent protein (GFP) did not show such a speckled cytosolic distribution, despite their possession of an NLS within the DBD. We also show that most of the RARα proteins in activated HSCs are insoluble. The possibility of using this cytosolic speckled distribution of RARα proteins as a marker for HSC activation is discussed.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA) and Vitromex (Vilshofen, Germany), respectively. Oligonucleotides were synthesized by Invitrogen. Alexa Fluor 546 phalloidin (Invitrogen), cytochalasin B, and cytochalasin D (Sigma-Aldrich; St. Louis, MO) were also purchased.
Plasmids
The cDNA fragments corresponding to aa 1–459 (full-length, or F), 1–181 (N-terminal half, or N), and 182–459 (C-terminal half, or C) of rat RARα were amplified by PCR from pCDNA3.1-ratRARα plasmid (Mezaki et al. 2007) with nucleotide overhangs for digestion by restriction enzymes (
Preparation of HSCs
Isolation of rat HSCs was performed as described previously (Senoo et al. 1984; Mezaki et al. 2007). Protocols for animal experimentation were approved by the Animal Research Committee, Akita University School of Medicine. All animal experiments adhered to the Guidelines for Animal Experimentation of the University. In brief, livers of male Wistar rats were perfused with bacterial collagenase solution (Wako Pure Chemical; Osaka, Japan), and dispersed cells were centrifuged at 50 × g to pellet parenchymal cells. The supernatant containing non-parenchymal cells was centrifuged at 600 × g, and pelleted cells were further purified by density gradient centrifugation with Percoll/RediGrad (GE Healthcare Bio-Sciences; Piscataway, NJ). The layer containing HSCs was collected, and cells were washed with DMEM supplemented with 10% FBS and seeded on plastic dishes or glass-bottom dishes.
Transfection
Transfections of plasmids into a human embryonic kidney cell line (HEK293T) or primary rat HSCs were done with Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Three to 5 days after seeding cells on 3.5-cm glass-bottom dishes, plasmids (0.75–3 μg) were transfected. Three hr after transfection, the medium was changed to DMEM supplemented with 10% FBS and further cultured until day 7.
Immunocytochemistry
Cells were washed and fixed with 2% (w/v) paraformaldehyde in PBS for 5 min at room temperature and then treated with 100% methanol for 5 min at −20C for permeabilization. Nonspecific binding was blocked by incubating cells with 1% bovine serum albumin (BSA; Sigma-Aldrich) in PBS for 30 min at room temperature. Cells were then incubated with polyclonal rabbit anti-RARα antibody (1:50 dilution, sc-551, Santa Cruz Biotechnology; Santa Cruz, CA) or monoclonal mouse anti-RARα antibody (50 μg/ml, clone Rα10, Affinity BioReagents; Golden, CO) in PBS plus 2% heat-inactivated normal goat serum (Kohjin Bio; Saitama, Japan) and 1% BSA for 1 hr at room temperature. After washing, secondary antibody (Alexa Fluor 546 goat anti-rabbit IgG, A-11035, or Alexa Fluor 488 goat anti-rabbit IgG, A11034, or Alexa Fluor 546 goat anti-mouse IgG, A-11030; Invitrogen) in PBS plus 1% BSA was added (1:200 dilution) for 30 min at room temperature. When needed, cells were further stained with monoclonal mouse anti—α-smooth-muscle actin (αSMA) antibody (1:200 dilution, clone 1A4; Sigma-Aldrich). Finally, cells were washed and immersed in 1% 1,4-diazabicyclo-[2,2,2]octane (Sigma-Aldrich) in PBS to prevent fluorescence decay. Cells were treated with 0.5 μM TO-PRO-3 (Invitrogen) and observed with an LSM510 laser scanning microscope (Carl Zeiss; Jena, Germany).
Organelle Visualization
Activated rat HSCs were treated for 30 min with 100 nM MitoTracker Deep Red 633 (Invitrogen), 50 nM Lyso-Tracker Red DND-99 (Invitrogen), or 1 μM ER-Tracker Red (Invitrogen) for visualization of mitochondria, lysosomes, or endoplasmic reticulum (ER), respectively. For visualization of organelles with protein markers such as Rab5a for endosomes or Lamp1 for lysosomes, activated HSCs were treated with Organelle Lights Endosomes-GFP (Invitrogen) or Organelle Lights Lysosomes-RFP (Invitrogen) according to the manufacturer's instructions. For immunocytochemical staining with anti-RARα antibody (sc-551), cells treated for organelle visualization were further processed as follows: MitoTracker Deep Red 633-treated HSCs and Organelle Lights Endosomes-GFP-treated HSCs were fixed with 4% (w/v) paraformaldehyde, followed by treatment with 0.1% Triton X-100 (ICN Biomedicals, Inc.; Aurora, OH) in PBS for 5 min at room temperature for permeabilization. Organelle Lights Lysosomes-RFP—treated HSCs were fixed with 4% (w/v) paraformaldehyde plus 0.1% (w/v) glutaraldehyde, followed by treatment with 0.1% Triton X-100 in PBS for 5 min at room temperature. Fluorescent signals from LysoTracker Red DND-99 and ER-Tracker Red could not be retained after any fixation and permeabilization procedures tested.
HSC Fractionation and RARα Protein Solubilization
HSCs were collected and fractionated by ProteoExtract Subcellular Proteome Extraction Kit (Merck; Darmstadt, Germany) according to the manufacturer's instructions. The integrity and purity of each fraction were checked by Western blotting using anti—cellular retinol-binding protein I (CRBP-I) for cytosolic proteins [fraction (fr.) 1], anti—pan-cadherin for membranes and membrane organelles (fr. 2), anti—acetyl-histone H3 for nuclear proteins (fr. 3), and αSMA for cytoskeleton (fr. 4).
For RARα protein solubilization, HSCs were pelleted and lysed with 1% Nonidet P-40 (NP-40). After centrifugation at 8000 × g, supernatants were set aside (S1), and the pellet was treated with 1% NP-40 plus 0.5% sodium deoxycholate. After centrifugation, supernatants were set aside (S2), and the pellet was treated with 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS), which is identical to the composition of a RIPA buffer, a buffer widely used for cell lysis. The supernatant (S3) and pellet (P) were separated by centrifugation.
Western Blotting
Cell fractions were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Atto; Tokyo, Japan). Nonspecific binding was blocked with 5% skim milk in phosphate buffered saline with Tween 20 (PBST) [0.1% Tween 20 (Merck) in PBS]. Then, membranes were incubated with primary antibodies against RARα (1:200 dilution, sc-551; Santa Cruz Biotechnology), CRBP-I (1:200 dilution, sc-30106; Santa Cruz Biotechnology), lecithin:retinol acyltransferase (LRAT; 1:200 dilution, rabbit polyclonal antibody raised against aa 168–184 of LRAT of mouse origin, Immuno-Biological Laboratories, Gunma, Japan), proliferating cell nuclear antigen (PCNA; 1:200 dilution, sc-7907; Santa Cruz Biotechnology), β-tubulin (1:1000 dilution, clone TUB 2.1; Sigma-Aldrich), pan-cadherin (1:1000 dilution, clone CH-19; Sigma-Aldrich), acetyl-histone H3 (1:200 dilution, 06–599; Millipore, Billerica, MA), αSMA (1:1000 dilution, clone 1A4; Sigma-Aldrich), and β-actin (1:200 dilution, sc-1616; Santa Cruz Biotechnology) in PBST plus 1% skim milk for 1 hr at room temperature. After washing, secondary antibodies, peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H+L) (111-035-003; Jackson ImmunoResearch, West Grove, PA)for RARα, CRBP-I, LRAT, PCNA, and acetyl-histone H3, or peroxidase-conjugated AffiniPure goat anti-mouse IgG (H+L) (115-035-003; Jackson ImmunoResearch) for β-tubulin, pan-cadherin, and αSMA, and peroxidase-conjugated AffiniPure rabbit anti-goat IgG (H+L) (305-035-003; Jackson ImmunoResearch) for β-actin were applied to the PVDF membranes at a dilution of 1:10,000 in PBST for 30 min at room temperature. Bound antibodies were detected by enhanced chemiluminescence (GE Healthcare Bio-Sciences), and signals were recorded on X-ray film (Fujifilm; Tokyo, Japan).
Cytochalasin Treatments
Cytochalasin B and cytochalasin D were dissolved in DMSO and diluted with DMEM supplemented with 10% FBS. Spontaneously activated HSCs seeded on glass-bottom dishes were treated with 2 μM or 20 μM cytochalasin B or D for 1 hr just before fixation. The effective concentrations of cytochalasin B and D on stress fiber destruction were based on previous observations (Yahara et al. 1982).
Quantification of Western Blotting Signals
Signals from Western blotting bands on X-ray films were scanned and quantified using ImageJ software (National Institutes of Health; Bethesda, MD).
Results
Speckled Cytosolic Distribution of RARα Protein in Rat HSCs Activated In Vitro
We previously documented that RARα proteins show a punctate or speckled cytosolic distribution during rat HSC activation in vitro (Mezaki et al. 2007). In those studies, we used a rabbit polyclonal antibody (sc-551) raised against a peptide targeting the C terminus of human RARα (Figures 1A–1C). For confirmation, we used a mouse monoclonal antibody (Rα10) raised against a synthetic peptide corresponding to residues 444 to 462 of mouse RARα, a sequence identical to that in the rat (441 to 459). Almost the same distribution of RARα proteins was observed with this monoclonal antibody (Figures 1D–1F), verifying the speckled cytosolic distribution of RARα in activated HSCs. Because the sc-551 antibody could be used in both immuno-cytochemistry and Western blotting, it was used in the subsequent experiments.
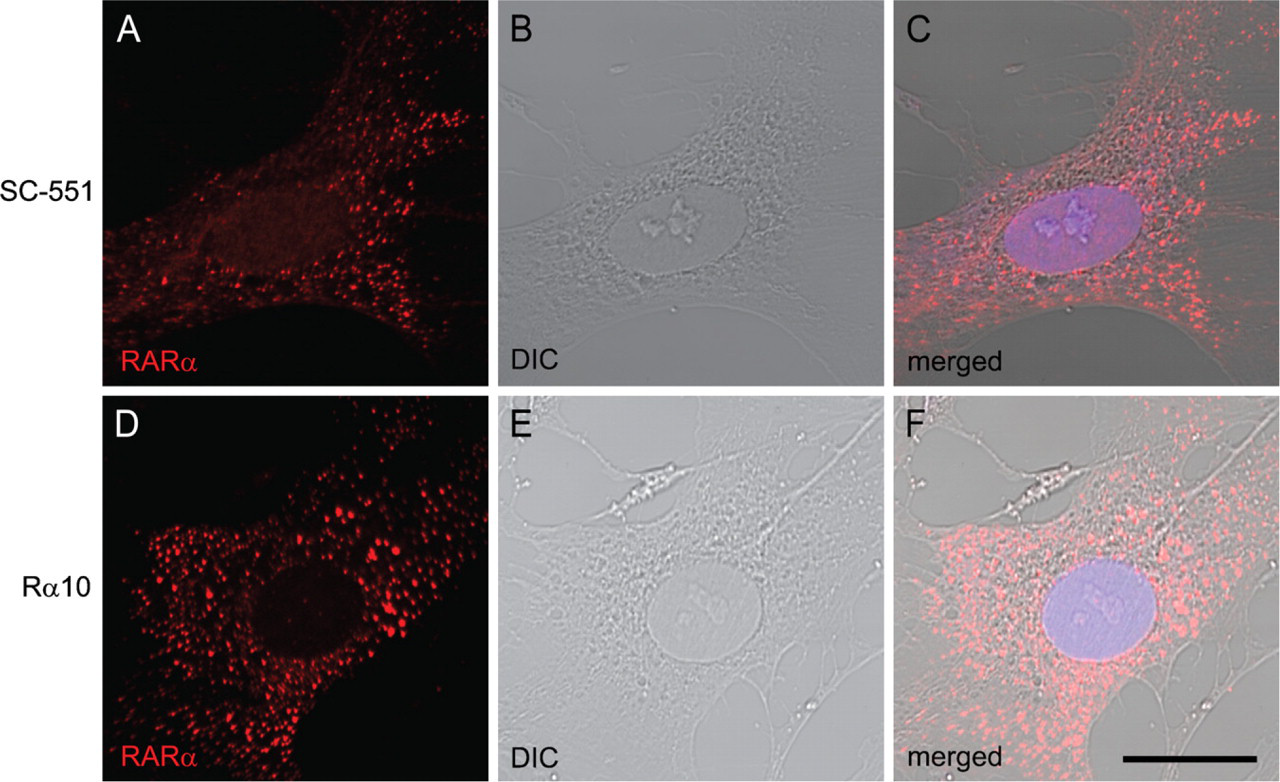
Cytosolic speckled distribution of retinoic acid receptor alpha (RARα) proteins in activated hepatic stellate cells (HSCs). Primary HSCs collected from rat liver were spontaneously activated by culturing on poly-
The NLS of RARα Proteins Resides in aa 161–164
We constructed plasmids containing cDNAs for full-length RARα (F) or their fragments [N-terminal half (N) and C-terminal half (C)] tagged with GFP (Figure 2). Two candidate NLS sequences are included in N and C fragments, respectively. These plasmids were transfected into a human embryonic kidney cell line, HEK293T, by a lipofection method. As shown in Figure 3, AcGFP (a plasmid control lacking an inserted fragment) and AcGFP-C were evenly distributed in both the nucleus and the cytosol (Figures 3A–3C and 3J–3L), whereas AcGFP-F and Ac-GFP-N were exclusively within the nucleus (Figures 3D–3I). The same localization was observed with C-terminally tagged constructs (Figures 3M–3P), indicating that localization of RARα protein fragments was not affected by the position of GFP tagging. These results indicate that the peptide fragment of aa 1–182 of rat RARα contain the NLS and strongly suggest that the RNKKKK sequence of aa 159–164 is the functional NLS, whereas the other candidate NLS sequence of aa 361–364 within the LBD is not.
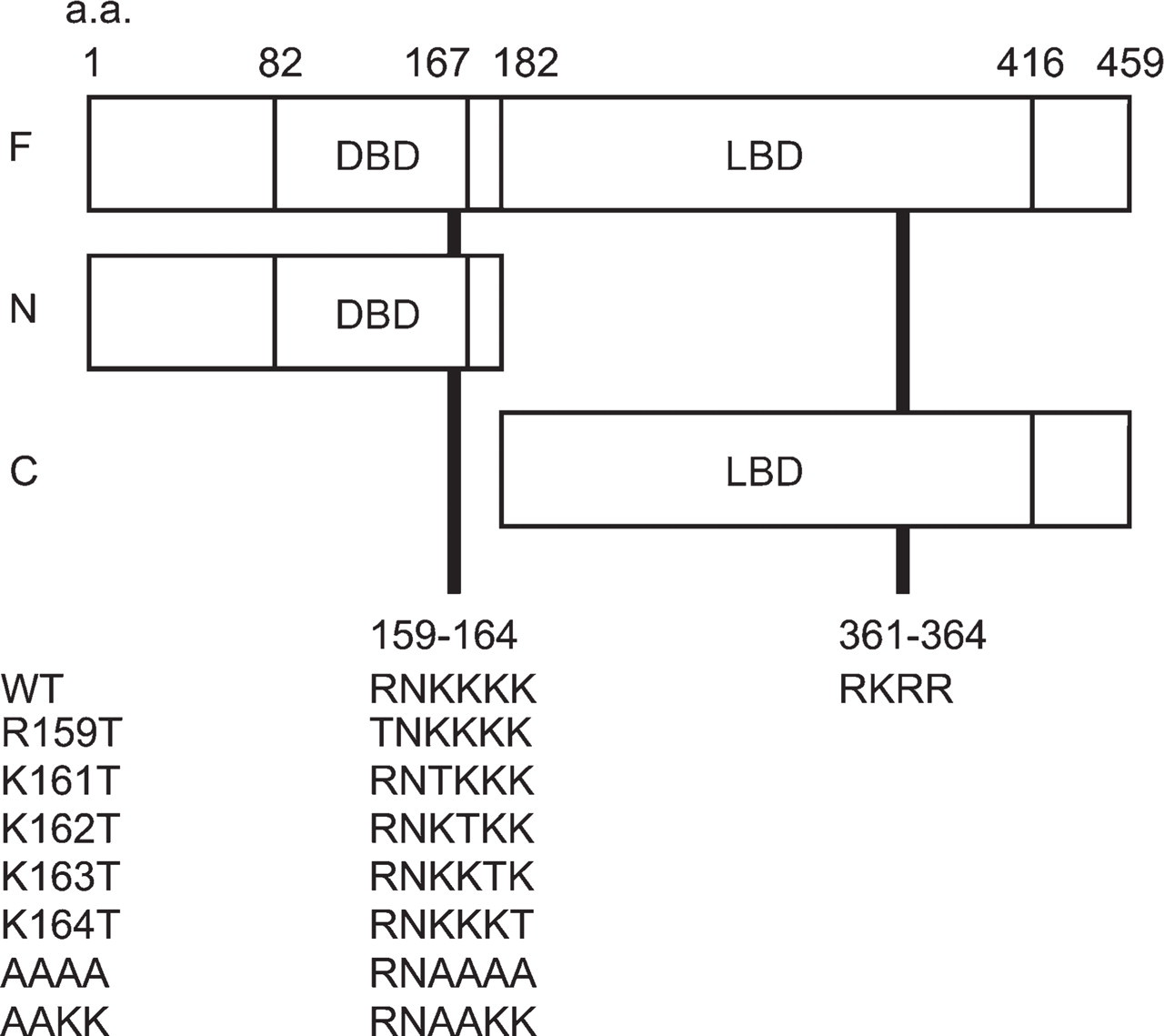
Domain structure of RARα protein fragments fused with a green fluorescent protein (GFP) tag and two possible nuclear localization signals (NLSs) located in N- and C-terminal halves of RARα protein. Numbers represent amino acid positions. Two candidate NLS sequences rich in basic amino acid residues are located in the DNA-binding domain (DBD) and the ligand-binding domain (LBD). Amino acid sequences of seven mutants used in this study are indicated at the lower part of the figure. a.a., amino acid.
To determine the exact location of the NLS, site-directed mutagenesis was applied to the F-EGFP vector (Figure 2), and cellular localizations of the mutated RARα proteins were observed. One amino acid substitution from arginine (R) or lysine (K) to threonine (T) within the RNKKKK sequence did not affect their localizations (Figures 3Q–3V). However, four sequential amino acid substitutions, from KKKK to AAAA of aa 161–164, resulted in the complete loss of NLS function (Figure 3W); they showed even distribution in both the nucleus and the cytosol, a pattern identical to that of the EGFP alone (Figure 3M). Two amino acid substitutions, from KK to AA of aa 161–162, seemed to have a marginal effect on NLS function (Figure 3X).
Exogenous and Endogenous RARα Proteins Showed Different Distributions Within the Activated HSCs
Next, these plasmids were transfected into rat HSCs 3 days after seeding, when endogenous RARα proteins begin to be translated from relatively abundant mRNA (Mezaki et al. 2007). Four days after transfection (a week after seeding), GFP fluorescence was observed. It showed the same cellular distribution as that observed in the HEK293T cell line: AcGFP-F, AcGFP-N, F-EGFP, and N-EGFP showed nuclear localization, whereas AcGFP, AcGFP-C, EGFP, and C-EGFP showed an even distribution within the cell (Figures 4A–4P). The AAAA mutant again showed the even distribution in both the nucleus and the cytosol (Figures 4Q–4S).
Transfection of F-EGFP plasmid followed by immunocytochemistry with an anti-RARα antibody, which detects both endogenous and exogenous RARα proteins, showed that cytosolic dots (RARα protein) did not include any of the exogenously expressed RARα proteins tagged with GFP (Figures 5D–5F, arrowheads). These results indicate that the NLS of endogenous RARα proteins is not functional within the activated HSCs, whereas the NLS of exogenous RARα proteins is functional in the same cells, suggesting that the NLS of endogenous RARα proteins is temporarily masked. Abrogation of the NLS by site-directed mutagenesis did not lead to the speckled distribution of the exogenous RARα proteins (Figures 4Q–4S), indicating that the suppression of the nuclear localization is not sufficient for speckled distribution of RARα proteins in activated HSCs.
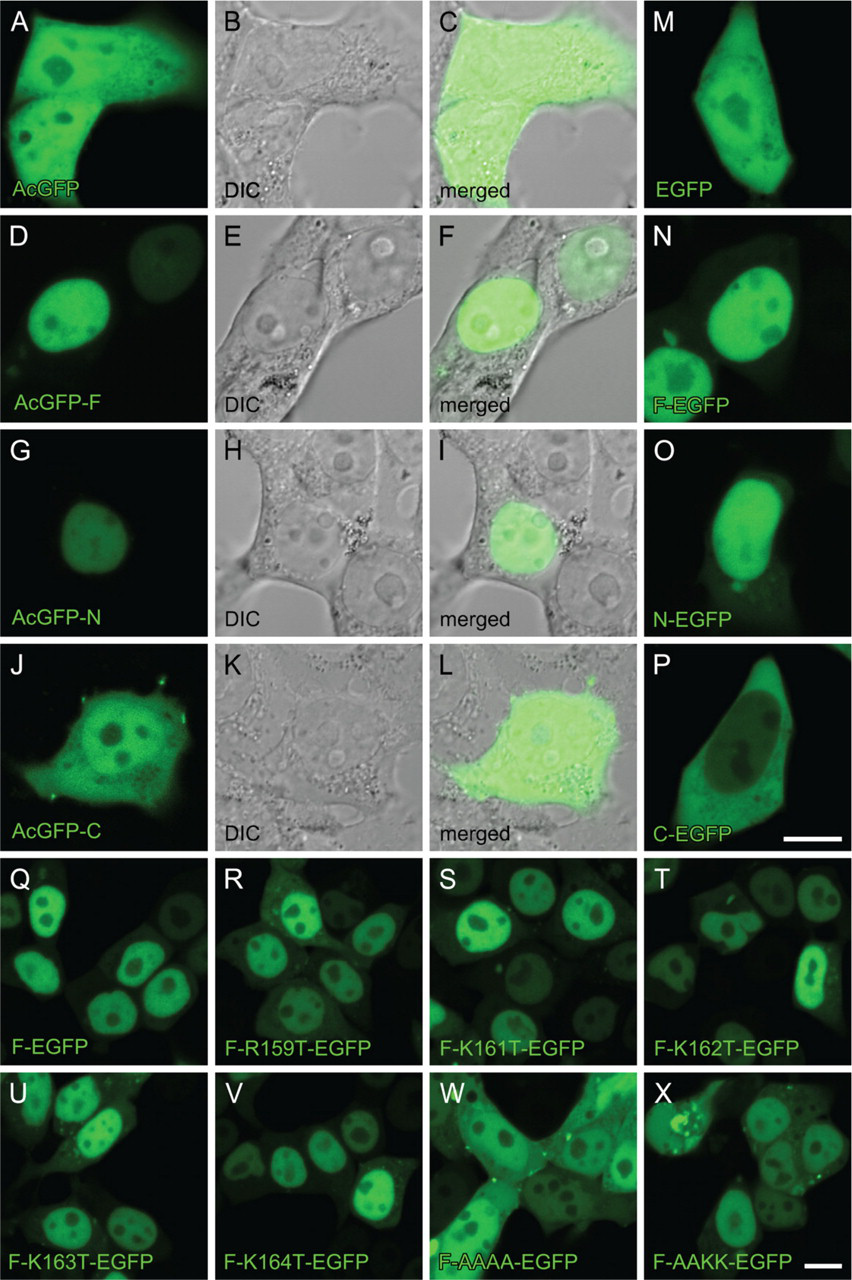
Cellular localization of RARα protein fragments or mutants fused with GFP in the HEK293T cell line. Cells were transfected with GFP-expressing plasmids with inserted cDNA for RARα fragments or mutants designated in Figure 2. The position of the GFP tag is at either the N terminus (
RARα Proteins in Activated HSCs Do Not Colocalize With Mitochondria, Endosomes, or Lysosomes
We investigated whether the cytosolic speckled distribution of RARα is colocalized with any known intracellular organelle. Visualization of mitochondria (Figure 6A), endosomes (Figure 6E), and lysosomes (Figure 6I) showed speckled patterns within the cytosol, whereas visualization of the ER showed a perinuclear diffuse distribution (Figure 6M). These cells were fixed and permeabilized for immunocytochemical staining with an anti-RARα antibody. Fluorescent signals from mitochondria were retained after fixation and permeabilization (Figures 6A and 6B). Some fluorescent signals from endosomes were lost (Figures 6E and 6F). Lysosomes seemed to be swollen after fixation and permeabilization (Figures 6J–6L). All fluorescent signals from the ER were lost after fixation and permeabilization (data not shown). In these conditions, no colocalization was observed between RARα proteins and intracellular organelles investigated (Figures 6B–6D, 6F–6H, and 6J–6L).
RARα Proteins in Activated HSCs Cofractionate With the Cytoskeleton but Do Not Colocalize With Microfilaments
The punctate distribution of cytosolic RARα proteins was further characterized by cell fractionation using a kit that enables the differential extraction of proteins according to their subcellular localization (Figure 7). Purity and selectivity of subcellular extraction was confirmed by using antibodies against CRBP-I for cytosolic proteins (fr. 1), pan-cadherin for membranes and membrane organelles (fr. 2), acetyl-histone H3 for nuclear proteins (fr. 3), and αSMA for the cytoskeleton (fr. 4) (Figure 7). RARα proteins fractionated exclusively with the cytoskeleton.
Because RARα and αSMA proteins cofractionated, we next examined the subcellular localization of those proteins by immunocytochemical methods. We also asked whether disruption of the stress fiber architecture [a characteristic feature of HSC activation (Enzan et al. 1994)], affected RARα protein localization. As shown in Figure 8A, no colocalization was observed between RARα and αSMA proteins. Treatment of the activated HSCs with cytochalasin B or D, the actin-depolymerizing agents, disrupted the architecture of microfilaments within the cells, but did not affect the cytosolic speckled distribution of RARα proteins (Figures 8B–8D). The same results were obtained by staining with phalloidin, which binds filamentous actins of all six isoforms (Vandekerckhove and Weber 1978) (Figures 8E–8H). These results indicate that the microfilament structure is not needed to maintain the speckled, cytosolic distribution of RARα proteins in the activated HSCs. It was also confirmed that almost all stress fibers within the activated HSCs contained αSMA, although the tips of each stress fiber seemed to contain a larger proportion of actin isoforms other than αSMA (Figure 8I).
RARα Proteins in Activated HSCs Are Largely Insoluble
We further analyzed RARα protein solubility by sequentially applying non-ionic (1% NP-40), mild ionic (0.5% sodium deoxycholate), and strong ionic (0.1% SDS) detergents (Figure 9A). Of RARα proteins, 0%, 5%, and 30% were present in NP-40—soluble, sodium deoxycholate—soluble, and SDS-soluble fractions, respectively. Final composition of the detergents in this solubilization experiment is equivalent to that of RIPA buffer, suggesting that 65% of RARα proteins in activated HSCs are insoluble in RIPA buffer (Figure 9B). Most cellular protein other than RARα was solubilized by 1% NP-40, as was shown by Coomassie Brilliant Blue R-250 staining, whereas RARα proteins seemed to be a major component of the RIPA-insoluble fraction (Figure 9C, arrowhead). The calculated molecular mass of unmodified rat RARα protein is ∼51 kDa.
Discussion
There have been several attempts to identify markers of HSC activation, by both transcriptomic (Sancho-Bru et al. 2005; Boers et al. 2006; Jiang et al. 2006; De Minicis et al. 2007) and proteomic (Kristensen et al. 2000) analyses. Despite these extensive efforts, RARα has never been identified as a marker for HSC activation. In fact, mRNA for RARα is decreased during HSC activation (Weiner et al. 1992; Ohata et al. 1997; Mezaki et al. 2007); therefore, RARα expression has been considered as a quiescent marker. We previously reported that RARα upregulation is achieved at the posttranscriptional level (Mezaki et al. 2007). In this report, we demonstrated that a large proportion of RARα protein in activated HSCs is insoluble. These two characteristic features of RARα protein expression and localization in activated HSCs may have made it difficult to identify RARα as a marker for HSC activation at both mRNA and protein levels. Moreover, RARα proteins in the cytosol showed a speckled distribution, which, to the best of our knowledge, has not previously been reported as a feature of RAR localization in any cell type. Therefore, we propose that insoluble, speckled cytosolic distribution of RARα protein is a marker of HSC activation, at least, in vitro. It is of great interest whether these cytosolic RARα dots could be observed in HSCs activated in vivo, because several lines of evidence indicate that HSC activation in vivo and activation in vitro have different features (Sancho-Bru et al. 2005; De Minicis et al. 2007). The difficulty of studying HSCs activated in vivo is that the activated HSCs cannot be collected by conventional centrifugal methods that rely on the density differences between HSCs and other cell types. That is, activated HSCs lose vitamin A—containing lipid droplets from their cytoplasm, increasing their density (Desmoulière 2007). Immunohistochemical identification of RARα protein dots in activated HSCs of liver sections from rats treated with carbon tetrachloride will give some information about the differences between HSC activation in vitro and in vivo.
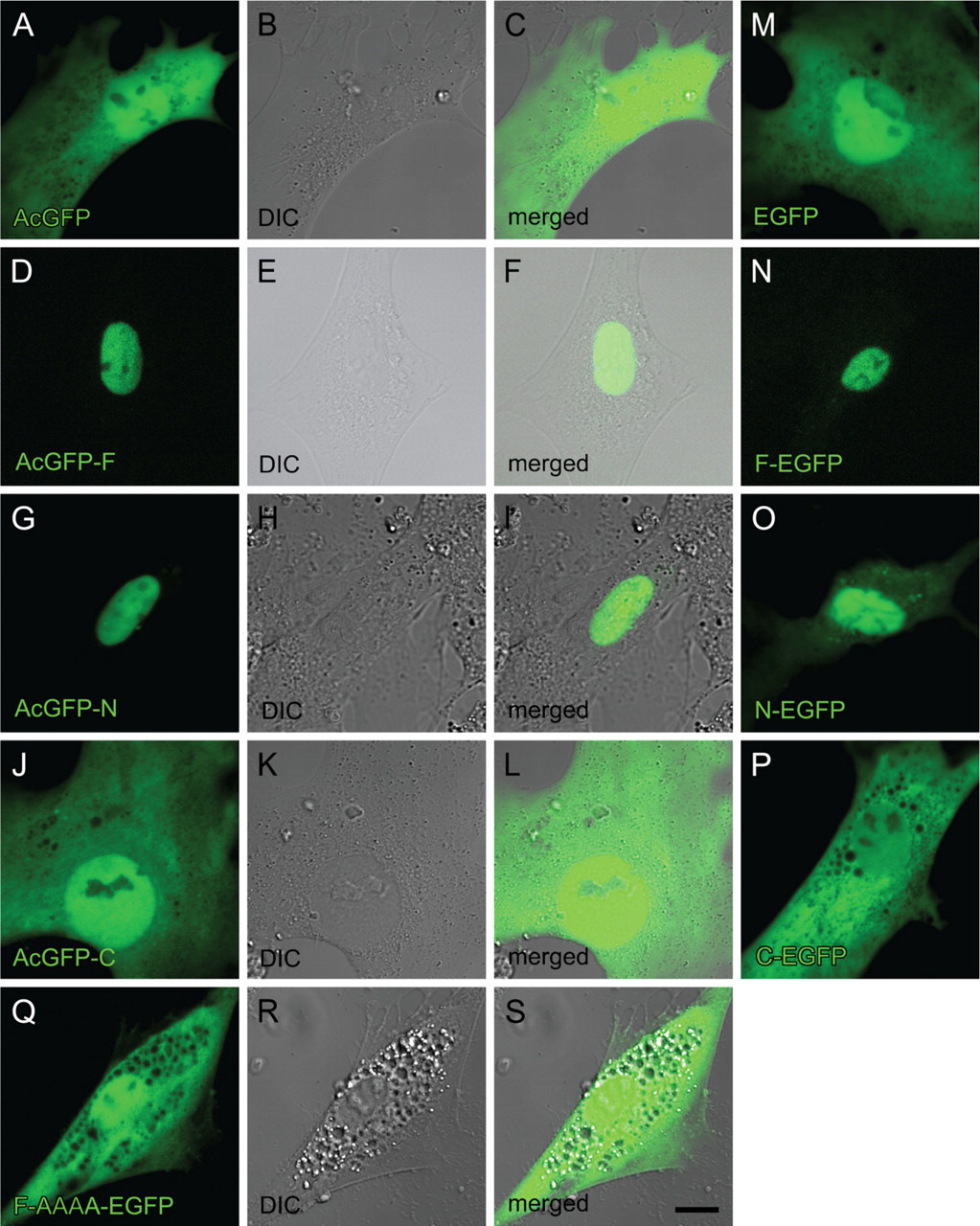
Cellular localization of RARα protein fragments or a mutant fused with GFP in activated primary rat HSCs. Cells were transfected with GFP-expressing plasmids inserted with cDNA for RARα fragments or a mutant designated in Figure 2 3 days after seeding. The position of the GFP tag is either at the N terminus (
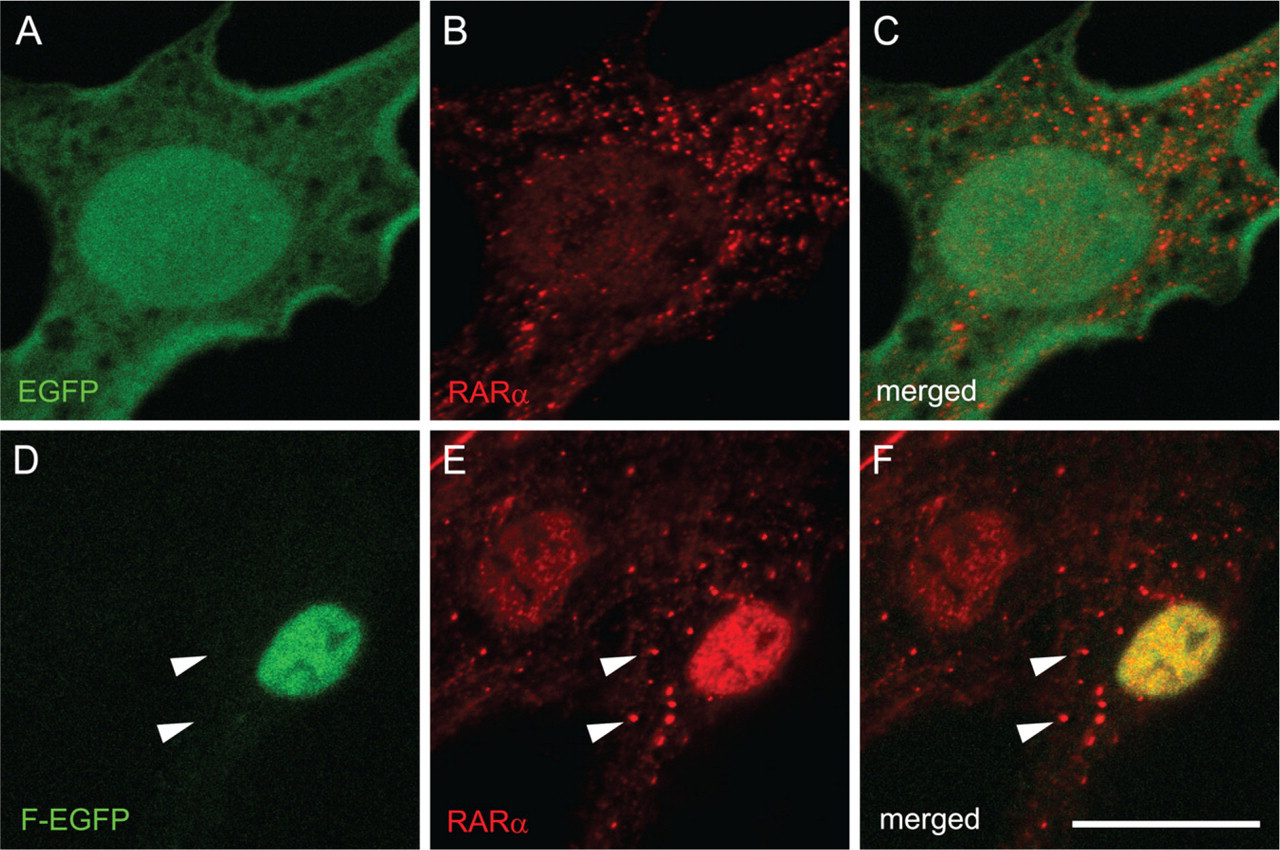
Cellular localization of exogenous and endogenous RARα proteins in activated primary rat HSCs. Cells were transfected with GFP-expressing plasmids with (
Two types of nuclear localization and transcriptional regulation among the nuclear receptor super-family are known (Evans 1988; Stunnenberg 1993). The type I nuclear receptors, which include steroid hormone receptors such as glucocorticoid receptor and estrogen receptor, reside in the cytosol without ligands. Upon ligand binding, they enter the nucleus, bind to their responsive DNA elements as homodimers, and activate transcription of their target genes (Picard and Yamamoto 1987; Ylikomi et al. 1992). On the other hand, type II nuclear receptors, which include vitamin D, vitamin A, and thyroid hormone receptors, are known to be nuclear in the absence of their ligands. They form heterodimers with RXRs on their responsive DNA sequences and actively repress the transcription of their target genes (Fondell et al. 1993). Upon ligand binding, the conformational changes within the LBD lead to the exchange of cofactors, resulting in the enhancement of gene transcription (Kishimoto et al. 2006). The RARα protein fragments containing an NLS fused with GFP were localized in the nucleus without ligands, which is consistent as a feature of type II nuclear receptors. However, the endogenous RARα proteins in activated HSCs did not localize to the nucleus, implying that the NLS of endogenous RARα proteins is masked by an unknown mechanism. Our previous report demonstrated that retinoic acid addition during HSC activation reduced cytosolic RARα protein levels (Mezaki et al. 2007), implying that cytosolic RARα proteins enter the nucleus upon ligand binding. This feature of RARα protein shuttling from cytosol to nucleus is rather similar to that of the type I nuclear receptors. The NLSs of type I nuclear receptors in the cytosol are known to be masked by heat shock proteins (Denis et al. 1988). Therefore, it is of interest whether heat shock proteins are involved in the cytosolic distribution of endogenous RARα proteins in activated HSCs.
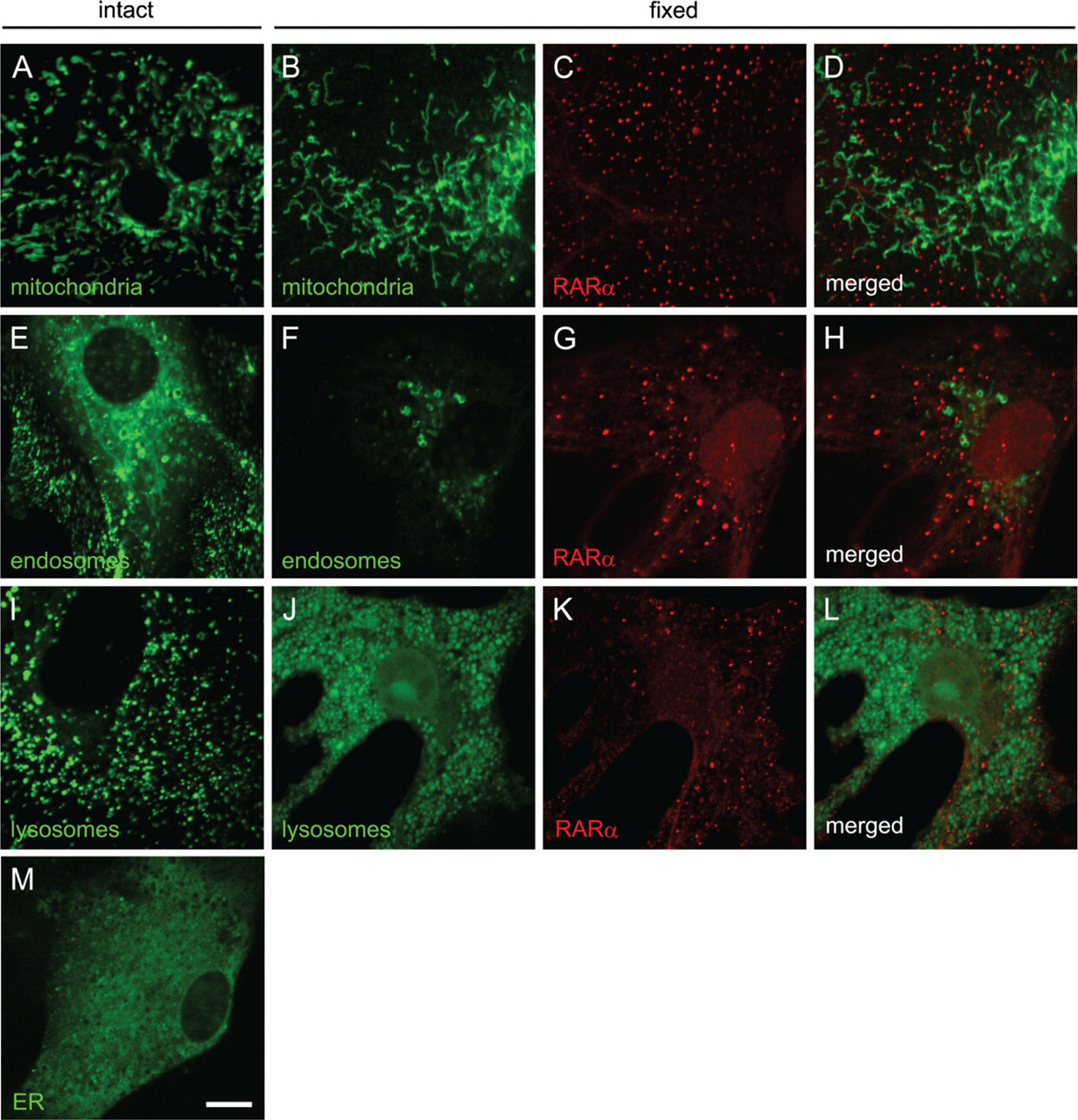
RARα proteins in activated HSCs do not colocalize with mitochondria, endosomes, or lysosomes. Mitochondria (
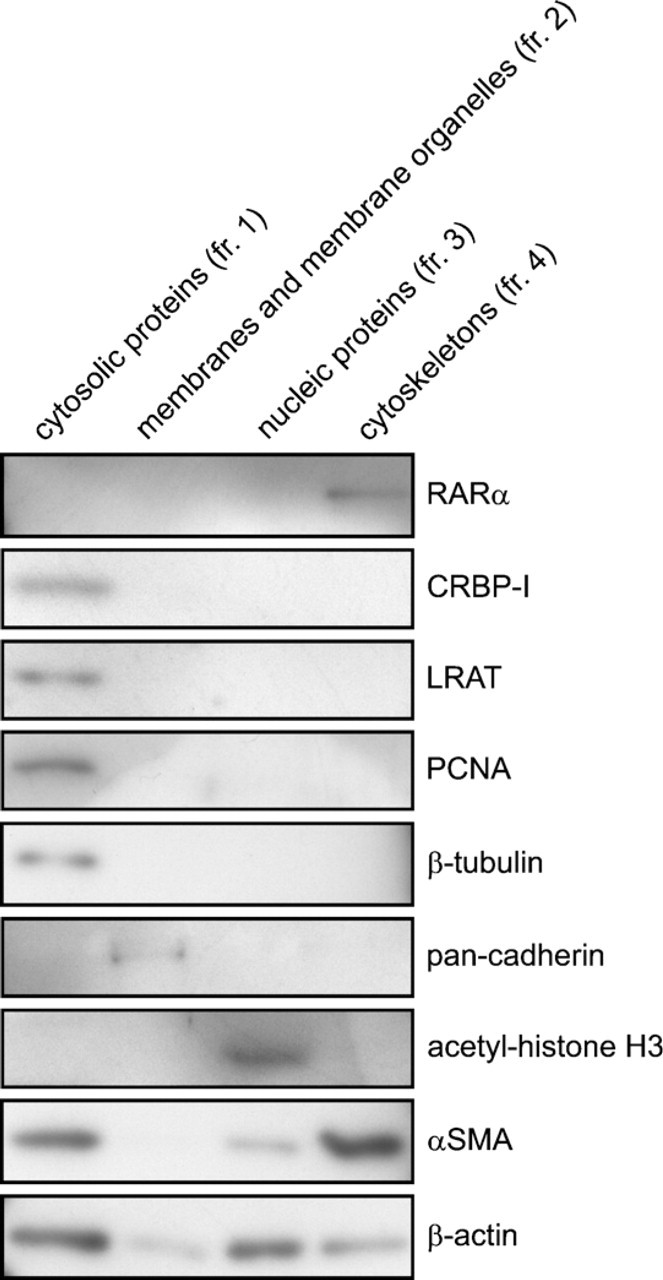
Cofractionation of RARα proteins with the cytoskeleton. Primary rat HSCs were cultured on plastic dishes for 7 days for spontaneous activation. Cells were collected and fractionated into four cellular components. Each fraction derived from the same amount of cells was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting using several antibodies indicated in the figure. CRBP-I, cellular retinol-binding protein I; fr., fraction; LRAT, lecithin:retinol acyltransferase; PCNA, proliferating cell nuclear antigen; αSMA, α-smooth-muscle actin.
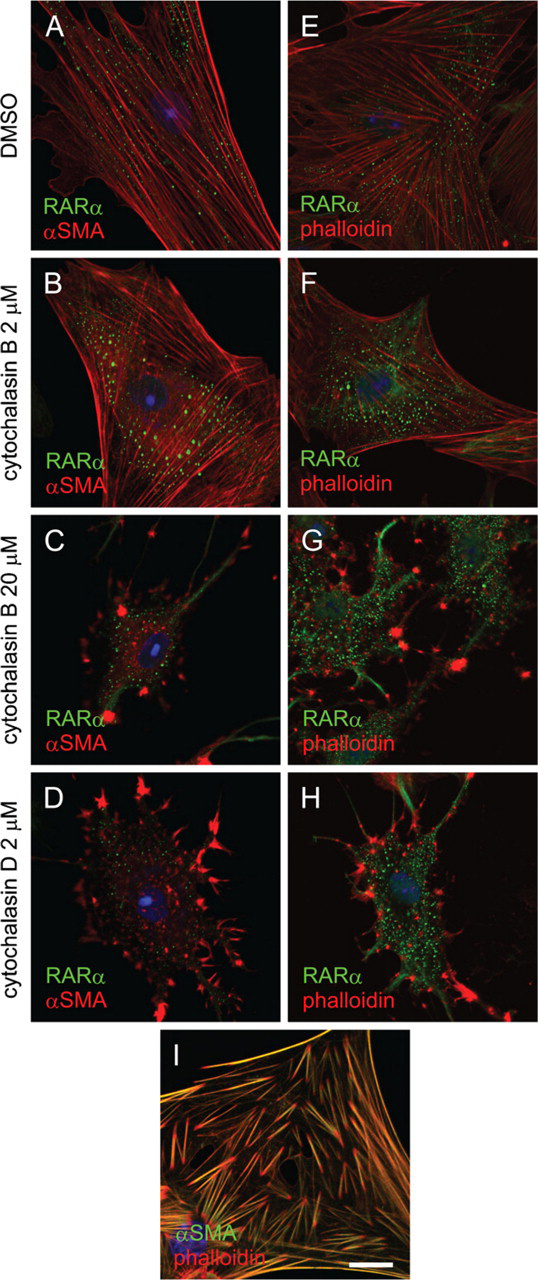
Disruption of stress fiber structure in activated HSCs has no effect on RARα protein localization. Activated HSCs were treated with DMSO (
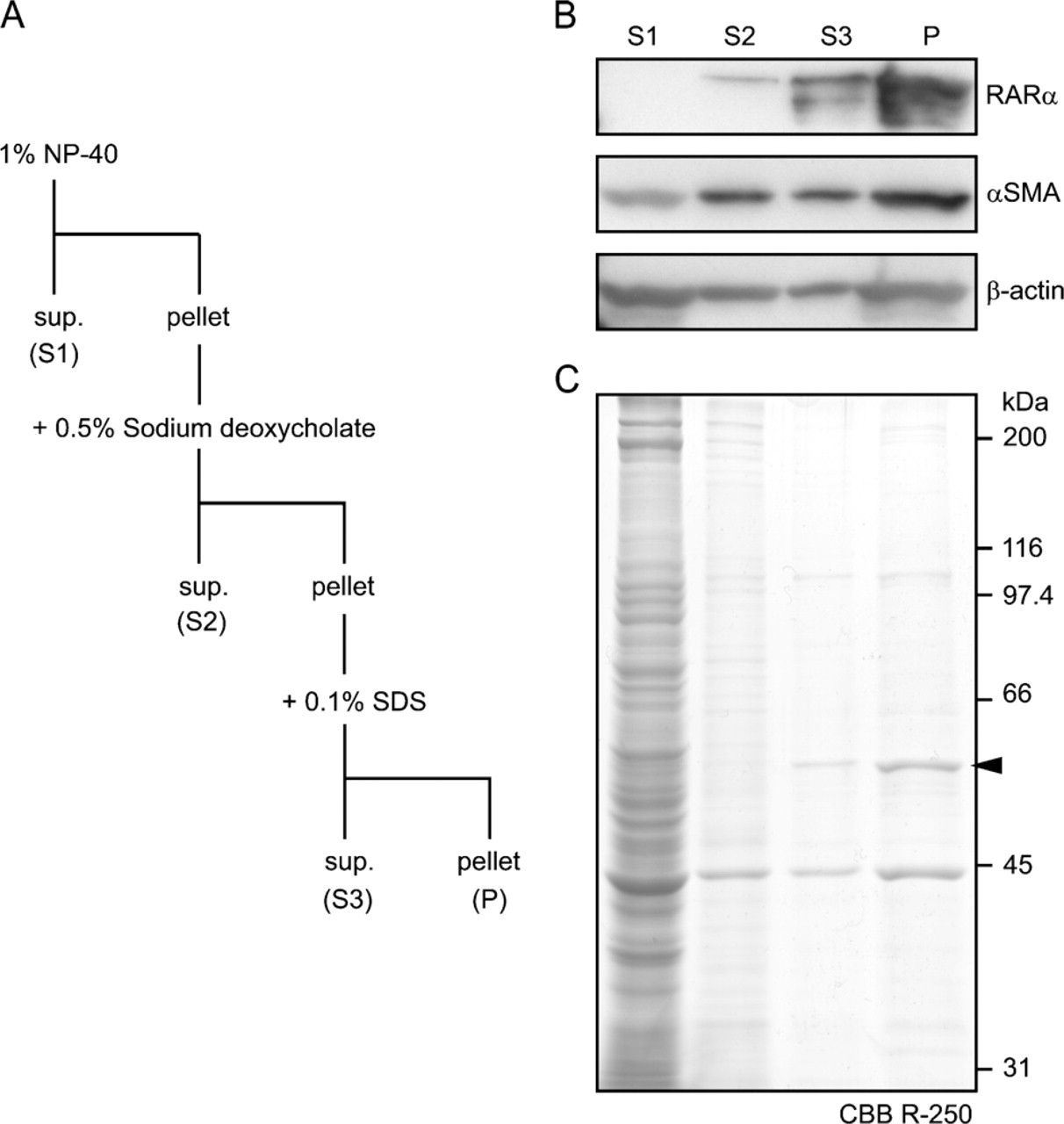
Solubility of RARα proteins in activated HSCs. Activated HSCs were collected and sequentially solubilized by non-ionic (NP-40), mild ionic (sodium deoxycholate), and strong ionic (SDS) detergents (
RARα proteins in activated HSCs were exclusively detected in the cytoskeleton fraction (Figure 7). The cytoskeleton includes three types of elements: micro-filaments, intermediate filaments, and microtubules. Microtubules seemed to be depolymerized during cell fractionation, because β-tubulin, a component of micro-tubules, was detected in cytosolic proteins (Figure 7). A smooth-muscle—specific actin isoform, αSMA, was cofractionated with RARα. However, an involvement of microfilaments in the maintenance of speckled distribution of RARα proteins was ruled out, because disruption of microfilaments had no effect on the distribution of RARα proteins. It is well known that the composition of intermediate filaments in HSCs changes during activation. For example, glial fibrillary acidic protein is expressed in quiescent HSCs, whereas desmin is predominantly expressed in activated HSCs (Jiroutová et al. 2005). Therefore, it is interesting whether intermediate filaments and/or the alteration of their composition are involved in the cytosolic speckled distribution of RARα proteins.
In summary, we have analyzed cytochemical and biochemical features of RARα proteins in activated HSCs by using RARα protein fragments or mutants tagged with GFP. The data demonstrate that the NLS of RARα proteins resides in the KKKK sequence of aa 161–164. Although exogenous RARα proteins with an intact NLS were localized in the nuclei of activated rat HSCs, endogenous RARα proteins showed speckled distribution in the cytosol, suggesting that the NLS of endogenous RARα proteins is suppressed. Finally, a considerable proportion of RARα proteins are resistant to solubilization by a mixture of non-ionic, mild ionic, and strong ionic detergents. From these results, we propose that the insoluble, cytosolic speckled distribution of RARα proteins in activated HSCs may represent a marker for HSC activation, at least in vitro.
Footnotes
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (20790158 to Y.M.).
The authors thank Dr. Tomokazu Matsuura (Jikei University, Japan) for providing anti-LRAT antibodies.