
Abstract
Members of the Bcl-2 family include pro- and antiapoptotic proteins that regulate programmed cell death of developing tissues and death in response to cellular damage. In developing mice, the antiapoptotic Bcl-xL is necessary for survival of neural and hematopoietic cells, and consequently, bcl-x-deficient mice die around Day 13.5 of embryogenesis. Furthermore, adult bcl-x +/- heterozygous male mice have reduced fertility because of testicular degeneration. Bax, a multi-BH (Bcl-2 homology) domain proapoptotic member of the Bcl-2 family, is regulated by Bcl-xL and is required for the neuropathological abnormalities seen in bcl-x-deficient embryos. The BH3 domain only subgroup of the Bcl-2 family includes proapoptotic members that are essential for the initiation of apoptotic signaling. In this study, we investigated the role for Bim, a BH3 domain only protein, in the embryonic lethality and increased developmental cell death in bcl-x-deficient animals and the perturbed testicular function in bcl-x +/- adults. Our studies show that bim deficiency attenuates hematopoietic cell death in the fetal liver of bcl-x-deficient animals, indicating that Bim contributes to programmed cell death in this cell population. In addition, we found that testicular degeneration of adult bcl-x +/- males was rescued by concomitant Bim deficiency. However, concomitant Bim deficiency had no effect on the embryonic lethality and widespread nervous system abnormalities caused by bcl-x deficiency. Our work identifies Bim as an important regulator of bcl-x deficiency-induced cell death during hematopoiesis and testicular development.
M
Members of the proapoptotic BH3 domain only subgroup of the Bcl-2 family are essential for the initiation of apoptotic cell death and are thought to act by activating proapoptotic molecules (e.g., Bax or Bak) either directly or indirectly by binding and inhibiting antiapoptotic Bcl-2-like proteins, thereby unleashing Bax or Bak (Huang and Strasser 2000). The interactions between the different members of the Bcl-2 family are highly cell type and death stimulus-specific and seem to link a diverse number of proapoptotic stimuli to the apoptosis effector machinery. The BH3-only protein Bim, which can interact with Bcl-xL (O'Connor et al. 1998), is required for developmentally regulated programmed death of autoreactive B and T cells (Bouillet et al. 2002; Enders et al. 2003), as well as leukocyte apoptosis induced by cytokine deprivation, ER stress, or other cytotoxic insults (Bouillet et al. 1999; Puthalakath et al. 2007). In addition, Bim expression is increased in neurons in response to a variety of apoptotic insults (Harris and Johnson 2001; Putcha et al. 2001; Biswas and Greene 2002; Linseman et al. 2002), and Bim loss partially protects sympathetic neurons from nerve growth factor deprivation in vitro (Putcha et al. 2001). Interestingly, loss of even one allele of bim prevents the fatal polycystic kidney disease and lymphopenia seen in Bcl-2-deficient mice, and loss of both alleles also prevents the premature graying of hair seen in Bcl-2-deficient mice (Bouillet et al. 2001).
We hypothesized that Bim provides a critical proapoptotic stimulus that causes the neurological, hematopoietic, and gonadal abnormalities seen in bcl-x -/- and bcl-x +/- mice, respectively, and tested this hypothesis by intercrossing bim -/- with bcl-x +/- mice. We found that concomitant Bim deficiency does not prevent embryonic lethality or neuropathology associated with bcl-x deficiency. However, we show that bim is critical for the abnormal hematopoietic cell death in bcl-x -/- embryos, and concomitant bim deficiency rescues testicular degeneration seen in bcl-x +/- adult mice. Our studies identify Bim as an important regulator of testicular and hematopoietic development and highlight the complexity of bcl-x-dependent survival pathways.
Materials and Methods
Mice
The generation of mice with gene disruptions in bcl-x and bim has been described previously (Motoyama et al. 1995; Bouillet et al. 1999). The two lines were backcrossed six and eight times, respectively, onto the C57BL/6 background. Endogenous and disrupted genes were detected by PCR analysis of DNA extracts from limb or tail samples as described previously (Shindler et al. 1997; Bouillet et al. 1999). The morning on which a vaginal plug was seen was designated as embryonic Day 0.5 (E0.5). Pregnant mice were anesthetized with pentobarbital and killed on gestational Day 12.5 by cervical dislocation. Adult male mice were similarly anesthetized and sacrificed for testis analysis. Mice were cared for in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Immunohistochemistry
Embryos and testes were fixed at 4C in 4% paraformaldehyde overnight. Tissues were dehydrated and paraffin embedded, and 5-μm sections were cut. Sections were deparaffinized and stained with hematoxylin and eosin (H&E) as described previously (Shindler et al. 1997). For terminal deoxynucleotidyltransferase-mediated dUPT nick end labeling (TUNEL) staining, sections were deparaffinized, and endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in PBS. Sections were permeabilized for 10 min in PBS containing 0.3% Triton X-100. Sections were hybridized with Dig-11-dUTP (Roche Applied Science; Indianapolis, IN) for 1 hr at 37C according to the manufacturer's instructions. Sections were blocked for 20 min at room temperature in PBS-BB (PBS with 0.1% BSA, 0.3% Triton X-100, and 0.2% non-fat powdered dry milk). Mouse anti-digoxigenin monoclonal antibody (Abcam; Cambridge, MA) conjugated with horseradish peroxidase was diluted in PBS-BB and applied to sections overnight at 4C. After washes with PBS, biotin-labeled tyramide was deposited using a tyramide signal amplification system (PerkinElmer Life Sciences; Boston, MA) according to manufacturer's instructions. After three washes with PBS, sections were incubated for 45 min at room temperature with streptavidin-conjugated horseradish peroxidase (Jackson ImmunoResearch; West Grove, PA) diluted in PBS-BB. Immunostaining was detected using DAB-metal (Pierce; Rockford, IL) according to the manufacturer's instructions. TUNEL-stained sections were counterstained with hematoxylin. H&E- and TUNEL-stained sections were imaged using a Zeiss (Oberkochen, Germany) Axioscop equipped with an Axiocam MRc camera. On H&E-stained sections, apoptotic nuclei were defined as nuclei appearing condensed and hyperchromic, fragmented, and/or exhibiting a marginated chromatin staining pattern. Apoptotic nuclei were counted in multiple fields from each animal using a X100 oil-immersion objective.
Statistics
All data points represent mean ± SEM. At least three animals per group were analyzed in all experiments. Statistical significance was established by one-way or two-way ANOVA, followed by Bonferroni's test for all pairwise comparisons.
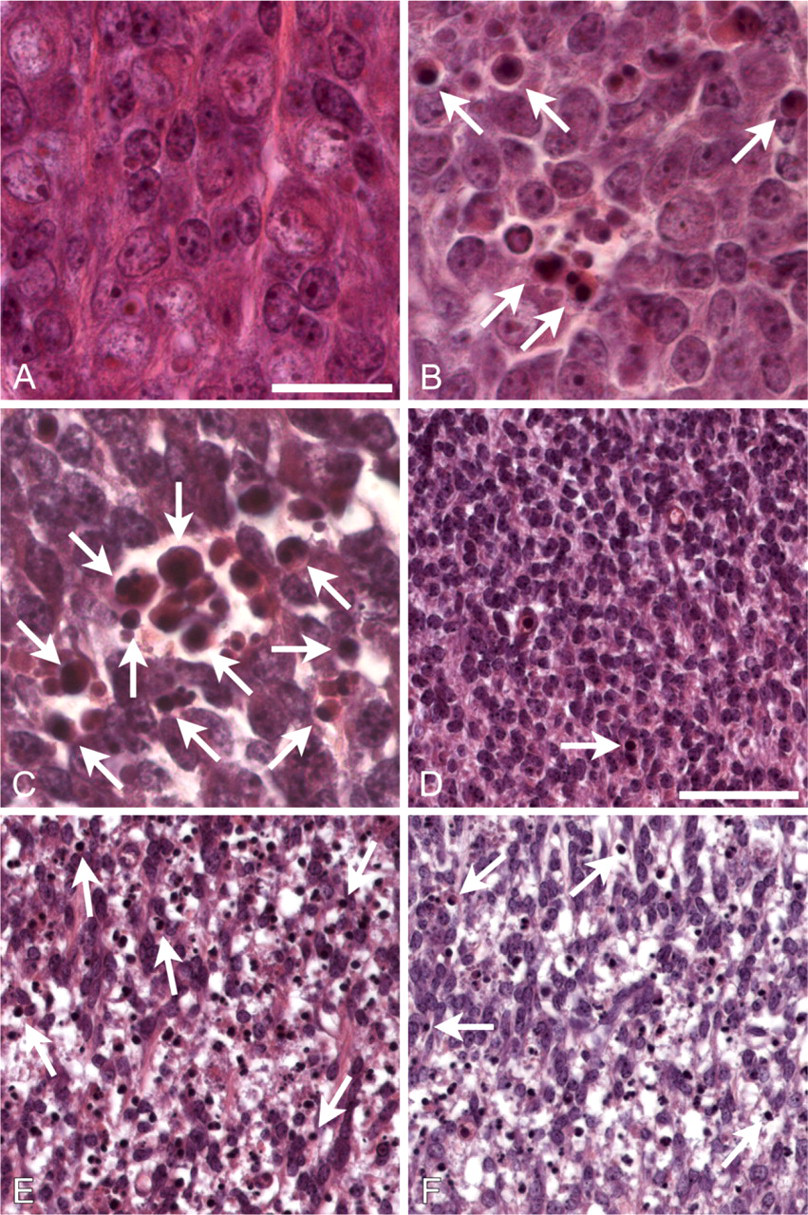
Bim loss has no effect on neurodegeneration caused by bcl-x deficiency.
Results
Generation of bcl-x-/- bim-/- Embryos
The bim and bcl-x genes are both located on mouse chromosome 2 (Eppig et al. 2005). Therefore, double-deficient mice can only be generated if mutated alleles of both genes are recombined on the same chromosome during gametogenesis. To begin, bcl-x +/- bim +/- mice were intercrossed, and a small number of bcl-x +/- bim -/- offspring were identified. These animals were crossed with wild-type (wt) mice to isolate the bcl-x - bim - chromosome, and such bcl-x +/- bim +/- progeny were intercrossed to determine whether loss of bim could prevent the embryonic lethality caused by bcl-x deficiency. In 15 litters with 80 total living offspring, 21 wt mice (26.25%; 25% expected frequency), 43 bcl-x +/- bim +/- mice (53.75%; 50% expected frequency), and no live-born bcl-x -/- bim -/- mice (25% expected frequency) were identified. In addition, germ cell recombination in a single parent led to six bcl-x +/- bim +/+ live-born mice, two bcl-x +/- bim -/- mice, and eight bcl-x +/+ bim +/- mice, but no live-born bcl-x -/- bim +/- mice were found. To confirm that bcl-x-deficient embryos were generated from these crosses, 61 mice were harvested at E12.5 from the F1 crosses described above. Fourteen wt embryos (22.95%; 25% expected frequency), 26 bcl-x +/- bim +/- embryos (42.62%; 50% expected frequency), and 7 bcl-x -/- bim -/- embryos (11.48%; 25% expected frequency) were identified. Parental recombination events generated embryos with a variety of genotypes for bcl-x and bim; one bcl-x +/- bim +/+ embryo, three bcl-x +/- bim -/- embryos, six bcl-x +/+ bim +/- embryos, three bcl-x -/- bim +/- embryos, and one bcl-x +/+ bim -/- embryo were identified. Thus, although bcl-x -/- bim -/- and bcl-x -/- bim +/- embryos were viable at E12.5, none survived to birth.
Bim Loss Reduces Hematopoietic Cell Death in bcl-x -/- Embryos but Has No Effect on Neuronal Degeneration
Embryos (12.5) from the crosses described above were prepared for histological and immunohistochemical analysis and assessed for the abundance of apoptotic nuclei. As expected, few apoptotic cells were found in DRG (Figure 1A) or ventral thoracic spinal cord (Figure 1D, example indicated by arrow) of bcl-x +/o bim +/o mice (the +/o designation includes both +/+ and +/- genotypes). Furthermore, there was no significant increase in the number of apoptotic cells in bcl-x +/o bim -/- mice (data not shown). In contrast, numerous apoptotic cells were detected in these regions in bcl-x -/- bim +/o mice (Figures 1B and 1E), consistent with previous analysis of bcl-x -/- embryos (Motoyama et al. 1995). Loss of both alleles of bim did not reduce the number of apoptotic cells in the DRG (Figure 1C) or spinal cord (Figure 1F) in bcl-x -/- mice. TUNEL staining and immunohistochemical staining for activated caspase-3 confirmed that bcl-x +/o bim +/o (Figure 2A) and bcl-x +/o / bim -/- (data not shown) embryos had only few apoptotic cells in their spinal cords. In contrast, large numbers of TUNEL-positive cells were detected in spinal cords of bcl-x -/- bim +/o (data not shown) and bcl-x -/- bim -/- (Figure 2B) embryos. Quantification of TUNEL-positive cells showed no significant difference between bcl-x -/- bim +/o and bcl-x -/- bim -/- animals (Figure 3). These findings indicate that bim and bax do not possess equivalent proapoptotic function in this context, because Bax loss attenuates bcl-x deficiency-induced embryonic neuropathology (Shindler et al. 1997), whereas Bim loss does not.
Next, the effect of Bim loss on hematopoietic cell death in bcl-x -/- embryos was examined. bcl-x +/o bim +/o and bcl-x +/o bim -/- embryos had only low numbers of TUNEL-positive cells in the liver, but bcl-x-deficient mice had abnormally increased numbers of apoptotic cells (Figure 3), as previously reported (Motoyama et al. 1995). Bim deficiency led to a reduction of TUNEL-positive nuclei in the liver of bcl-x-deficient embryos (Figure 3), showing that Bim is an important initiator of the abnormal death of hematopoietic cells that lack antiapoptotic Bcl-xL.
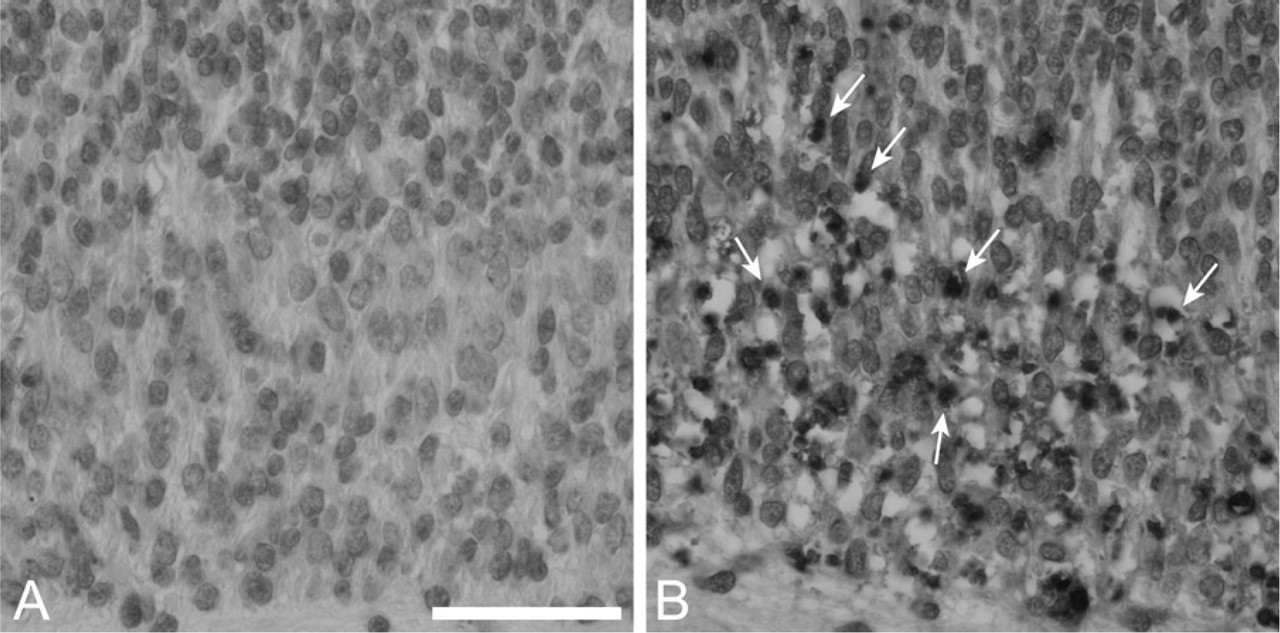
Bim loss does not alter terminal deoxynucleotidyltransferase-mediated dUPT nick end labeling (TUNEL) reactivity in bcl-x-deficient spinal cord.
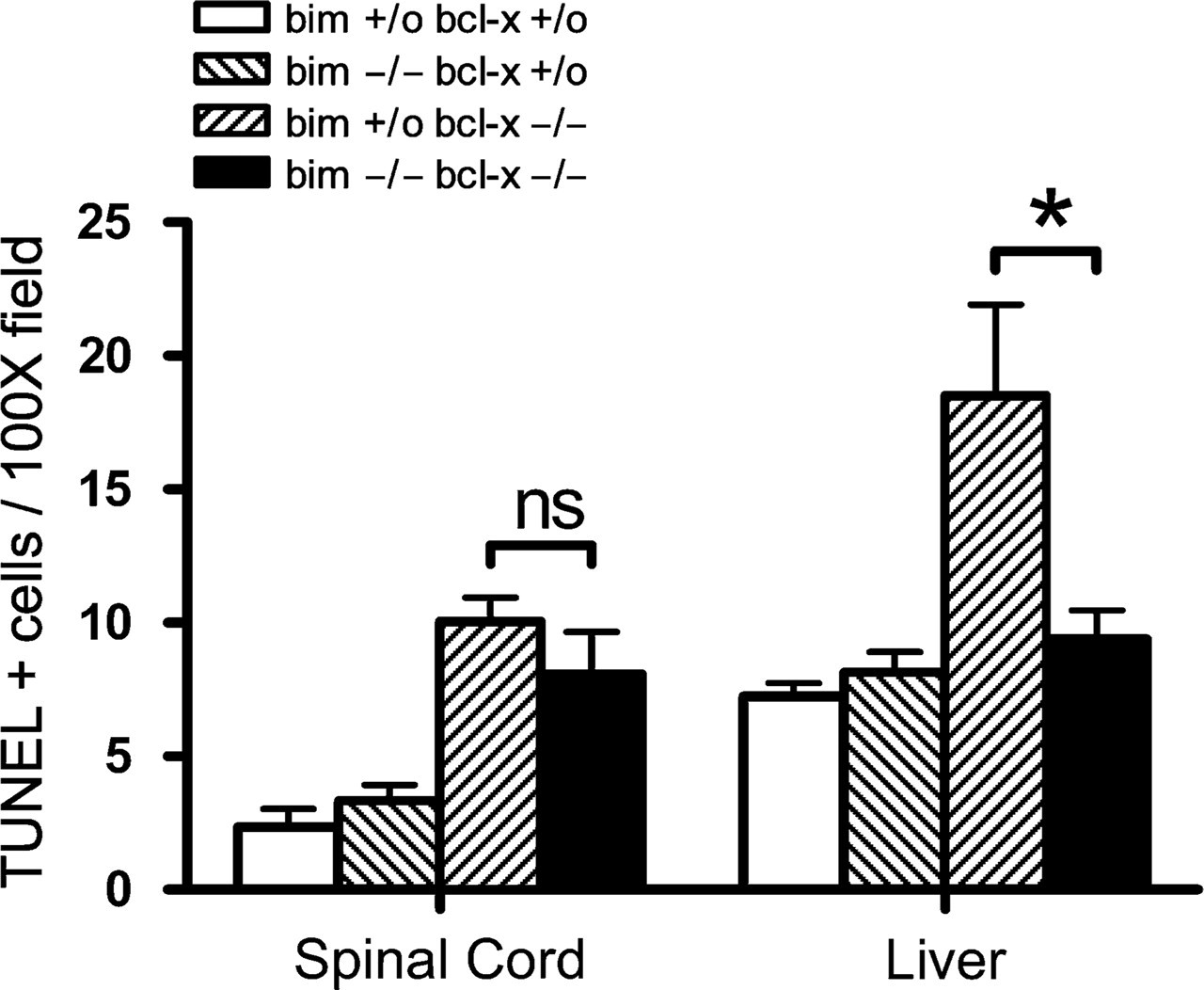
Bim loss significantly reduces the abnormal apoptosis of hematopoietic cells in the fetal liver but not spinal cord caused by bcl-x deficiency. TUNEL staining was performed on sections from E12.5 embryos, and TUNEL-positive cells within multiple X100 fields were quantitated. Fields were assessed in spinal cord (left) and liver (right). ns, not significant. ∗p<0.005.
Bim Loss Rescues Testicular Degeneration Seen in Adult bcl-x+/- Animals
Bim is expressed during spermatogenesis (O'Reilly et al. 2000) and combined loss of the two BH3-only proteins Bim and Bik inhibited apoptosis of immature germ cell progenitors as did loss of Bax (Coultas et al. 2005). Although embryos were generated for the studies described above, adult male bcl-x +/- bim +/- and bcl-x +/- bim -/- mice displayed improved fertility compared with bcl-x +/- bim +/+ animals (data not shown), and we hypothesized that Bim may be essential for the testicular atrophy observed in bcl-x +/- adult males. The average adult (90 days old) testicular weight did not differ significantly between bcl-x +/+ bim -/-, bcl-x +/+ bim +/-, and wt males (Figure 4), consistent with previous observations that bim disruption alone does not affect testicular size (Coultas et al. 2005). In contrast, in bcl-x +/- bim +/+ males, average testicular weight was <40% of that seen in wt animals. Concomitant bim deficiency restored normal testes weight in bcl-x +/- mice (bcl-x +/- bim -/- mice) and even loss of a single allele of bim (bcl-x +/- bim +/- mice) provided a partial rescue (Figure 4). In accordance with a previous report (Kasai et al. 2003), histological analysis showed degenerative changes in the testes of bcl-x +/- bim +/+ males (Figure 5A). Consistent with the data on testes weights, loss of one allele of bim led to a partial rescue of the testicular atrophy seen in bcl-x +/- mice (Figure 5B), and complete deficiency of bim restored normal testicular morphology (Figure 5C). These findings showed that Bim is essential for the testicular degeneration caused by loss of Bcl-xL.
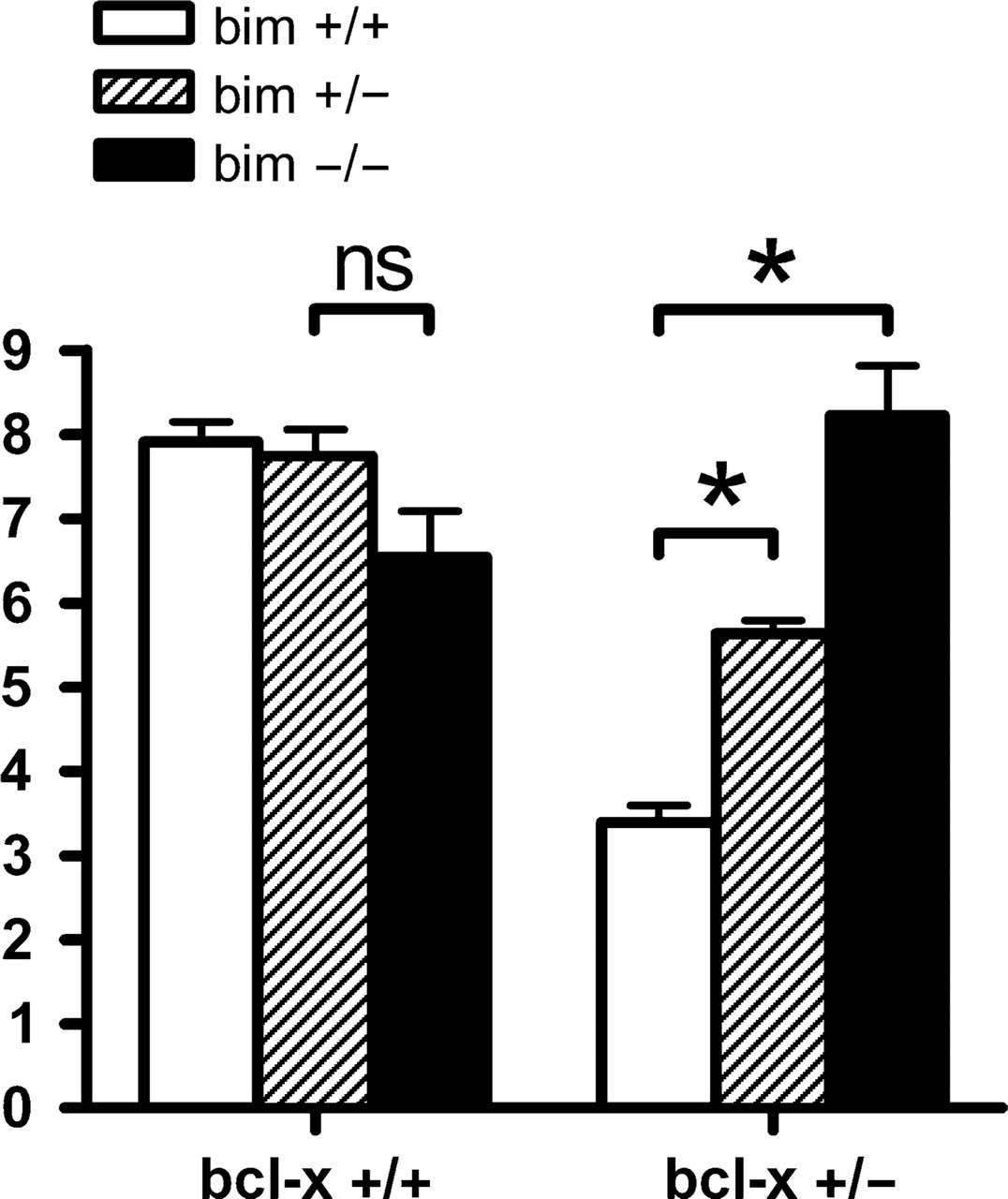
Bim loss reduces testicular hypoplasia in bcl-x happloinsufficient adult mice. Testis weights from multiple adult male mice of the indicated genotypes were assessed, and average testis weight (g)/total body weight (kg) was calculated. Testes of adult bcl-x +/- mice were 57% smaller than those of bcl-x +/+ mice. This size difference was reduced to 27% in bcl-x +/- bim +/- adults and eliminated in bcl-x +/- bim -/- adults. ns, not significant. ∗p<0.005.
Discussion
In this report, we assessed the role of the proapoptotic BH3-only Bcl-2 family member Bim in the abnormal cell death caused by deficiency of the antiapoptotic bcl-x. This aim was accomplished by generating mice that lacked both bim and bcl-x and analyzing the consequences in neural, hematopoietic, and germinal tissues. Because both bim and bcl-x reside on the same chromosome, we identified and bred mice that underwent gametal recombination. These breeding experiments produced embryos that lacked both bim and bcl-x at E12.5, but none of these animals survived to birth. Analysis of these embryos showed that bim deficiency reduced the abnormal death of hematopoietic cells but had no effect on neurodegeneration or embryonic lethality caused by loss of Bcl-xL. We also found that adult bcl-x +/- animals displayed reduced fertility and significant testicular degeneration and showed that this defect was rescued by concomitant bim deficiency. Overall, our results showed that the Bim/Bcl-xL interaction regulates cell fate in a cell type-specific manner.
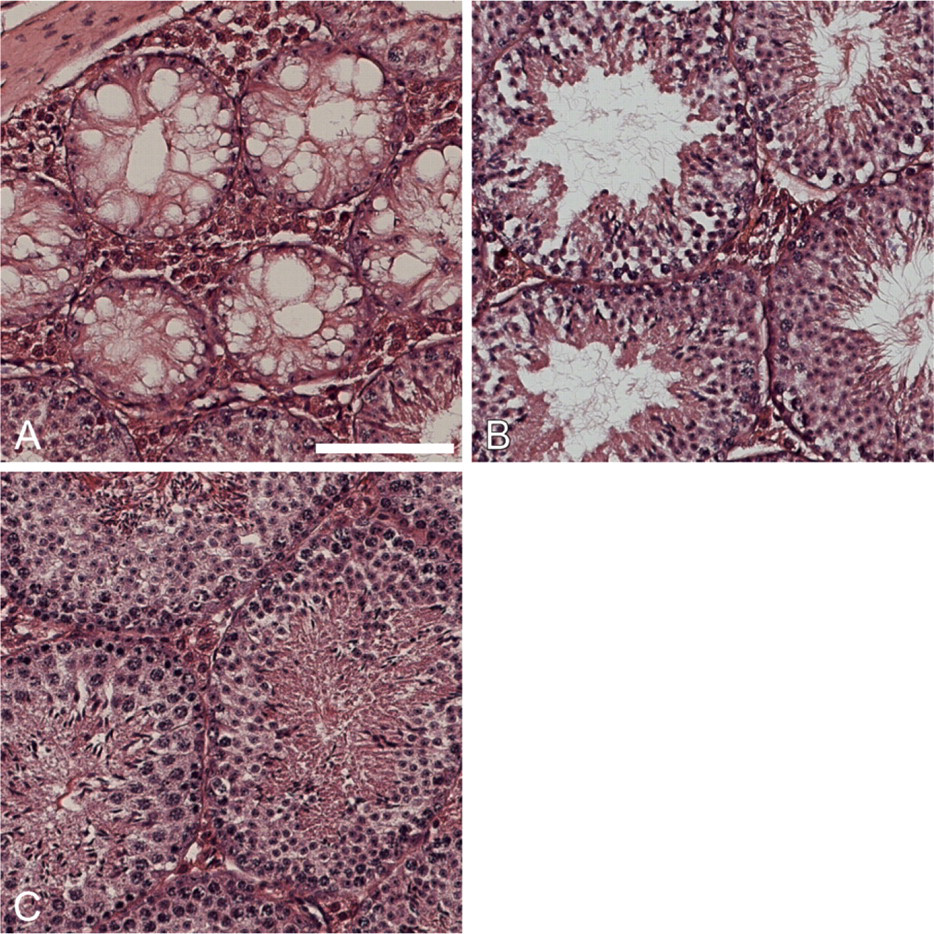
Bim loss rescues testicular degeneration caused by loss of one allele of bcl-x.
Deficiency of bcl-x results in embryonic lethality and abnormally increased apoptosis of neuronal cells in the brain stem, DRG, ventral spinal cord, and erythroid progenitors in the fetal liver. It remains unclear whether neurodegeneration, fetal anemia, or both abnormalities cause embryonic lethality. A number of double knockout mice lacking Bcl-xL plus any one of the proapoptotic factors Bax, caspase-3, or caspase-9 have been generated, but none of these mice survive to birth (Shindler et al. 1997; Roth et al. 2000; Zaidi et al. 2001; Klocke et al. 2002; Cecconi et al. 2004). However, Bax deficiency prevents neurodegeneration seen in bcl-x-deficient animals, and as seen in this report, Bim deficiency protects hematopoietic cells. It therefore seems that abnormal death of either neuronal cells or erythroid progenitors alone is sufficient to cause embryonic lethality in bcl-x -/- mice. Notably, embryonic lethality is seen in mice lacking erythropoietin (Wu et al. 1995), which have defective erythropoiesis but no neuronal abnormalities, and also in mice lacking XRCC4 (a component of the non-homologous DNA end-joining complex), which have abnormal neurogenesis but normal erythropoiesis (Gao et al. 1998). One may therefore predict that combined loss of Bax and Bim might prevent embryonic lethality of bcl-x -/- mice. However, we cannot exclude the possibility that still other cell types, such as hepatocytes, may be affected by Bcl-xL deficiency at later developmental stages and contribute to embryonic lethality in bcl-x -/- animals.
Although Bim has been shown to play a critical role in nerve growth factor deprivation-induced apoptosis of certain neuronal populations (Harris and Johnson 2001; Putcha et al. 2001; Biswas and Greene 2002; Linseman et al. 2002), Bim deficiency, unlike loss of Bax, did not rescue the degeneration of neuronal cells in the DRG and ventral spinal cord caused by loss of Bcl-xL. This indicates that another BH3-only protein may be critical for this death. Puma is a potential candidate, because, like Bim, it binds all prosurvival Bcl-2 family members (Chen et al. 2005) and its loss protects neural cells against certain apoptotic stimuli (Akhtar et al. 2006; Wyttenbach and Tolkovsky 2006). Because BH3-only proteins exhibit significant functional overlap (Coultas et al. 2005; Erlacher et al. 2006), we speculate that Bim and Puma may together cause the neurodegeneration seen in bcl-x -/- mice.
Primordial gonocytes populate the genital ridge before E11.5 and some undergo programmed cell death around E13.5 (Coucouvanis et al. 1993). A variety of hypomorphs for bcl-x have been generated (Rucker et al. 2000; Kasai et al. 2003) that demonstrate the requirement of bcl-x in determining the number of spermatogenic cells that survive during this period. The abnormal death of these cells caused by loss of Bcl-xL seems to require proapoptotic bax (Rucker et al. 2000), and our studies showed that bim is also required for mediating the apoptosis of these cells. Collectively, our studies showed a heretofore undescribed cell type-specific interaction between Bim and Bcl-xL. Additional studies are needed to identify yet other BH3-only proteins that contribute to the increased neuronal apoptosis in bcl-x-deficient embryos.
Footnotes
Acknowledgements
This work is supported by grants from the National Institutes of Health (NS35107 and NS41962). R.S.A. is supported by the University of Alabama-Birmingham (UAB) Medical Scientist Training Program (GM008361). A.S. is supported by the National Health and Medical Research Council (Canberra, Australia), the Leukemia and Lymphoma Society of America, and the Virtual Research Institute of Ageing.
We thank the UAB Neuroscience Core Facilities (NS47466 and NS57098) and Cecelia B. Latham for technical assistance.