
Abstract
Pathology repositories worldwide store millions of celloidin-processed human brain and temporal bone (TB) sections vital for studying central nervous system diseases and sensory organs. However, accessing these sections for modern molecular-pathological research, like immunohistochemistry, is hindered by the challenge of removing celloidin without damaging tissue. In this study, we explored the use of polyethylene glycols (PEGs), a class of non-hazardous, ethylene glycol oligomers, combined with an improved section mounting technique, to gently and effectively dissolve celloidin from sections archived for up to 40 years. Optimizing our protocol involved exploring celloidin dissolution kinetics in PEGs of varying molecular weights and terminations, as well as different temperatures. Low molecular weight PEGs, particularly PEG 200, were the most efficient celloidin solvent. Nuclear magnetic resonance (NMR) spectroscopy of celloidin-PEG 200 dissolution products revealed no chemical alterations, suggesting pure solvation without chemical modification. Because the solvation of celloidin in PEG was inhibited by proteins, we further developed a protein-free mounting protocol allowing complete celloidin removal in 30 to 60 minutes by immersing in PEG 200. In summary, our approach overcomes major methodological hurdles, rendering decades-old archival celloidin sections viable for immunohistochemical and other molecular biological techniques, while enhancing safety and workflow efficiency.
Introduction
Celloidin, a cellulose nitrate polymer, has been a favored embedding medium for histological studies of human brain tissue,1–3 and the human auditory and vestibular sense organs encased in the temporal bone (TB)4,5 for over a century. Its superior qualities over other embedding media, such as paraffin, include thorough penetration of the dense, lipid-rich brain tissue and the compact (decalcified) bone matrix of the TB. This ultimately results in high-quality serial tissue sections with minimal processing artifacts from these challenging specimens. Another notable advantage is celloidin’s permeability to popular histology dyes such as cresyl violet (Nissl stain) and hematoxylin and eosin (HE), facilitating vibrant and background-free staining without needing first to remove the celloidin. However, celloidin’s presence becomes problematic for applications such as immunohistochemistry and nucleic acid extraction. Owing to its strong protein affinity, 6 celloidin binds antibodies and protein-bound DNA, resulting in unspecific (background) staining in immunohistochemical applications7,8 and lower DNA extraction yields. 3 Although existing protocols effectively dissolve celloidin from recently produced sections,7–9 they proved ineffective for completely dissolving “aged” celloidin from older archived sections. Moreover, the harsh chemicals (e.g., acetone, ether/ethyl alcohol, or methanol/sodium hydroxide) used may damage tissue sections that may become increasingly delicate over time. Considering that pathology repositories worldwide hold millions of celloidin sections from human brain and TB specimens, some stored for decades, their compatibility, due to aging celloidin, for modern molecular analysis methods is already limited and is set to reduce further.
To address these challenges, we explored more efficient methods for dissolving celloidin from archival tissue sections. Drawing from prior studies that demonstrated celloidin’s high solubility in polyethylene glycol (PEG) compounds,10–12 we here assessed various PEGs for dissolving celloidin from tissue sections, delved into the underlying chemistry, and developed a streamlined protocol. This protocol not only surpasses the efficiency of previous celloidin removal protocols but also offers a safer and more streamlined workflow, opening new possibilities for accessing and using archival celloidin-embedded specimens in modern molecular analyses.
Materials and Methods
Assessing Celloidin Dissolution in PEGs of Different Molecular Weights and Chemical Formulations
Uniform celloidin pellets were crafted from a recently solidified 12% celloidin block, the standard for embedding human TBs, using a 2-mm biopsy punch (#33-31, Miltex GmbH, Rietheim-Weilheim, Germany). Pellets were left to air-dry overnight, subsequently weighed with a digital analytical balance (AE 240S, Mettler Toledo, Greifensee, Switzerland) and submerged for 10 minutes in 50 ml of either pure diethylene glycol (PEG 106; Thermo Fisher Scientific Inc., Waltham, MA), PEG 200, PEG 300, PEG 400, or PEG 600 (TCI America, Portland, OR). Then, the pellets were rinsed in 1x phosphate-buffered saline (PBS) for 10 min, air-dried overnight, and reweighed. Using the before and after immersion weights (with five pellets tested for each PEG compound), we computed the percentage of dissolved celloidin. Using this experimental setup, we further determined celloidin dissolution in (1) diethylene glycol and PEG 200–600 of equimolar concentrations by diluting diethylene glycol (1:4.56, v/v), PEG 200 (1:2.03, v/v), PEG 300 (1:1, v/v), and PEG 400 (1:0.49, v/v) in distilled water, and thus normalizing their molar concentrations to the free hydroxyl groups per unit volume of PEG 600, (2) PEG 200 at the temperatures 4°C, 22°C, 30°C, 40°C, and 50°C, (3) for PEG 200 using celloidin pellets that were pre-soaked in either pure albumin (Mayer’s fixative, #R03203-70, Merck Millipore, St. Louis, MO), or varied albumin dilutions in PBS (ratios of 1:10, 1:100, 1:1000), and subsequently air-dried overnight before immersing them in PEG 200, and (4) for PEG dimethyl ether (PEG 240; #P224125G, TCI America), a PEG terminated with methoxy instead of hydroxyl groups.
NMR Spectroscopic Analysis of Celloidin Dissolution in PEG
A celloidin piece of approximately 5 × 10 × 30 mm was soaked in 80% ethanol overnight to clean them from cedarwood oil residues, a standard supplement introduced during celloidin block hardening, and air dried for 1 day. The prepared celloidin was immersed in PEG 200 at 20 wt% and stored at room temperature for 3 days to allow for complete dissolution of the celloidin. Proton nuclear magnetic resonance spectroscopy ( 1 H-NMR), carbon nuclear magnetic resonance spectroscopy ( 13 C-NMR), and correlated nuclear magnetic resonance spectroscopy (COSY) spectra in deuterated acetone of PEG 200, celloidin, and celloidin dissolved in PEG 200 were acquired on a Bruker Avance III 400 NMR spectrometer (Bruker BioSpin Corp., Billerica, MA). One sample was used for each NMR measurement. Chemical shifts were reported relative to the respective solvent peak (C3D6O, δ = 2.05 ppm for 1H-NMR and δ = 29.84 and δ = 206.26 for 13C-NMR and COSY).
Human Brain and TB Celloidin Sections
This study received approval from the Institutional Review Board at Massachusetts Eye and Ear (protocol no. 2020P000508). Celloidin-embedded human brain and TB sections from a total of seven tissue donors that were archived in the Otopathology Laboratory at Massachusetts Eye and Ear were used in this study. All specimens were initially embedded in celloidin (parlodion strips, Mallinckrodt Laboratory Chemicals, Phillipsburg, NJ) following established methods. 5 These sections, cut to a thickness of 20 µm, were archived in 80% ethyl alcohol at room temperature for 5 to 40 years prior to their utilization in this study.
Protein-Free, Heat-Facilitated Mounting of Celloidin Sections
The equipment and consumables required for this method are listed in Fig. 1A. The section, immersed in a petri dish with 80% ethanol, is floated onto ungummed cigarette paper (#M1689; OCB, Republic Technologies Int., Perpignan Cedex, France) using an angled glass rod (Fig. 1B). The cigarette paper is trimmed to a size slightly larger than the section to provide a thin border of celloidin around the tissue (Fig. 1C), placed face (section) down onto a SuperFrost Plus microscope slide (#12-550-15, Electron Microscopy Sciences, Hatfield, PA) (Fig. 1D), and then covered with a piece of bibulous paper (Fisherbrand, Fisher Scientific, Waltham, MA) soaked in distilled water (Fig. 1E) and a second cigarette paper (Fig. 1F), carefully avoiding any wrinkles and trapped air bubbles. The layered papers are then wetted with a few drops of distilled water before placing a 500-g lead weight with dimensions of 25.4 mm [W] × 50.8 mm [L] × 50.8 mm [H]) on top (Fig. 1G). After approximately 30 minutes, a few more drops of distilled water are added to the layered paper around the lead weight to prevent the sections beneath from drying out. After 1 hour, the weight and the papers are carefully removed one after the other (Fig. 1H to J). The mounted section is air-dried for approximately 10 minutes and covered with a sheet of polytetrafluoroethylene (PTFE) membrane (thickness 0.1 mm), which prevent detachment of adhesions of the histologic sections when storing multiple slides pressure in a mounting press (Fig. 1K). The slide is placed in a slide mounting press (#1465315, Technovit mounting press, Electron Microscopy Sciences) (Fig. 1L). The press is firmly tightened (Fig. 1M) and stored in a dry heat environment (e.g., paraffin oven) at 50°C (Fig. 1N). If multiple sections are mounted at a time, the slides covered with PTFE membrane can be stacked upon each other in the slide mounting press. The next morning, the mounting press is removed from the oven and cooled to room temperature before releasing its pressure (Fig. 1O), removing all slides from the press, and carefully peeling off the PTFE membrane from each slide (Fig. 1P). The mounted sections (Fig. 1Q) can be subsequently used for celloidin removal and immunohistochemistry (see next paragraphs), or stored at room temperature, for example, in a microscope slide box, for weeks to months before further use. This method is referred to as “protein-free mounting.”

Step-by-step instructions for mounting archival celloidin sections for PEG 200-mediated celloidin removal. The detailed protocol, (A) to (Q), is given in the “Methods” section.
To compare the effectiveness of celloidin removal following protein-free mounting and the standard method that uses protein adhesive slide coating (“protein-mediated mounting”), some sections were mounted using gelatin-subbed glass slides smeared with albumin adhesive solution and dried for 1 hour at room temperature, as previously described. 8
Celloidin Removal From Celloidin Sections Using PEG 200
Mounted celloidin sections are immersed in a Coplin staining jar filled with PEG 200 (Fig. 2A) that is placed on a rocker shaker (Fig. 2B). Celloidin is completely dissolved after approximately 30 minutes in recently produced sections, and after approximately 60 min in archival (storage times above 15 years) sections. The slides are then transferred to a Coplin staining jar filled with distilled water (Fig. 2C). Residual, that is, incompletely removed, celloidin will eventually become visible as white (“foggy”) residue after approximately 3 minutes of immersion in distilled water (Fig. 2D). Slides with residual celloidin are transferred back to PEG 200 for another 15 to 30 min, depending on the amount of residual celloidin, before checking again for completeness of celloidin removal in distilled water (Fig. 2D). Changes between PEG 200 and distilled water can be repeated until celloidin is completely removed. PEG 200 can be recycled; that is, 50 ml of solution (approximately volume of one Coplin jar) can be used to dissolve the celloidin from approximately eight sections (2 × 1 inches) before the solution appears to saturate, as suggested by progressively longer times needed for complete celloidin removal.

PEG 200-mediated celloidin removal from TB sections. (A) Mounted TB sections are immersed in a glass Coplin staining jar filled with PEG 200. (B) The jar is placed on a rocker shaker for 30 to 60 minutes. (C) To check for completeness of celloidin removal, the slides are then transferred to a Coplin staining jar filled with distilled water. (D) Incomplete celloidin removal will result in residual celloidin becoming eventually visible as white (“foggy”). No residual celloidin can be seen after complete celloidin removal.
For experiments comparing the effectiveness of celloidin removal between the method introduced here and the standard method, celloidin was dissolved from a few mounted sections using methanol saturated with sodium hydroxide, as described previously. 8
A detailed step-by-step protocol of the PEG 200-mediated celloidin removal is provided as Appendix. Possible solutions of issues that may be encountered are summarized in Table 1.
Troubleshooting Table With Issues That May Be Encountered With the PEG 200-Mediated Celloidin Removal and Possible Solutions.

HE Staining
Celloidin sections were processed either by (1) HE staining free-floating without prior celloidin removal, then mounting and coverslipping with Permount (#SP15-500, Fisher Scientific, Pittsburgh, PA) on microscope slides (standard method), (2) mounting on microscope slides using protein-free mounting (see Fig. 1), followed by HE staining and coverslipping in Permount without prior celloidin removal, or (3) protein-free mounting followed by celloidin removal (see Figs. 1 and 2) prior to HE staining and coverslipping in Permount. To assess whether protein-free heat-facilitated section mounting and PEG 200-mediated celloidin removal methods introduce any tissue damage or other artifacts, we qualitatively compared the light-microscopic histomorphology of celloidin sections processed with these three approaches.
Immunohistochemistry
Celloidin sections were either mounted using the standard method (protein-mediated mounting) or the protein-free mounting method, followed by celloidin removal using sodium methoxide 8 or PEG 200 (present protocol). Sections from which celloidin had been removed were subsequently used for enzyme immunohistochemistry with 3,3ʹ-diaminobenzidine (DAB) and immunofluorescence labeling.
For DAB immunohistochemistry, sections were incubated with 5% normal horse serum (NHS) in PBS for 30 min. Next, a primary antibody raised in rabbit and directed against ADAM metallopeptidase with thrombospondin type 1 motif 18 (ADAMTS18; #NBP1-91651, Novus Biologicals, Centennial, CO) was applied overnight. The sections were then incubated with a biotinylated secondary antibody raised in goat and directed against rabbit IgG (#111-065-144, Jackson Immunoresearch, West Grove, PA) for 1 hour, followed by application of a horseradish peroxidase-based avidin-biotin (ABC) reagent (#PK-4000, Vector Laboratories, Newark, CA) for 1 hour. Then, the sections were incubated in 4% DAB/hydrogen peroxide in PBS for 5 minutes to develop the brown reaction product. Between all steps, the sections were washed in PBS for 5 min. Hematoxylin was used to counterstain cell nuclei. The slides were then dehydrated in an ascending ethanol series, cleared with Histoclear (#HS2001GLL, National Diagnostics, Atlanta, GA), mounted with Permount (#SP15-500, Fisher Scientific), and air-dried.
A negative control experiment was performed by immunolabeling human fetal otic capsule tissue using an anti-Adamts18 antibody with pre-absorbing the antibody overnight at room temperature with the control peptide (10:1; #NBP1-91651PEP, Novus Biologicals; Fig. A1).
For immunofluorescence labeling, sections were either incubated overnight with a primary antibody directed against osteoprotegerin (OPG; #ab73400, Abcam, Waltham, MA; dilution 1:100) and raised in rabbit, or with two primary antibodies that were directed against neurofilament 200 (NF200; #N4142, MilliporeSigma, Burlington, MA; dilution 1:500) and α-tubulin (#T5168, MilliporeSigma; dilution 1:100) and that were raised in rabbit and mouse, respectively. The primary antibodies were visualized by incubating sections for 1 hour with secondary antibodies conjugated with Alexa 488 (#A21206, Invitrogen/ThermoFisher Scientific, Carlsbad, CA; dilution 1:400) and Alexa 568 (#A10037, Invitrogen/ThermoFisher Scientific; dilution 1:400) respectively, both of which were raised in donkey, before coverslipping in Vectashield mounting medium with DAPI (#H-1200, Vector Laboratories, Burlingame, CA). All antibodies were diluted in 1% NHS in PBS. All incubation steps were performed at room temperature.
Image Acquisition
DAB-stained sections were analyzed using an Olympus BX51 microscope (Olympus, Tokyo, Japan) equipped with an Olympus DP70 digital camera (Olympus). Analysis of fluorescent-labeled sections was performed using a Leica TCS SP8 confocal microscope (Leica, Mannheim, Germany).
Statistics
Statistical analyses were performed using Prism for Apple Macintosh, version 9.5.1 (GraphPad Software, Inc., La Jolla, CA). To test whether means of several experimental groups are different, a one-way analysis of variance (ANOVA) with post-hoc multiple comparisons was used. To correct for multiple comparisons, Tukey’s test was used for data with normal distribution, and Dunn’s test was used for data with non-normal distribution. The significance level was set to p < 0.05.
Results
Celloidin Dissolution With Low Molecular Weight PEG
PEGs are linear polymers of the monomer ethylene glycol and are categorized according to their average number of monomers (Fig. 3A), determining their molecular weight (MW) and chemical features. 13 We explored the celloidin dissolution rates for diethylene glycol (PEG 106; average MW = 106 g/mol), PEG 200 (MW = 200 g/mol), PEG 300 (MW = 300 g/mol), and PEG 400 (MW = 400 g/mol), and PEG 600 (MW = 600 g/mol), as well as for PEG dimethyl ether (PEG 240). Those, in contrast to higher MW PEG compounds (PEG ≥ 800), are in a liquid state at room temperature and thus, are, in principle, suited for a celloidin removal protocol at room temperature. Here, we determined the celloidin dissolution rates (kinetics) for diethylene glycol—PEG 600 by immersing standardized celloidin pellets (Fig. 3B) in neat solutions of these compounds and measuring the pellets’ dry weight loss after 10 minutes of immersion. We measured a significant weight loss—corresponding to high celloidin dissolution rates—for diethylene glycol, and PEG 200 to 400, while we did not observe a significant weight change for PEG 600 (Fig. 3C). Among the PEGs terminated with hydroxyl groups, the highest celloidin dissolution rate was determined for PEG 200, with decreasing dissolution rates for increasing PEG MW; except for the lowest-molecular-weight oligomer, diethylene glycol, which dissolved slightly (mean difference 8.7 percentage points, p = 0.03) less celloidin as compared with PEG 200 with more variable data (diethylene glycol < PEG 200 > PEG 300 > PEG 400 > PEG 600; Fig. 3C). PEG 240 terminated with methoxy instead of hydroxyl groups dissolved significantly more celloidin that PEG 200. To equalize the molar concentration of hydroxyl functionalities, that is, the quantity of reactive OH groups (Fig. 3A) per volume, which decreases with increasing MW of the PEG compound in neat solutions as used in the experiments above, we prepared solutions of each compound with a fixed molar concentration of hydroxyl groups (relative to neat PEG 600). After diluting diethylene glycol, PEG 200, PEG 300, and PEG 400 to match the molar concentration of hydroxyl groups in PEG 600, we found markedly reduced celloidin dissolution rates for all compounds (Fig. 3D). Nevertheless, a significant reduction in pellet weight was still observed for diethylene glycol, PEG 200, PEG 300, and PEG 400.
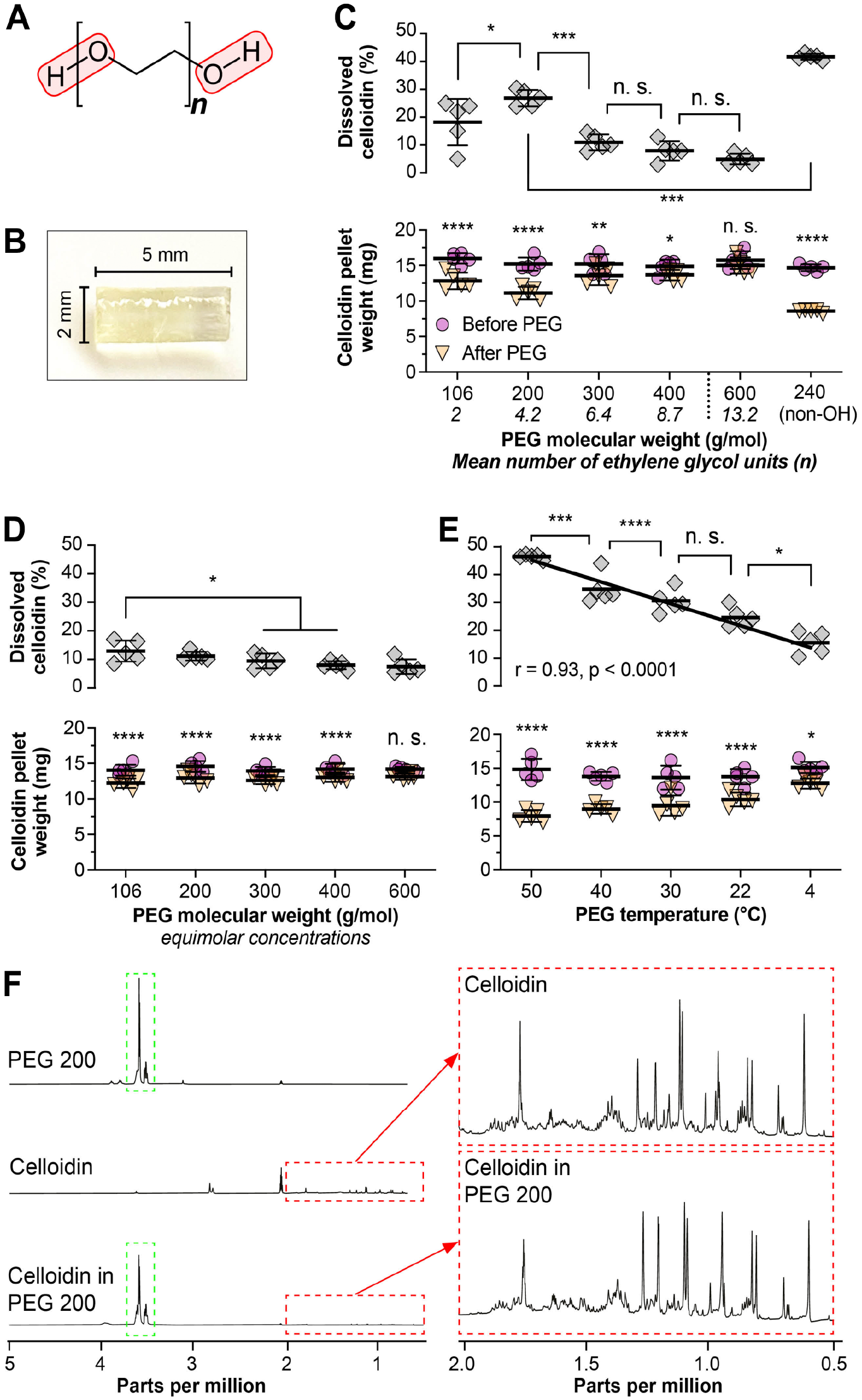
Determining the relative and absolute celloidin solubility for PEGs. (A) General chemical structure of PEG with varying number (n) of repeating units of ethylene glycol. The polar hydroxyl groups are outlined in red. (B) Celloidin pellet used for determining the celloidin solubility in solutions of PEGs. (C) Celloidin solubility in PEGs of different molecular weight. (D) Celloidin solubility in PEGs of different molecular weight at equimolar concentrations of hydroxyl groups normalized to PEG 600. (E) Celloidin solubility in PEG 200 at different temperature levels. The upper graph shows the correlation between temperature und relative celloidin solubility. (F) Comparative NMR spectroscopic analysis of nitrocellulose before and after dissolution in PEG 200, with spectra displayed for PEG 200 (top left spectrum), celloidin (middle left spectrum), and celloidin dissolved in PEG 200 (bottom left spectrum). The persistence of key spectral features in the celloidin and celloidin in PEG 200 spectra (red dashed box) indicates that celloidin does not undergo detectable changes in its chemical structure when dissolved in PEG 200. Green dashed box, characteristic spectral region of PEG 200. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; n. s., not statistically significant; r, correlation coefficient. Solid line indicates linear regression line. In Fig. 3C to E, the y-axis applies to both the upper (dissolved celloidin) and lower (celloidin pellet weight) panel.
Celloidin Dissolution in PEG Exhibits Temperature-Dependent Kinetics
We tested the effect of different temperatures on the celloidin dissolution rates in PEG 200, the compound that demonstrated (one of) the highest celloidin dissolution rates, rendering it the preferred candidate for celloidin removal from tissue sections. Within the tested temperature range, that is, from 4°C to 50°C, we found a strong negative correlation between decreasing temperature and the amount of dissolved celloidin (r = −0.93, p < 0.0001; Fig. 3E), although increasing the temperature from 22°C (room temperature) to 30°C had no significant effect on the amount of dissolved celloidin. At 40°C, significantly more celloidin was dissolved in PEG 200, but exposing mounted sections to 40°C, in our experience, leads to their partial or complete detachment from the glass slides.
Celloidin dissolution in PEG did not result in changes in the molecular structure of the polymers
To explore the chemistry underlying celloidin dissolution in PEG 200, we investigated samples of celloidin, PEG 200, and the solution of celloidin in PEG 200, using NMR (Fig. 3F). In 1H-NMR and 13C-NMR spectroscopic measurements, the peaks corresponding to PEG 200 and celloidin did not change substantially in all three samples, indicating that the dissolution was not mediated by molecular changes to the celloidin polymer. In addition, COSY analysis, which indicates protons that are in close proximity of each other via chemical binding, of the dissolved mixture did not indicate any coupling between hydrogens of PEG 200 and as would appear at potential sites of chemical binding. This finding further argues against the formation of intermolecular species between PEG 200 and celloidin.
Celloidin-Protein Interactions Abolish the Dissolution of Celloidin in PEGs
In pilot experiments, we observed that coating microscope slides with the protein adhesives gelatin and albumin, as is part of the protein-mediated mounting method,5,8 prolonged complete celloidin removal from approximately 1 hour to several (3–4) weeks. To systematically investigate the chemical interactions between celloidin, proteins, and PEG and to identify avenues to optimize the celloidin removal protocol for tissue sections, we soaked celloidin pellets in albumin before immersion in PEG 200, which completely abolished celloidin dissolution (no significant pellet weight change) within the 10-min immersion period for all tested albumin concentrations (1:1, 1:10, 1:100, 1:1000; Fig. 4A). We further assessed the kinetics of celloidin removal more qualitatively on toluidine blue-stained TB sections after protein-mediated mounting5,8 and protein-free mounting (see material and methods section, Fig. 1) and immersion in PEG 200 for 60 min. Light microscopic and confocal (tissue and celloidin autofluorescence; excitation filter: 560/40 nm; emission filter: 610/75 nm) analyses demonstrated complete removal of the lightly toluidine blue-stained celloidin matrix from sections after protein-free mounting (Fig. 4B and B’) and an incomplete celloidin removal with abundant, patchy residual celloidin on sections after protein-mediated mounting (Fig. 4C and C’).
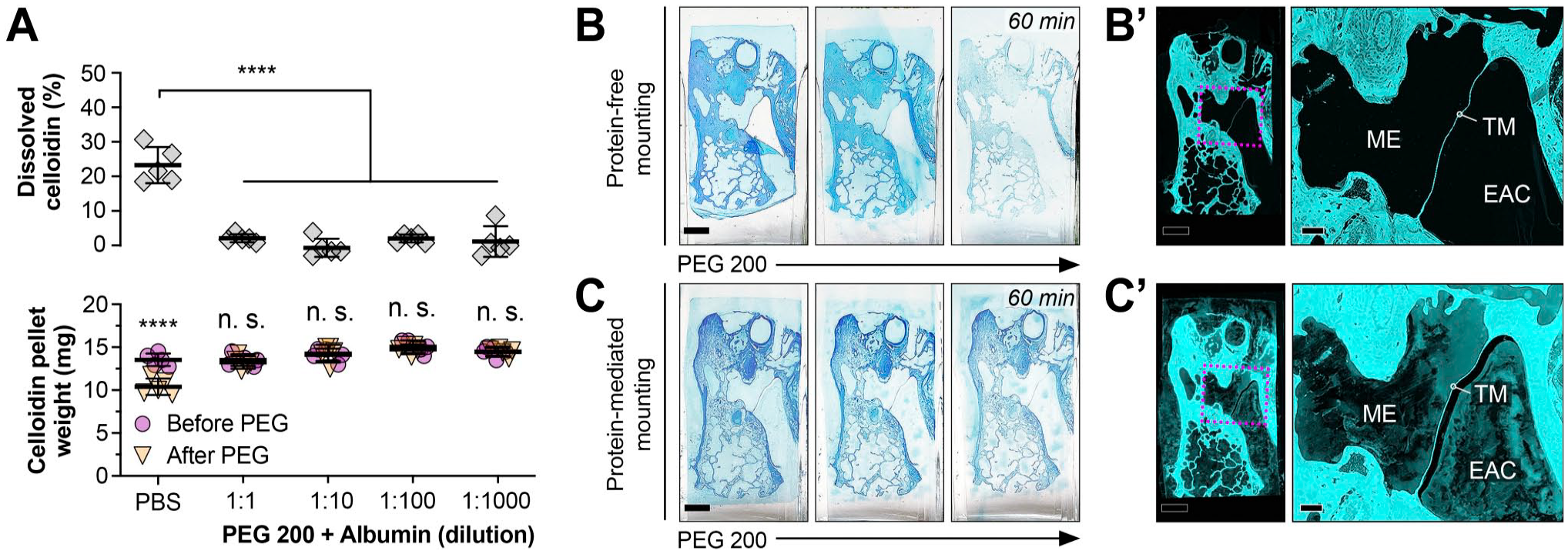
Interaction of PEG 200-mediated celloidin removal with albumin. (A) Solubility in PEG 200 of pellets that were pre-soaked in albumin. ****, p < 0.0001; n. s., not statistically significant. (B–C’) Qualitative assessment of PEG 200-mediated celloidin removal from TB sections after protein-free (B–B’) and protein-mediated (C–C’) mounting. In each row, the first three images show PEG 200-mediated celloidin removal of a toluidine blue stained-celloidin TB section over 60 min at intervals of 20 min. The last two images (B,’ C’) show laser scanning-based tissue and celloidin autofluorescence at 594 nm after 60 min of PEG 200-mediated celloidin removal of the respective TB section (dashed square indicates area shown in higher magnification on the right). Note the residual autofluorescent celloidin in the middle ear (ME) and the external auditory canal (EAC) as well as in the human TB tissue. TM, tympanic membrane. Scale bars: B/C, B’/C’ (left panel) 500 μm; B’/C’ (right panel) 100 μm.
Protein-free mounting and PEG-based celloidin removal preserve tissue morphology and enhance tissue antigenicity of archival celloidin sections
We mounted celloidin sections either with the standard method using protein adhesives (protein-mediated mounting) or with our heat-facilitated protein adhesive-free mounting technique (protein-free mounting), followed by celloidin removal using either sodium methoxide or PEG 200 to compare the effects of the different mounting and celloidin removal methods in subsequent histochemical and immunohistochemical applications.
HE-stained TB sections processed with the novel protein-free mounting technique followed by PEG 200-mediated celloidin removal demonstrated excellent morphological preservation with only very minor artefactual damage (Fig. 5). Since HE staining was performed on mounted sections instead of free-floating (Fig. 5A and A’), the staining intensity was overall weaker (Fig. 5B to C’), necessitating adjustments in staining times.
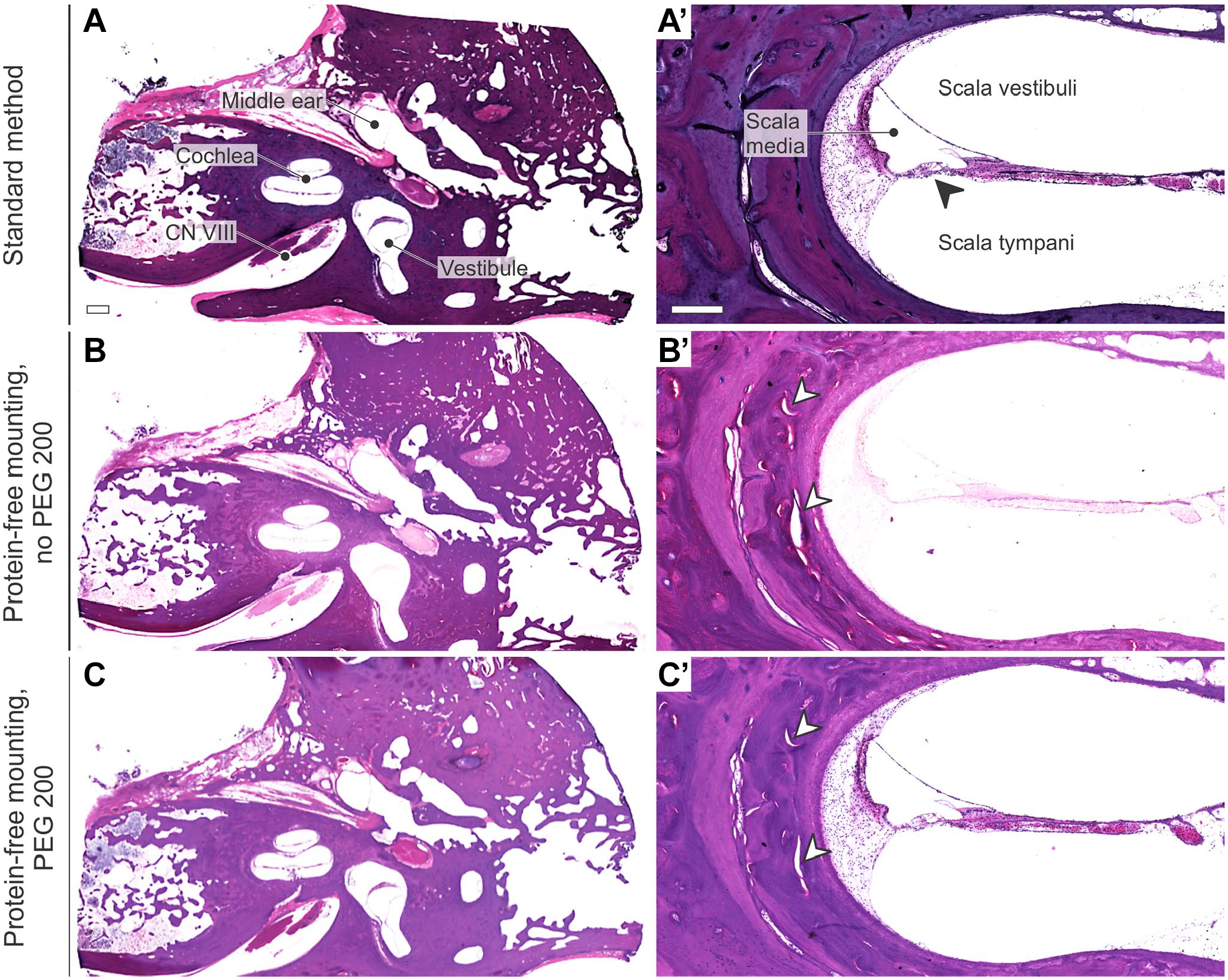
Histomorphology (HE staining) of TB sections following (A–A’) free-floating HE staining, without prior section mounting or celloidin removal, and coverslipping in Permount (reference method), (B–C’) protein-free mounting without (B–B’) and with (C–C’) subsequent PEG 200-mediated celloidin removal (protocols in Figs. 1 and 2). Note the different staining intensities due to free-floating (A–A’) vs. mounted (B–C’) HE staining. Arrowheads, artefactual damage; CN VIII, cranial nerve eight (vestibulocochlear nerve). Scale bars: A 1 mm; A’ 200 µm.
After protein-mediated mounting of celloidin sections, neither sodium methoxide (five rinses) nor PEG 200 (2 hours immersion) sufficiently removed the celloidin matrix. This resulted in nonspecific staining of residual celloidin that obscured most of the sections when applying enzyme immunohistochemistry with DAB for visualizing an anti-Adamts18 primary antibody (Fig. 6A and B). In contrast, after protein-free mounting, both sodium methoxide and PEG 200 achieved virtually complete removal of the celloidin matrix, enabling specific and strong immunolabeling of ADAMTS18 in the spiral ligament and the encapsulating otic capsule, as demonstrated in TB sections from a human fetus at 19 weeks gestational age (Fig. 6C and D). Similarly, when applying immunofluorescence labeling for OPG, NF200, and α-tubulin on sections after protein-mediated mounting and treatment with sodium methoxide, extensive celloidin residues caused strong unspecific autofluorescence signals and made interpretation of any specific immunolabeling impossible (Fig. 6E and G). In contrast, TB sections processed using protein-free mounting and treated with PEG 200 demonstrated specific, background-free fluorescence labeling of OPG in cells in the spiral limbus, supporting cells on the basilar membrane, and osteocytes in the otic capsule (Fig. 6F). Moreover, brain sections processed using the same protocol demonstrated specific, background-free fluorescence labeling of NF200 and α-tubulin (Fig. 6H). The immunostaining patterns for ADAMTS18 and OPG in human TB sections, achieved using the new protocol described here, will be described and discussed in separate publications.
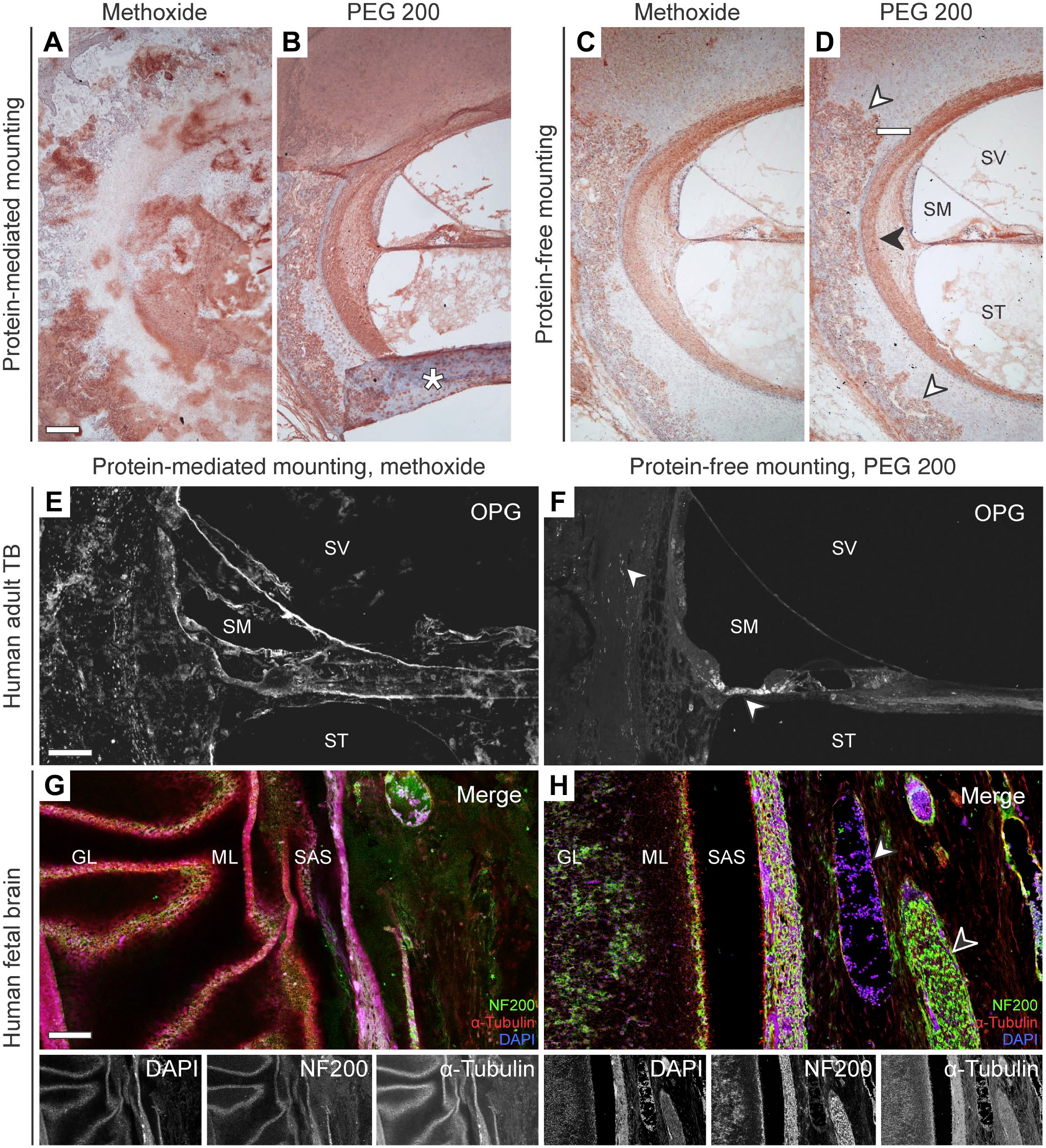
Enzyme immunohistochemistry with DAB and immunofluorescence labeling of TB and brain sections following celloidin removal using either methoxide or PEG 200 and either protein-mediated or protein-free mounting. (A–D) DAB immunolabeling for ADAM Metallopeptidase With Thrombospondin Type 1 Motif 18 (ADAMTS18) in the cochlea and adjacent otic capsule. The white arrowhead indicates ADAMTS18-positive chondrocytes within an expansion front of an ossification center in the otic capsule. The black arrowhead indicates ADAMTS18-positive lateral wall fibrocytes. Asterisk, artefactually folded tissue area; SM, scala media; ST, scala tympani; SV, scala vestibuli. (E–F) Immunofluorescence labeling for Osteoprotegerin (OPG) in the cochlea and adjacent otic capsule after celloidin-removal using either methoxide (E) or PEG 200 (F). Arrowheads, OPG-positive cells near the organ of Corti. SM, scala media; ST, scala tympani; SV, scala vestibuli. (G–H) Immunofluorescence labeling for the 200 kD neurofilament (NF200), α-Tubulin, and DAPI after celloidin-removal using either methoxide (G) or PEG 200 (H) on brain sections. Note the patchy, unspecific staining of the remaining celloidin in (G). GL, granular cell layer; ML, molecular layer; SAS, subarachnoid space; black arrowhead, cranial nerve; white arrowhead, blood vessel. Scale bars: A/E 100 μm; G 200 μm.
Discussion
Here, we introduce a novel approach using PEG compounds along with a protein adhesive-free heat-facilitated section mounting technique to remove celloidin effectively, gently, and rapidly from archival celloidin-embedded tissue (brain and TB) sections. This method enables an easier workflow as compared to previous celloidin removal protocols and unlocks the potential of historic celloidin-embedded tissue collections for contemporary (molecular) research methods.
The popularity of celloidin as a histological embedding media throughout the last century, particularly for human brain1–3 and TB specimens,4,5 has led to the establishment of extensive celloidin-embedded brain14,15 and TB tissue archives. 16 These archives provide invaluable resources for studying the pathologies of brain diseases, as well as hearing and balance disorders. Using these historic resources with modern molecular biology applications becomes increasingly significant, as the availability of fresh tissue samples for research declines due to an ongoing decrease in postmortem tissue donations. 17 However, significant challenges arise when using archival celloidin sections for immunohistochemical experiments. First, the “chemical aging” process of celloidin over decades-long storage, involving hydrolysis, ester cleavage, oxidation, and polymer chain scission, effectively “locks in” the tissue within a progressively brittle and poorly soluble celloidin matrix. in addition, the use of protein adhesive slide coatings in previous protocols for mounting celloidin sections 8 creates strong chemical bonds with both the tissue and the 6 celloidin matrix, which exhibits a strong chemical affinity to proteins, 6 reducing the effectiveness of subsequently applied celloidin solvents, as determined in this study. To address these challenges, our new method omits protein adhesives and instead “bakes” celloidin sections onto SuperFrost Plus microscope glass slides. Under these “protein-free” conditions, the celloidin matrix becomes more susceptible to chemical interactions with its solvents, aiding in its rapid dissolution. An additional benefit of the new mounting technique is the flattening of the celloidin section by pressure applied during “baking,” in principle enabling more consistent immunostainings and easier microscopic analysis (no “wavy tissue sections”), both of which could be difficult to accomplish using standard wet mounting techniques.
Regarding the chemistry of PEG-mediated celloidin dissolution, our measurements of celloidin dissolution kinetics and NMR analysis did not indicate any chemical changes in the celloidin or PEG after dissolution, suggesting that dissolution was not mediated by chemical reactivity, nor did it require the presence of hydroxyl groups (e.g., in PEG dimethyl ether, i.e. PEG 240). Based on this new data, we hypothesize that the amphiphilic properties of PEG 18 are critical to mediate celloidin dissolution. Celloidin forms a solid material due to hydrogen bonding and van der Waals forces between disparate celloidin chains. 19 As PEG exhibits both hydrophilic and hydrophobic properties, PEG may be able to interact with celloidin and disrupt the hydrogen bonds or van der Waals interactions between individual celloidin chains. Of the samples tested in our work, PEG 200 dissolved celloidin most efficiently, indicating that it may possess the right MW and amphiphilicity to interact with and dissolve the celloidin chains. PEG dimethyl ether (PEG 240) showed a comparable efficiency; however, it is less suited due to its flammable, irritant and hazardous properties as well as its higher price compared to PEG 200. 20 Diethylene glycol resulted in decreased dissolution perhaps due to its increased hydrophilicity relative to PEG 200. 18 Higher MW PEG molecules also exhibited decreased dissolution, which may be attributed to the increased size or changes in the amphiphilicity relative to PEG 200.
Importantly, PEG is a widely used polymer in the biomedical sciences with a proven track record of safety and compatibility with biological specimens. 10 Furthermore, PEG has been explored as a “Green solvent” in the context of sustainable, “Green” chemistry focusing on minimizing the generation and application of hazardous substances. 21 This combined with the evidence that there are no hazardous reaction products resulting from dissolving celloidin in PEG 200, makes PEG 200-based celloidin dissolution an effective and safe procedure. On a note, diethylene glycol, another compound that showed favorable celloidin dissolution kinetics in our study, is hazardous to human health,22,23 and, when interacting with celloidin, may form explosive reaction products, such as diethylene glycol dinitrate (“blasting oil”). 24 Furthermore, for PEG 3350, a higher MW PEG not used in this study, rare but severe allergic reactions are described after ingestion, 25 underscoring the need for safety awareness and adherence to standard good laboratory practices.
Finally, it is crucial to acknowledge that the challenges associated with using archival celloidin sections for molecular analysis methods go beyond the necessity for comprehensive removal of the celloidin-embedding matrix. The celloidin-embedding procedure, during which the tissue is exposed to various chemicals over rather long time periods, partially compromises the preservation and accessibility of tissue proteins and nucleic acids (DNA, RNA). Furthermore, during long-term storage, usually in aqueous alcohol (80% v/v), the degradation of these crucial biomolecules in celloidin sections further occurs. This presents a significant problem that requires further exploration and the development of solutions to ensure the integrity and reliability of molecular analysis conducted on archival celloidin-embedded specimens.
Footnotes
Appendix
Acknowledgements
The authors are grateful for the technical expertise of Barbara J. Burgess, Diane D. Jones, Clarinda C. Northrop, MengYu Zhu, and Anbuselvan Dharmarajan in preparing the human temporal bone specimens. They thank Garyfallia Pagonis for expert assistance in photography and image processing, resulting in Figs. 1 and ![]() .
.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AHE conceived and designed the study; DB, JTO, MW, and AHE acquired the data; DB, JTO, MW, SB, MWT, and AHE analyzed and interpreted the data; AHE, DB, MW, and MCL drafted the manuscript; all authors critically reviewed and revised the manuscript; all authors approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: By the National Institute on Deafness and Other Communication Disorders (NIDCD) grant no. U24DC020849.