
Abstract
The large family of transient receptor potential (TRP) channels are integral membrane proteins that function as environmental sensors and act as ion channels after activation by mechanical (touch), physical (heat, pain), and chemical stimuli (pungent compounds such as capsaicin). Most TRP channels are localized in the plasma membrane of cells but some of them are localized in membranes of organelles and function as intracellular Ca2+-ion channels. TRP channels are involved in neurological disorders but their precise role(s) and relevance in these disorders are not clear. Endothelial cells of the blood–brain barrier (BBB) express TRP channels such as TRP vanilloid 1–4 and are involved in thermal detection by regulating BBB permeability. In neurological disorders, TRP channels in the BBB are responsible for edema formation in the brain. Therefore, drug design to modulate locally activity of TRP channels in the BBB is a hot topic. Today, the application of TRP channel antagonists against neurological disorders is still limited.
Introduction
Blood vessels in larger organisms are organized as a vascular tree to deliver oxygen and nutrients to tissues and to remove carbon dioxide and waste products from tissues and to facilitate activity of hormones and the immune system.1–4
The final vascular component of the arteriolar tree, in communication with the venous components for draining blood from tissues, 5 is classified as: (1) continuous non-fenestrated capillaries, (2) continuous fenestrated capillaries, and (3) discontinuous capillaries 6 (Fig. 1). “Con-tinuous non-fenestrated capillaries” are present in skin, lung, and brain; these capillaries strictly regulate transport over the capillary wall. “Continuous fenestrated capillaries” are present in regions where high filtration or trans-endothelial transport is required; these capillaries are found in the choroid plexus, glomeruli, renal tubules, and glands.
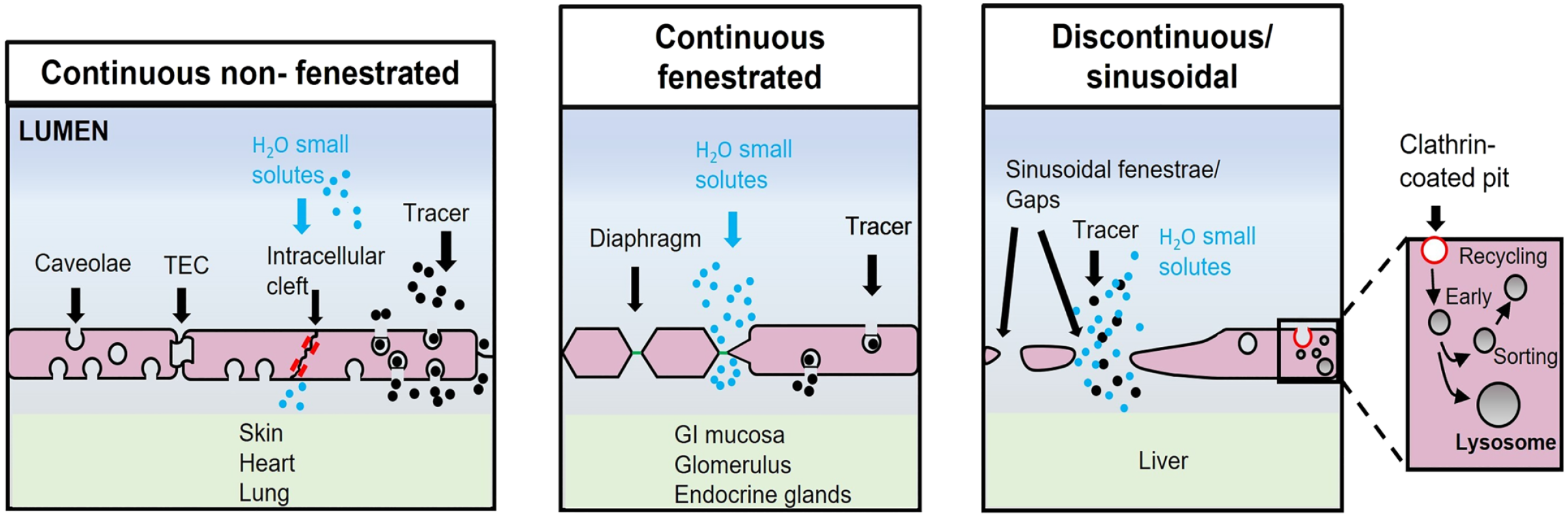
Different types of capillaries and their permeability. Capillaries allow the transfer of fluids and solutes between the blood and underlying tissues. Continuous non-fenestrated capillaries: Water and small solutes go directly through endothelial cells (ECs), whereas larger solutes pass via trans-endothelial channels mediated by caveolae. Continuous fenestrated capillaries: more permeable to water and solutes than continuous non-fenestrated capillaries. Discontinuous/sinusoidal capillaries: characterized by a disorganized lamina basalis and the presence of fenestrae and gaps. The ECs contain many clathrin-coated pits, important for the receptor-mediated endocytosis (endosomal and lysosomal compartments are included). The image has been prepared based on the image in Aird. 6 Abbreviation: GI, gastrointestinal; TEC, transendothelial channel.
Continuous non-fenestrated capillaries have different effects on movement of solutes between blood and tissues and they are most restrictive compared with discontinuous vessels. 7 “Discontinuous vessels” or “sinusoidal capillaries” are found in the liver; they show many gaps among the cells and have an incomplete lamina basalis (LB; Fig. 1).6,8
The capillaries are the key components of the biological barrier at the blood–brain interface. 9 These barriers are determined by different cells at three levels: the “blood–brain barrier (BBB),” “blood–cerebrospinal fluid barrier,” and the “arachnoid barrier” (Fig. 2). 10
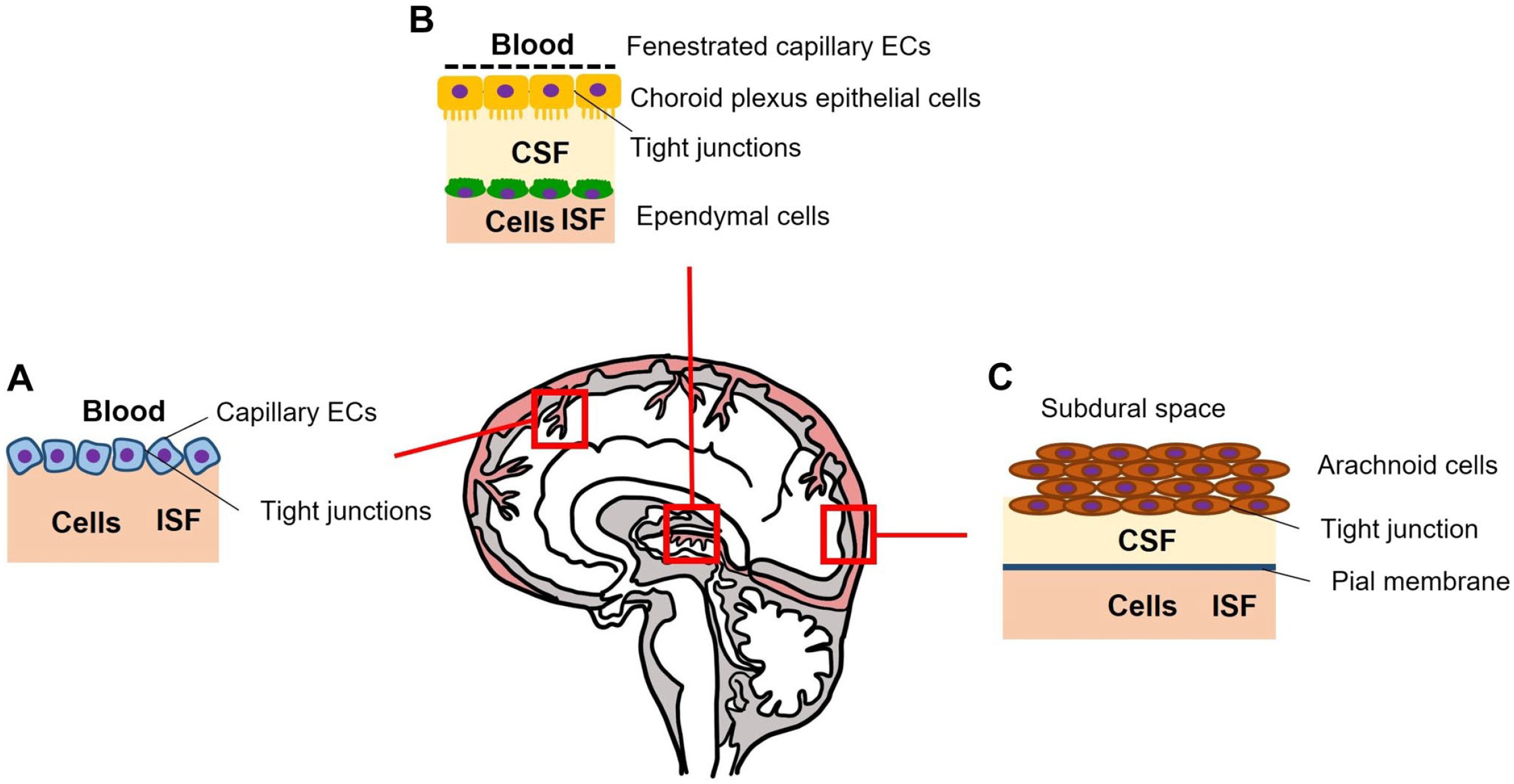
Barrier that protect the brain. (A) Blood–brain barrier; (B) blood–cerebrospinal fluid barrier; and (C) the arachnoid barrier. The image has been prepared based on the image in Abbott et al. 10 Abbreviations: ECs, endothelial cells; CSF, cerebrospinal fluid; ISF, interstitial fluid.
BBB and Its Components
The BBB is the main barrier contributing to central nervous system (CNS) protection and homeostasis of the brain. 10
Figure 3 shows the cell types in the BBB.
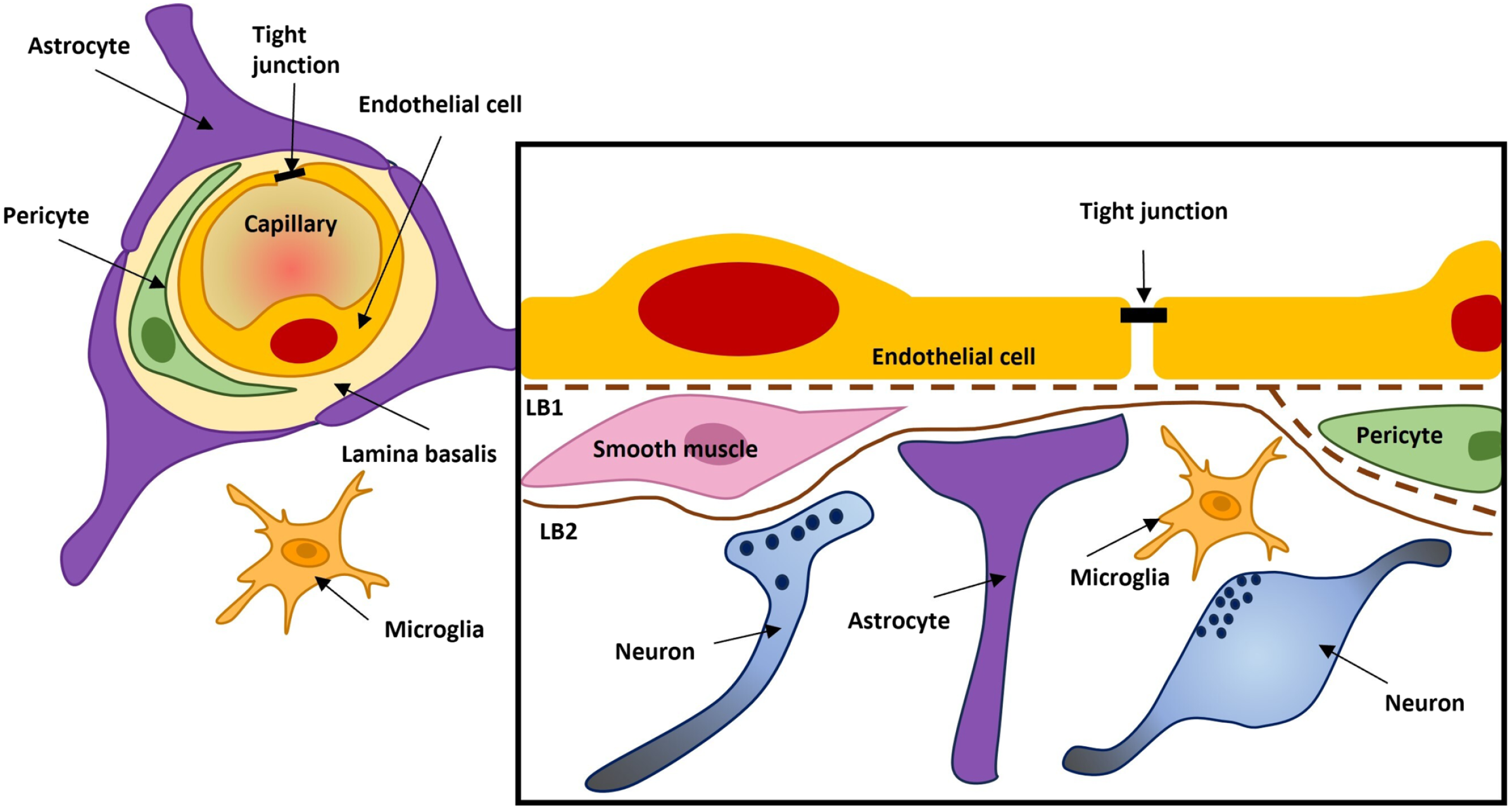
The cellular composition of the blood–brain barrier. The endothelial cells have tight junctions which seal off the diffusional pathway between the cells (paracellular transport). Pericytes are distributed along the vessels and partially cover the endothelium. The image has been prepared based on the image in Abbott et al. 10 Abbreviations: LB1, lamina basalis; LB2,, lamina basalis 2.
The end-feet of the astrocytes surround the capillaries, and they are involved in the integrity of BBB. They also enhance the level of the tight junction (TJ) proteins between endothelial cells (ECs) such as pentraxin 3; this has been demonstrated in animal models. Temporarily reduced astrocytes expression caused decreased expression of TJ proteins. 11 The end-feet regulate the transport of water and ions. They possess a high density of arrays of particles which contain the water channel aquaporin 4 and potassium channel Kir4.1. 12
Astrocytes are well studied for their immunological functions and capacities to induce autophagy in neurological diseases such as Alzheimer’s disease (AD). Their end-feet are altered by hyperphosphorylated tau protein and oligomer amyloid β (Aβ). 13
Microglia are resident macrophage-like cells of the spinal cord and brain and constitute 10–15% of all cells in the CNS.14,15 They were identified for the first time by the neuroanatomist Pio del Rio-Hortega in 1919, who suggested that these cells have phagocytic capacities, and they are of mesodermal origin. 16 Studies carried out in mice confirmed their origin; they are derived from hematopoietic precursors in the yolk sac that migrate into the parenchyma of the CNS, from embryonic day 8.5 until the development of the BBB is completed.17,18 In humans, microglia have the same origin, but invade the CNS around gestational week 4.5 and the progenitors require two transcriptional factors, Pu.1 and Irf8, that are specific biomarkers for this role of microglia which are lacking in macrophages.19,20
The life span of microglia cells is long; they have self-renewing capacity, and, during inflammatory status, they proliferate with a selected clonal expansion mechanism. 21 Microglia cells have a ramified morphology with long and motile processes with a potent role in the checking of their microenvironment. The electron microscopical image (Fig. 4) clearly shows their architecture. 22
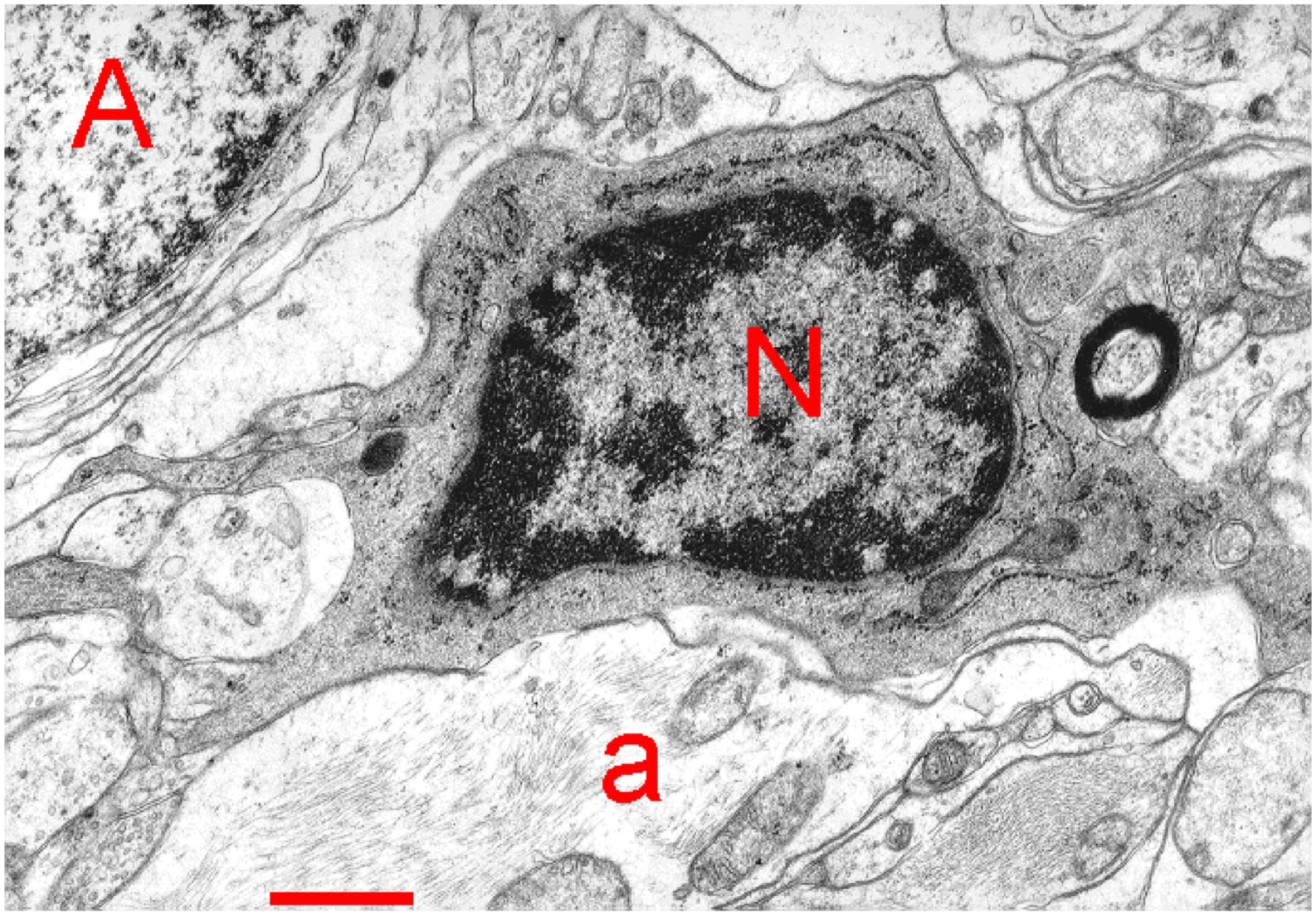
Electron microscopical image of a microglia cell with its nucleus (N) and its processes with gliofilaments (a) and an astrocyte (A) in the thalamic ventrobasal nucleus of the rat. The image has been published previously in Spacek. 22 “Bar = 1 µm.”
Under healthy conditions, microglia activity is important for synaptic network during development, 23 cell death and neurogenesis in adults, 24 synaptic plasticity, 25 as well as learning and memory. 15
Following BBB dysfunction in diseases such as AD and multiple sclerosis (MS), microglia cells have an ameboid architecture with short ramifications and large soma 26 and produce several pro-inflammatory cytokines, tumor necrosis factor-α (TNF-α), proteases, and reactive oxygen species (ROS), to repair injuries, but can have negative effects on BBB morphology when this condition persists for a long time. 15
The plasticity of microglia allows the identification of two functional active states of its components, the M1 and M2 states; M1 cells release pro-inflammatory cytokines like interleukin (IL)-1β, and TNF-α, whereas M2 cells are involved in tissue repair. 27 Considering their functions, M1 and M2 cells can be defined respectively inflammatory and non-inflammatory. In neurodegenerative diseases (NDs), the release of pro-inflammatory cytokines and ROS can induce BBB damage and permeability. 27 Thus, M1 cells are considered to have a non-proliferative inflammatory phenotype, whereas M2 is a protective phenotype. 28 Today, the definition of M1 and M2 for microglia is too strict although these terms are also used for macrophages that are identified in a similar way. 29
The present review consists of 2 parts: (1) the first part focuses on BBB in health and disease; and (2) the second part describes a family of the proteins, the transient receptor potential (TRP) members that are still not well studied, because their role in its member proteins has only been recently identified and not completely evaluated in neuronal cells. Maintenance of the integrity and homeostasis of the BBB can preserve the body in good health and protect it from comorbidities that are also caused by aging.
BBB in Health and Disease
All features of the BBB can become altered with aging and these alterations occur in many age-related disorders. Figure 5 shows the differences between the BBB in a healthy state and during its breakdown.
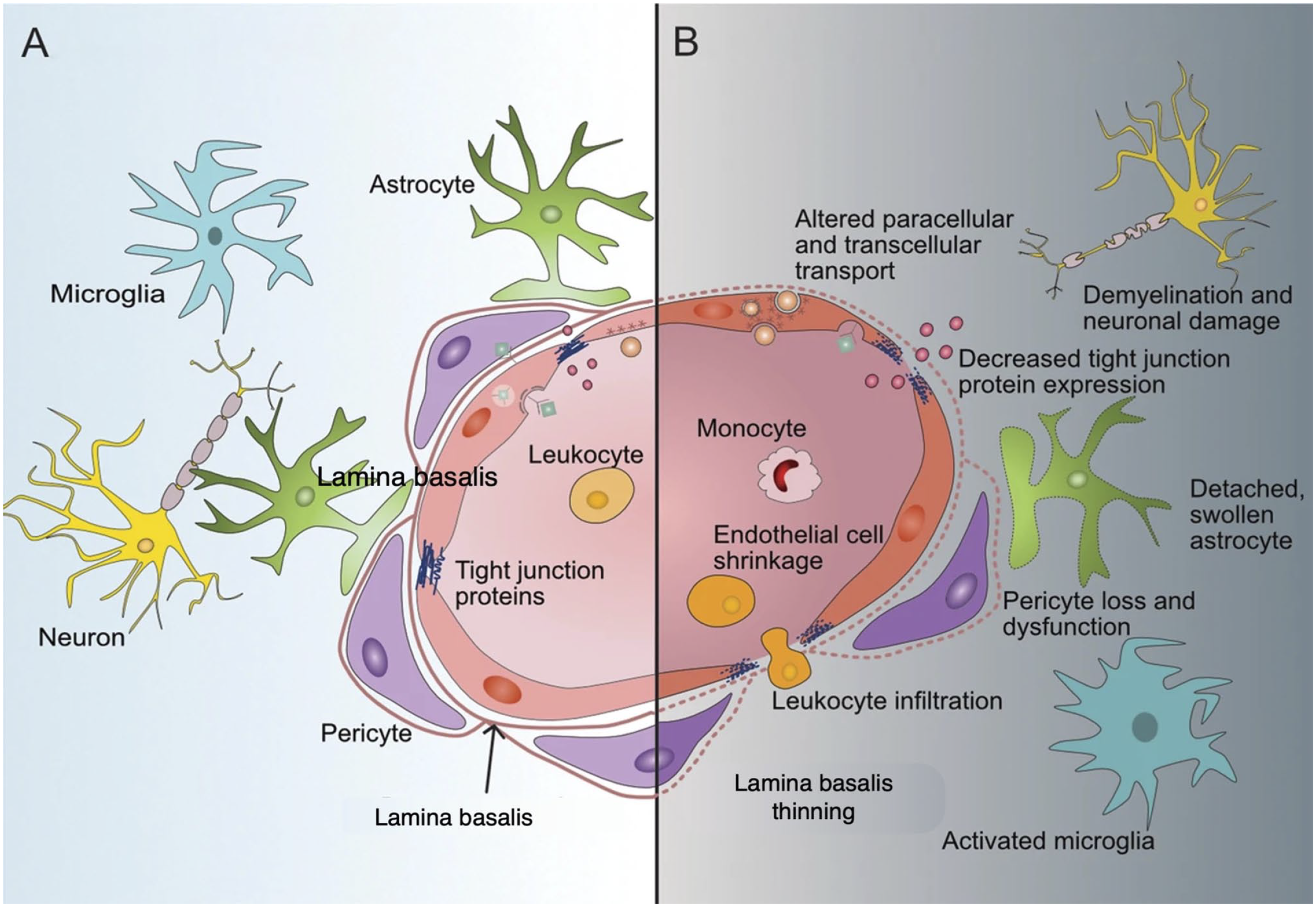
Blood–brain barrier (BBB) in a healthy state and during its breakdown. (A) Healthy BBB morphology and the key components that are essential for its integrity. (B) During BBB breakdown, its integrity becomes compromised at various levels and induces changes in components of microglial cells. The image has been prepared based on the image in Knox et al. 11
The alterations in the BBB can occur during aging as comorbidity associated with cognitive decline and dementia. 30 BBB alterations may be present even when the patients have no clinical manifestations. There are two hypotheses about the cognitive decline: (1) it can appear once the inflammation state results in cellular damage that causes epigenetic changes and the disruption of the BBB that may lead to the development of the clinical disease; and (2) it is just one of the features that characterizes the disorder and something else occurs before the BBB disruption and the pathology and these metabolic changes can induce BBB breakdown.11,31–34 However, the pathology is far from understood at present.
In this review, NDs are discussed that are not necessarily associated with genetic mutations, but rather are linked to BBB breakdown and cellular damage as occurs in acute NDs. In addition, we report evidence that the microbiome has an important role in CNS function, and its breakdown is responsible for neuroinflammation, neurodegeneration, and aging 35 (Fig. 6).
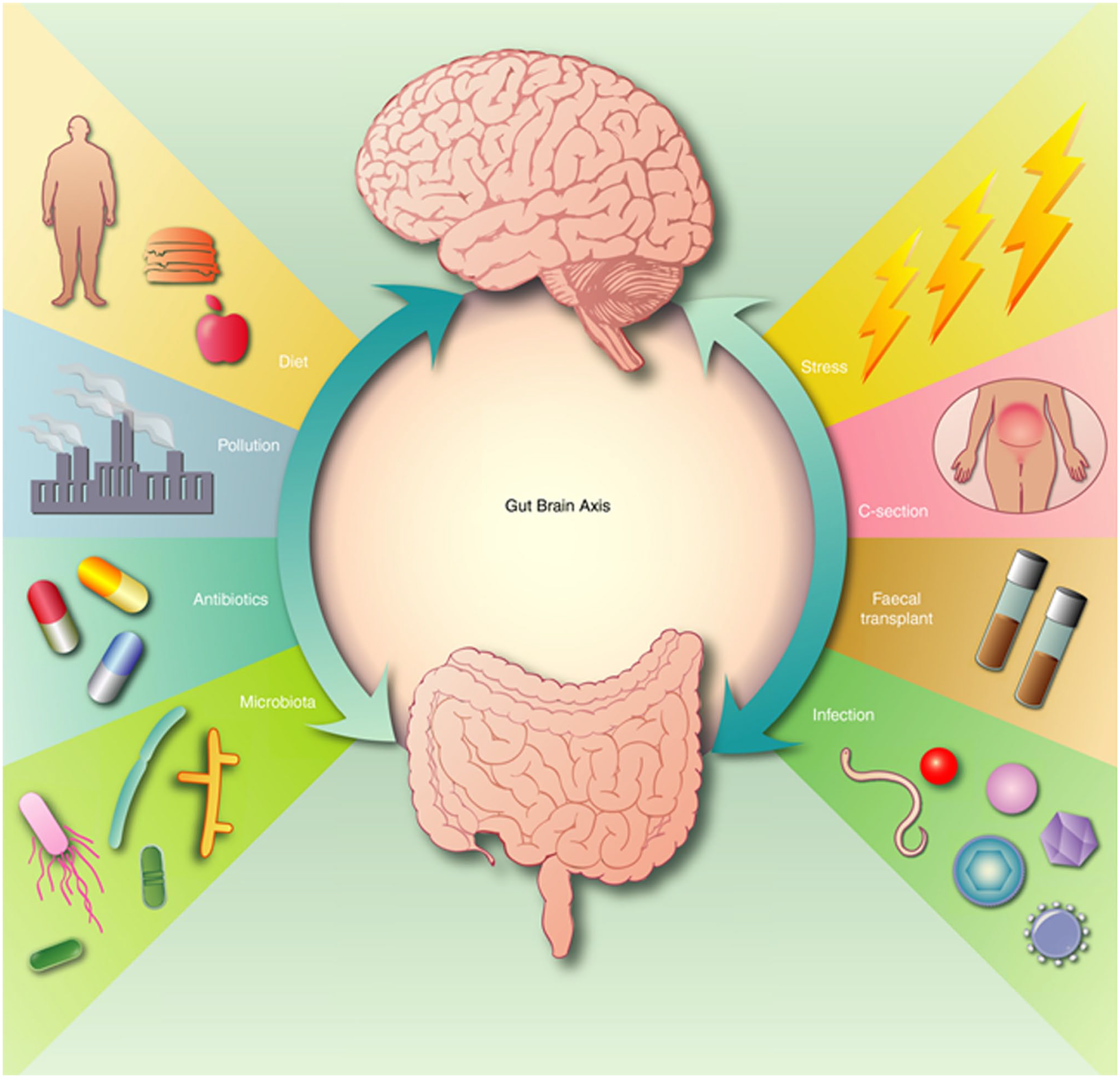
Neuroinflammatory disorders and possible mechanisms affecting the gut–brain axis. The arrows indicate that lifestyle and environment are important in brain functioning via the gut–brain axis. Changes in the gut microbiota, induced by stress or lifestyle, increase the possibility of developing NDA. Therapeutic approaches by controlled diet and the use of probiotics may help to maintain a healthy microbiome and therefore improve neurological health. The image has been published previously in Stephenson et al. 35 (license number: 5593480557561).
Alterations in the gut–brain axis may induce dys-biosis and changes in the permeability of the BBB affecting brain physiology; recent studies suggested that the alterations of gut microbiota (GM) can contribute to several NDs such as AD, amyotrophic lateral sclerosis (ALS) and MS, Parkinson’s disease (PD), autism, depression, and schizophrenia. 36 Diet and age are the main actors that are responsible for GM changes 37 ; untreated “leaky gut” stimulates the influx of luminal pathogens that induce pathological changes also occurring in the brain. 38 Usually, the brain is the last affected organ as compared with the intestine, liver, spleen, lungs, and vascular system, but when it occurs, the BBB is already disrupted when pathological changes become manifest. 39 We outline in Fig. 7 how the GM can induce neuroinflammation and neurodegeneration, especially during aging.
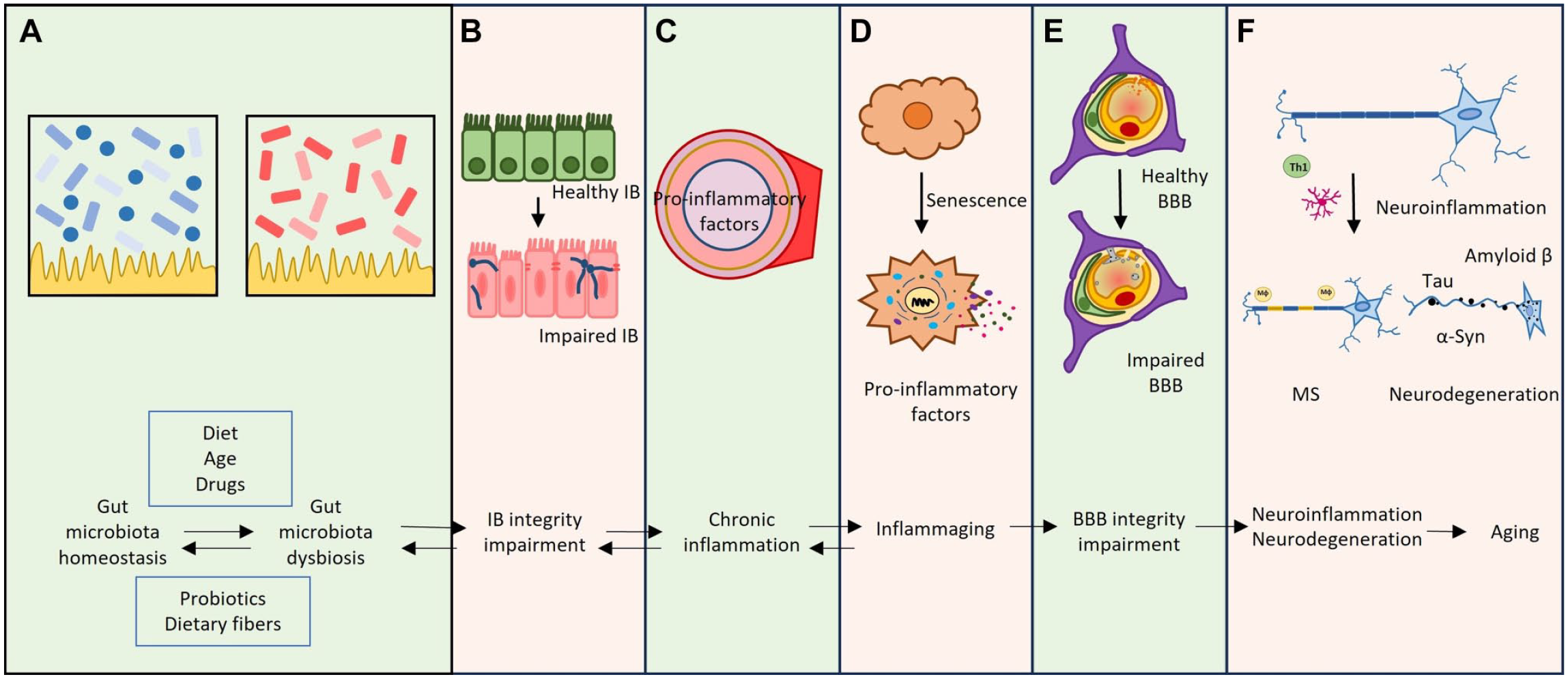
Relationship between gut microbiota, neuroinflammation, and neurodegeneration during aging. (A) diet and drugs disturb gut microbiota homeostasis inducing dysbiosis. (B, C) dysbiosis modifies the integrity of the intestinal barrier inducing chronic inflammation. (D, E) The inflammation causes cellular senescence and BBB impairment. (F) BBB breakdown induces many changes in neuronal cells including neuroinflammation and neurodegeneration. The image has been prepared based on the image in Mou et al. 34 Abbreviations: BBB, blood–brain barrier; IB, intestinal barrier; MS, multiple sclerosis; α-syn, α-synuclein; Th1, T-helper type 1.
Alzheimer’s Disease
The classical pathological hallmarks of AD are due to BBB breakdown and many markers have been studied that may be involved.40–43
Figure 8 shows BBB breakdown and some markers involved in the development of AD.
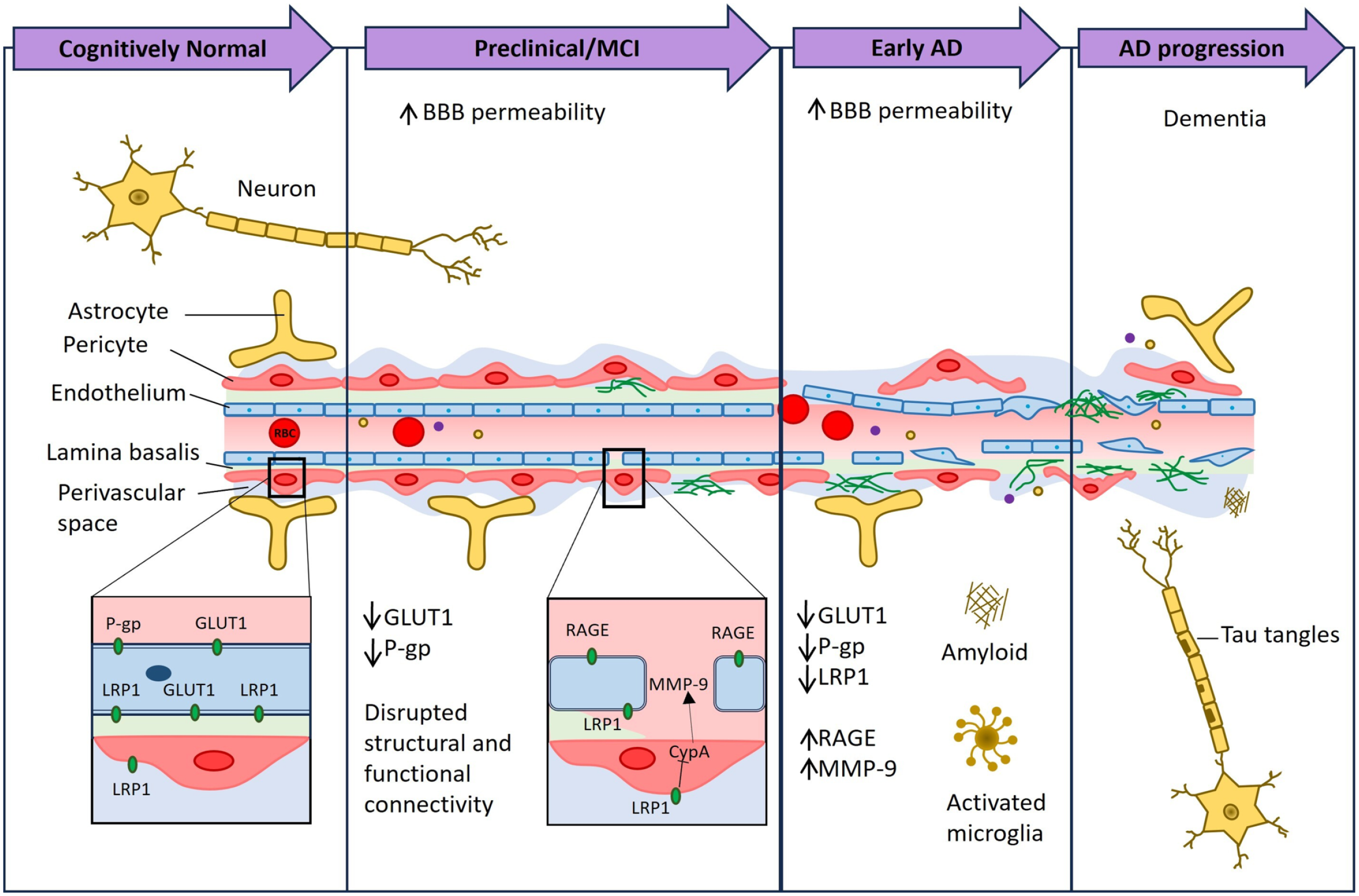
BBB breakdown and dysfunction in AD. New techniques demonstrated BBB breakdown in the hippocampus in individuals in early AD before brain atrophy and dementia occur. Diminished expression levels of many markers are involved in BBB breakdown and they are reported in this image. The image has been prepared based on the image in Sweeney et al. 41 Abbreviations: BBB, blood–brain barrier; MCI, mild cognitive impairment; AD, Alzheimer’s disease; GLUT1, glucose transporter; Aβ, amyloid-β; RBC, red blood cells; CypA, cyclophilin A; LRP-1, low-density lipoprotein receptor-related protein-1; MMP-9, matrix metalloproteinase-9; RAGE, receptor for advanced glycation end product.
Post-mortem human studies have suggested that BBB disruption is often observed in AD patients, and it is linked to reduced capillary length and vascular degeneration with low levels of protein expression in TJs, alterations in the LB of vessels, and brain EC degeneration. 44 In addition, AD patients present at BBB level low levels of many transporters, such as glucose transporter (GLUT1), low-density lipoprotein receptor-related protein-1 (LRP1), and increased levels of other receptors, such as those of advanced glycation end products.45,46
Parkinson’s Disease
PD has an unknown etiology like other NDs. 47 Vascular damage is responsible for the onset of PD and that cerebrovascular dysfunctions accelerate the progression of PD as described in Fig. 9.
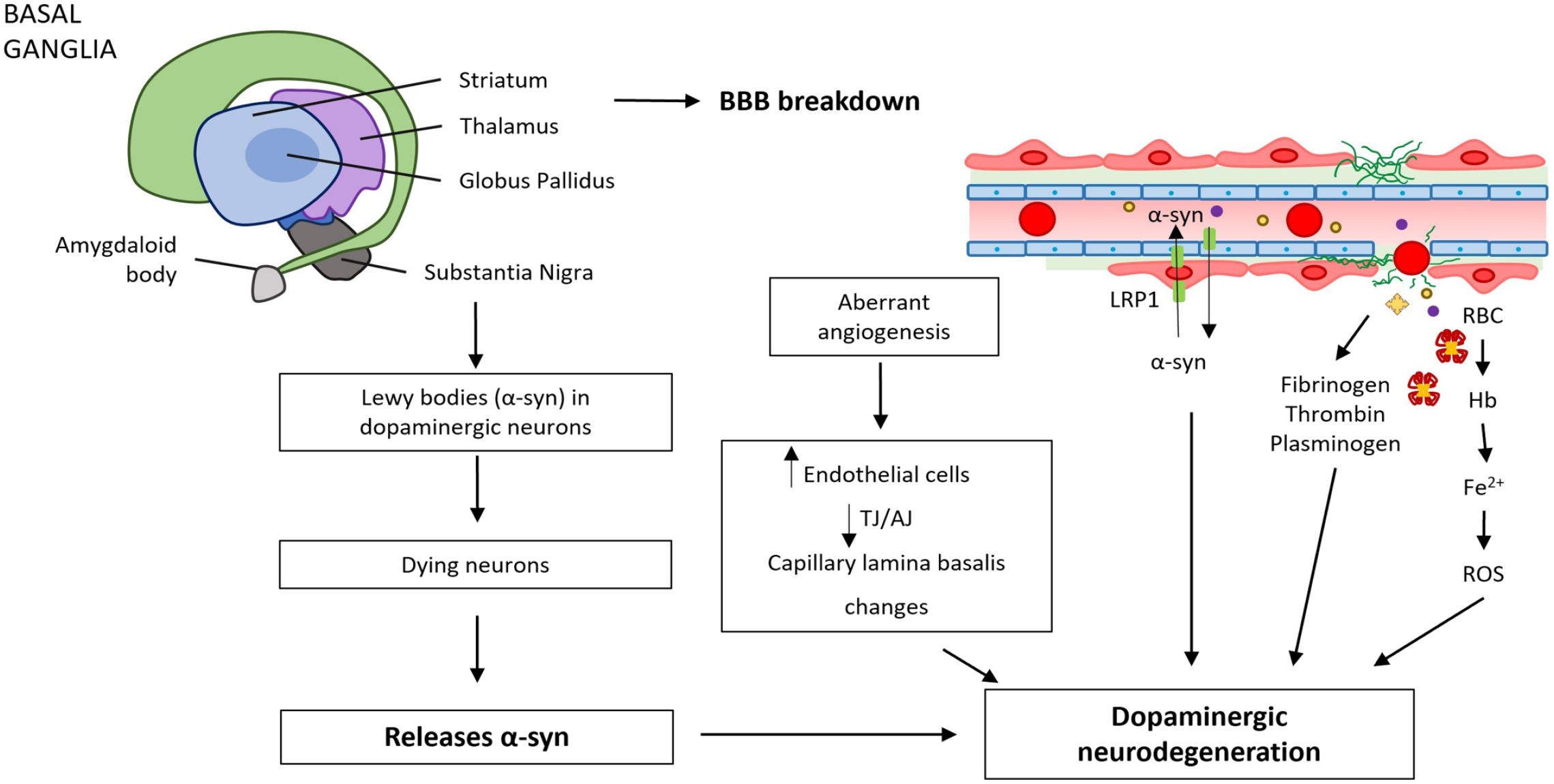
BBB breakdown and dysfunction in Parkinson’s disease (PD) with no clear etiology or genetic background. Vascular dysfunction occurs throughout the basal ganglia of PD patients, consisting of BBB breakdown and dysfunction. BBB breakdown can lead to neurotoxic accumulation of fibrinogen, thrombin, and plasminogen and RBC extravasation, release of Hb and Fe2+ causing ROS, which all can injure dopaminergic neurons.43,48,49 The image has been prepared based on the image in Sweeney et al. 43 Abbreviations: BBB, blood–brain barrier; TJ, tight junction; AJ, adherens junction; RBC, red blood cell; ROS, reactive oxygen species; α-syn, α-synuclein; LRP-1, low-density lipoprotein receptor-related protein-1; Hb, hemoglobin; Fe2+, iron (II).
BBB breakdown has been observed in basal ganglia in PD patients compared with controls.48,49,50
Moreover, it has been demonstrated that α-synuclein (α-syn) crosses the BBB in a bidirectional way and this may be an important marker of PD. Storck et al. 51 showed that LRP1, the transporter of α-syn, has been found in PD and in AD. 51 This may cause impaired clearance of BBB α-syn and its accumulation. It is known that α-syn enters the brain at high levels after lipopolysaccharide-induced breakdown of the BBB, which may also contribute to the development of PD. 48 Furthermore, disruption of BBB may induce increased levels of vascular endothelial growth factor (VEGF), placental growth factor, and decreased angiopoietin-2 expression. 52 These findings suggest that these are important biomarkers of PD progression. Of note, BBB breakdown induced by the intra-nigral injection of VEGF in rats induces dopaminergic neuron loss in the substantia nigra and this finding suggests that the BBB disruption can determine neurodegeneration. 53
Amyotrophic Lateral Sclerosis
ALS, a debilitating condition showing a progressive degeneration of motoneurons, shows a degeneration of BBB causing leakage of some proteins into the cerebrospinal fluid and brain parenchyma. 54 Among these proteins, hemoglobin is toxic to motoneurons by inducing oxidative stress. 55 Figure 10 shows a model how BBB breakdown can contribute to development of ALS disease.
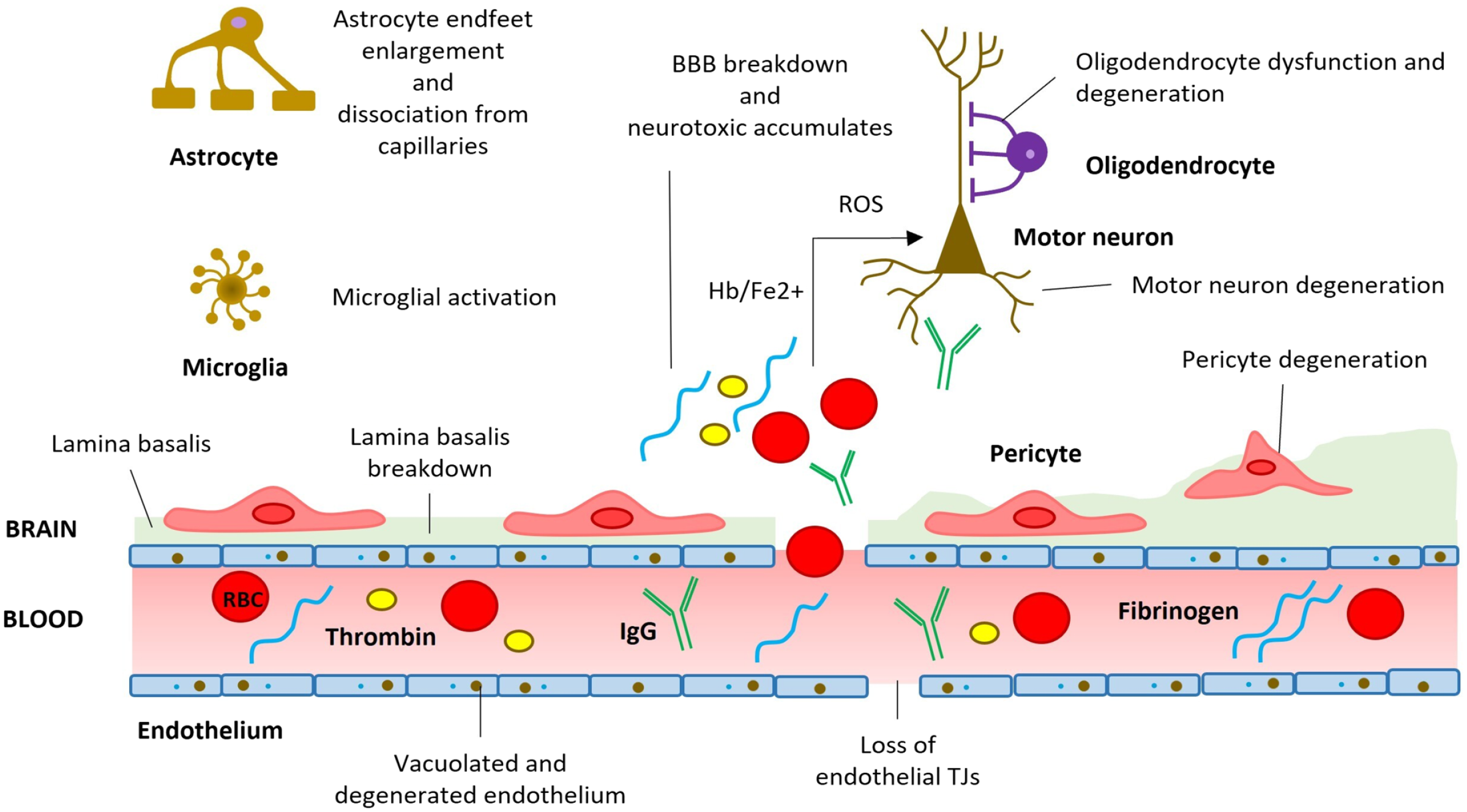
BBB breakdown and dysfunction in amyotrophic lateral sclerosis. BBB disruption induces loss of TJ proteins and endothelial cell and pericyte alterations. The image has been prepared based on the image in Sweeney et al. 43 Abbreviations: BBB, blood–brain barrier; ROS, reactive oxygen species; TJs, tight junctions; Hb, hemoglobin; Fe2+, iron (II); IgG, immunoglobulin G; RBC, red blood cell.
Multiple Sclerosis
MS destroys the myelin sheaths that surround nerves in the CNS. The etiopathology is unknown, but the involvement of immunological BBB in this disease has been determined; the BBB plays an important role in the exchanges of immune cells and their mediators between blood and brain. 56
Figure 11 illustrates BBB involvement in MS outlining that the inflammation state is due to leukocyte extravasation across the activated endothelium of vessels; the extravasation involves a series of steps: tethering/rolling, activation, adhesion, and transmigration. All these steps are dependent on specific markers and the fourth phase needs matrix metalloproteinases and chemokine–chemokine receptor interactions with leukocytes.
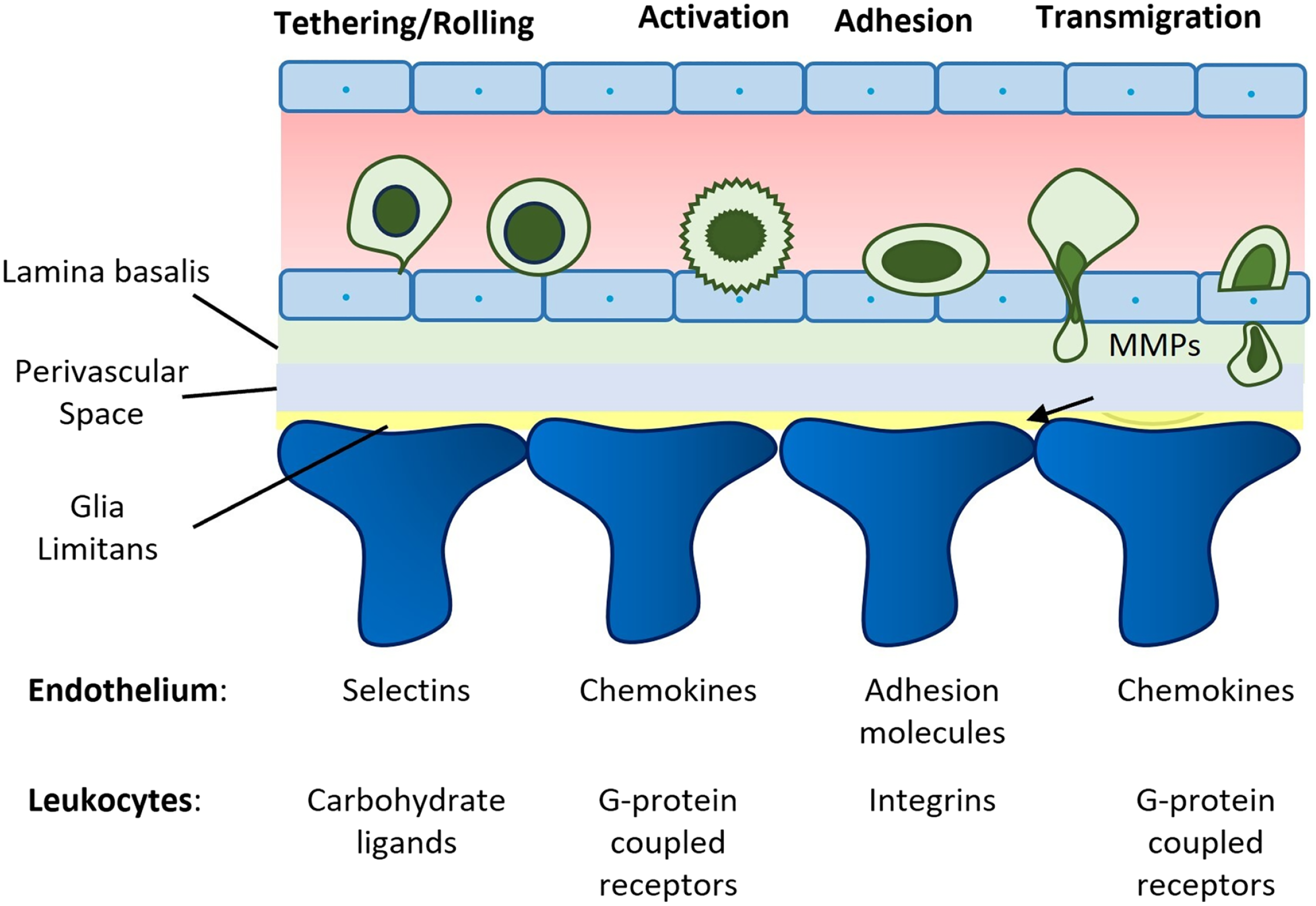
Schematic representation of blood–brain barrier (BBB) involvement in multiple sclerosis. Four steps are indicated that are responsible for extravasation of leukocytes and after the last step they can extravasate by paracellular or transcellular routes across BBB. The image has been prepared based on the image in Zlokovic. 56 Abbreviation: MMPs, matrix metalloproteinases.
Maintenance or Restoration of BBB Integrity
The breakdown of the BBB in neurological diseases makes it an important therapeutic target; the maintenance or the restoration of the BBB is a major future challenge when the BBB disruption can be repaired, and it may be possible to slow down the pathogenesis of the neurological diseases. As reported above, the increase of BBB permeability is due to the changes in expression and functions of BBB transporters and receptors such as GLUT1, LRP, or P-glycoprotein (Pgp) in ECs.56,57–59 These markers are still not well studied, and it remains an unanswered question whether other proteins are also involved in the impairment of the BBB.
The growing interest in evaluating the BBB requires the use of accurate and representative markers to study its integrity.9,60 These markers must be metabolically inert, non-toxic at the used doses, and not bound to other molecules such as proteins in blood plasma or tissues. Moreover, they should be preferably microscopically visualized and be reliably quantifiable. 9
Kadry et al. 9 proposed that there is not a single marker available that meets all the above criteria. When the BBB loses its integrity, large molecular mass markers enter the brain more easily which may be considered as an option to evaluate its integrity and its permeability. In fact, small molecular mass markers <400 Da are mostly used for examining small changes in BBB permeability that occur in many NDs.
Table 1 from Kadry et al.’s 9 studies reports several markers used for BBB permeability studies.
Summary of Different Markers That Can Be Used for BBB Permeability Studies.
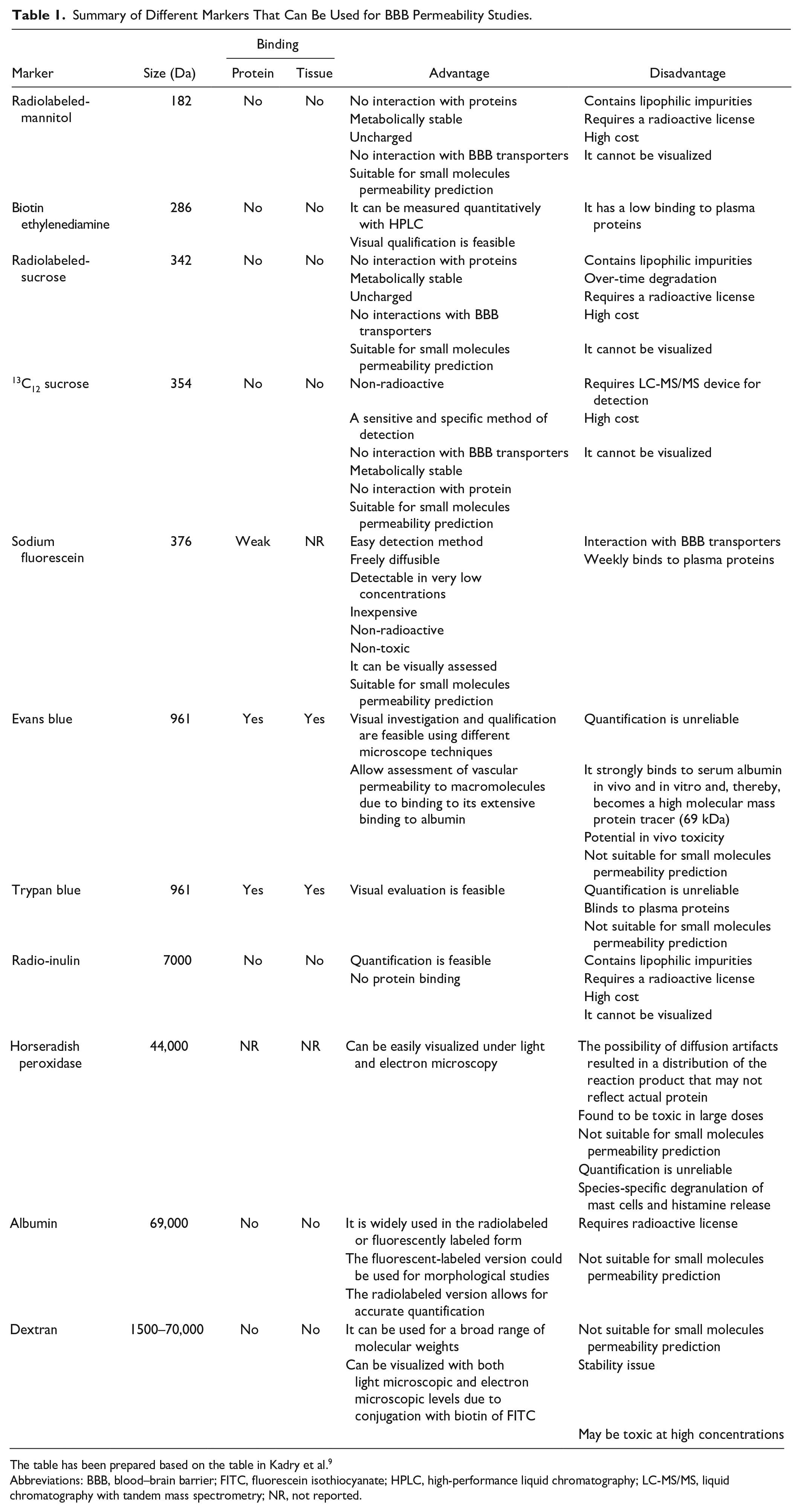
The table has been prepared based on the table in Kadry et al. 9
Abbreviations: BBB, blood–brain barrier; FITC, fluorescein isothiocyanate; HPLC, high-performance liquid chromatography; LC-MS/MS, liquid chromatography with tandem mass spectrometry; NR, not reported.
Some questions remain unanswered: Is there a common pathway for BBB damage in all NDs or are there several mechanisms inducing the BBB disruption for each disease?
Several signaling pathways have been shown to be important for BBB breakdown and many biomarkers are still not well identified. The important point is that the damage of BBB seems to be multimodal showing alterations of EC transport and metabolic processes with disruption of TJs. 3 Therefore, understanding of the molecular mechanisms regulating BBB disruption and prevention of BBB disruption are important issues to develop therapeutic strategies.
Considering these important clues for BBB disruption, an integrative and important outlook to explain why and when the BBB is dysregulated is very important. In the second part of our review, we focus on functions of the TRPCs and, in particular, of the TRP vanilloid (TRPV) channels whose expression or functionality in the BBB is less studied than in non-cerebral neuronal cells. 61
Transient Receptor Potential Channels: Biological Features and Functions
The superfamily of the TRP channels consists of seven families: TRP ankyrin (TRPA), TRP canonical (TRPC), TRP melastatin (TRPM), TRP mucolipin (TRPML), TRP polycystin (TRPP), TRPV, and TRP no mechanoreceptor potential (TRPN); the last one is found in fish and invertebrates and not in humans.62–65
Increasing numbers of studies provided evidence that many of these proteins are activated by mechanical, physical, and chemical stimuli; their functions are regulated by several mechanisms.
TRP channels were first described over 50 years ago. In 1969, Cosens and Manning 66 identified a visual mutant in Drosophila; this mutant was transient rather than a direct response to bright light stimuli. 67 This effect inspired Minke et al. 67 to define the mutant “trans receptor potential” trp following its electrophysiological phenotype. Then, other scientists cloned the trp gene and they identified it as a transmembrane protein.68,69
After this discovery, these channels became an important scientific topic. Minke and Sellinger, in 1992, established that TRP is a transmembrane protein oscillating between Ca2+ and non-Ca2+ transport states and other studies suggested that TRP and TRP-like proteins are a light-activated and Ca2+-permeable type of voltage-gated channels.70–72 Expression patterns and functions of TRP and TRP-like proteins in various cell lines provided evidence for important roles in healthy human tissues.73,74 These proteins have been studied in detail in the last two decades. 75 Furthermore, in 2021, the Nobel Prize in Physiology and Medicine was awarded to David Julius and Ardem Patapoutian for the identification of TRP and TRP-like protein functions. It was shown how thermal and mechanical stimuli are transduced by these proteins.
Progress in our understanding of TRP and TRP-like proteins is summarized in Fig. 12.
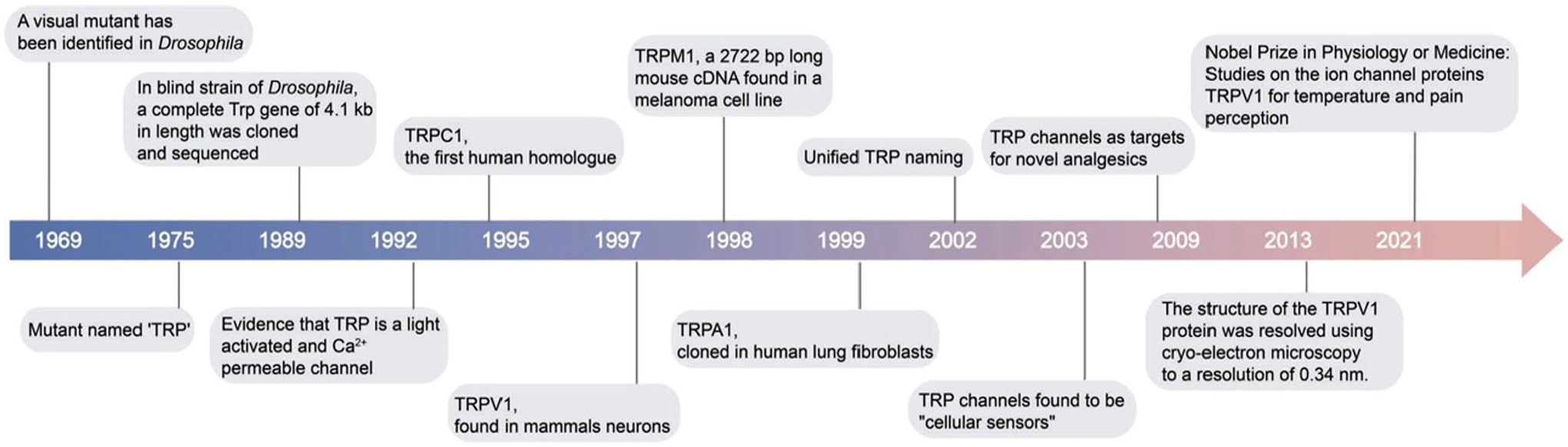
Major structural and functional findings of TRP channels. The image has been published previously in Zhang et al. 65 Abbreviations: TRP, transient receptor potential; TRPA, transient receptor potential ankyrin; TRPC, transient receptor potential canonical; TRPV, transient receptor potential vanilloid; TRPM, transient receptor potential melastatin; cDNA, complementary DNA. 66
The main roles and functions of different types of TRP channels in both CNS and peripheral nervous system (PNS) and other systems and organs are reported in Table 2. 65
Characteristics and Functions of Different Types of TRP Channels.
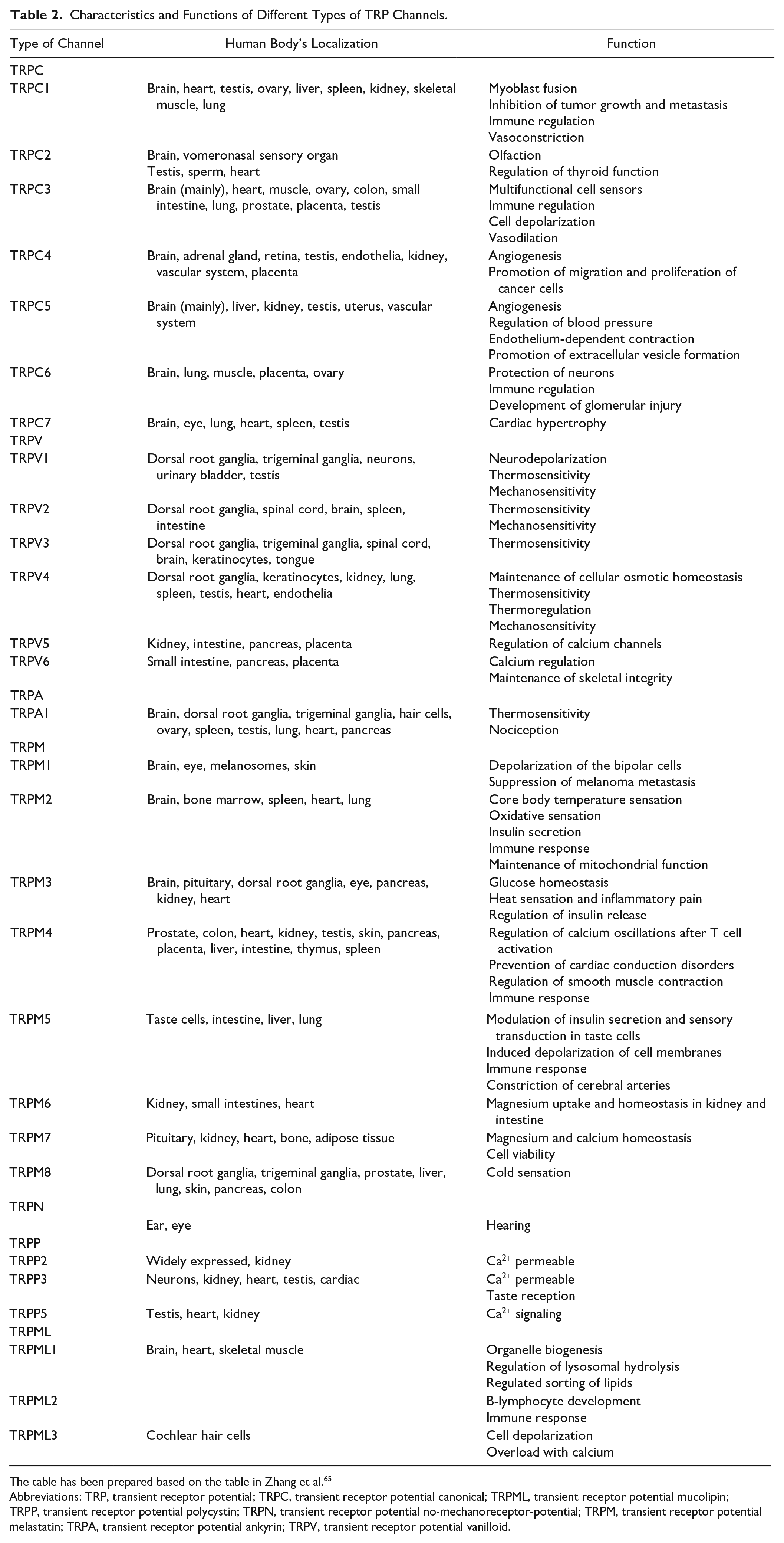
The table has been prepared based on the table in Zhang et al. 65
Abbreviations: TRP, transient receptor potential; TRPC, transient receptor potential canonical; TRPML, transient receptor potential mucolipin; TRPP, transient receptor potential polycystin; TRPN, transient receptor potential no-mechanoreceptor-potential; TRPM, transient receptor potential melastatin; TRPA, transient receptor potential ankyrin; TRPV, transient receptor potential vanilloid.
TRP channels are differently expressed in the CNS and PNS of mammals as indicated in Fig. 13. 76 Novel members have been identified in CNS and PNS in the recent years. This will be discussed in the next paragraphs.
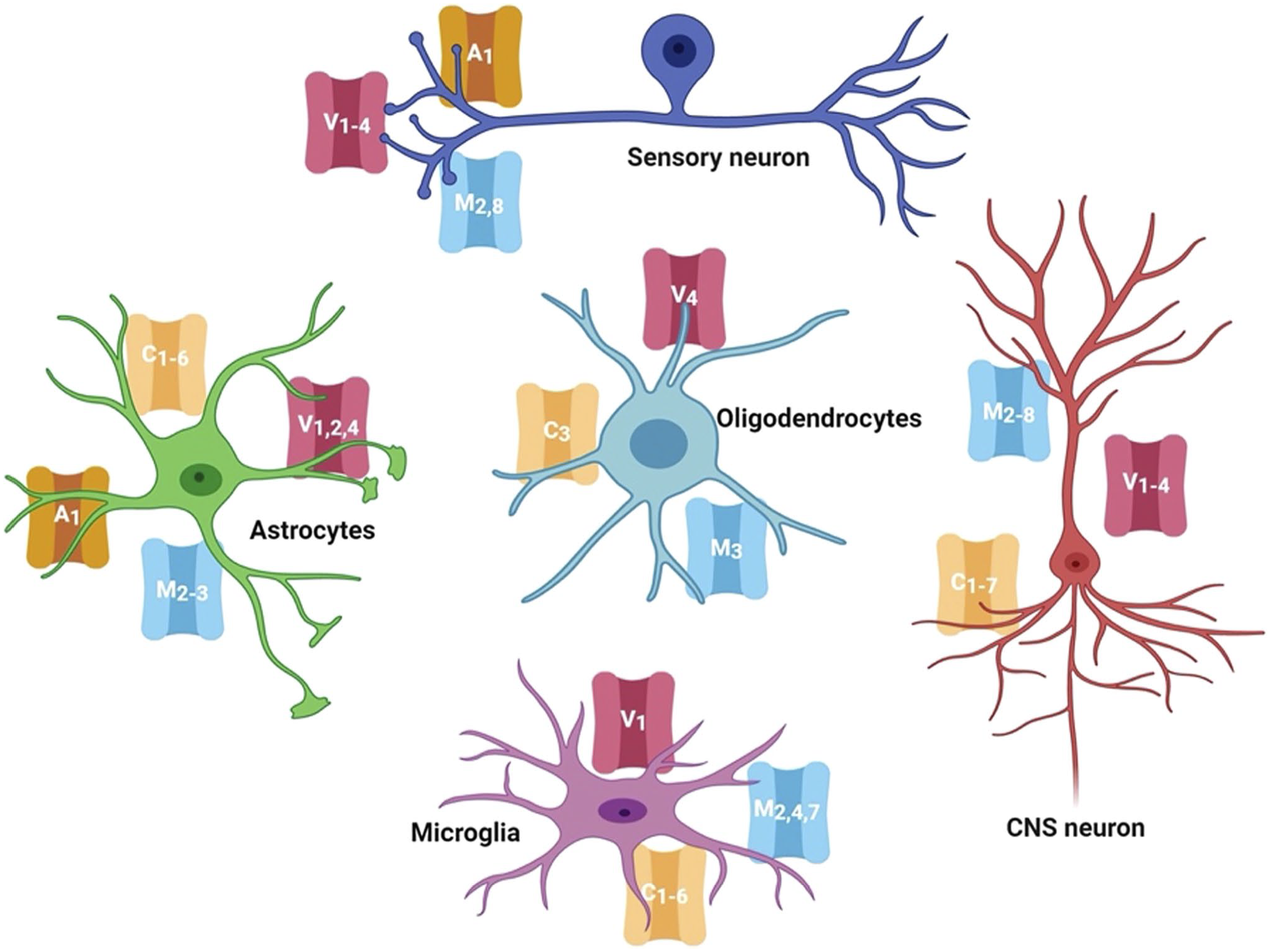
Transient receptor potential channels (TRP channels) expressed in the cells of central nervous system and peripheral nervous system. Several members are expressed in the neurons, astrocytes, oligodendrocytes, and microglia. The image has been published previously in Duitama et al. 76
In detail, TRPCs are divided into seven groups as reported in Table 2; some of these share functions (e.g., TRPC3/6/7 and TRPC4/5) and are considered to form only two groups; in this case, TRPC members are subdivided in only four groups and not seven. 64 They share their properties with respect to their sensitivity for the second messenger diacylglycerol (DAG). 77 They are activated by DAG via the link with phospholipase C regulating the release of Ca2+ from intracellular stores. This regulation of release of Ca2+ from intracellular stores occurs at the plasma membrane 78 (Fig. 14).
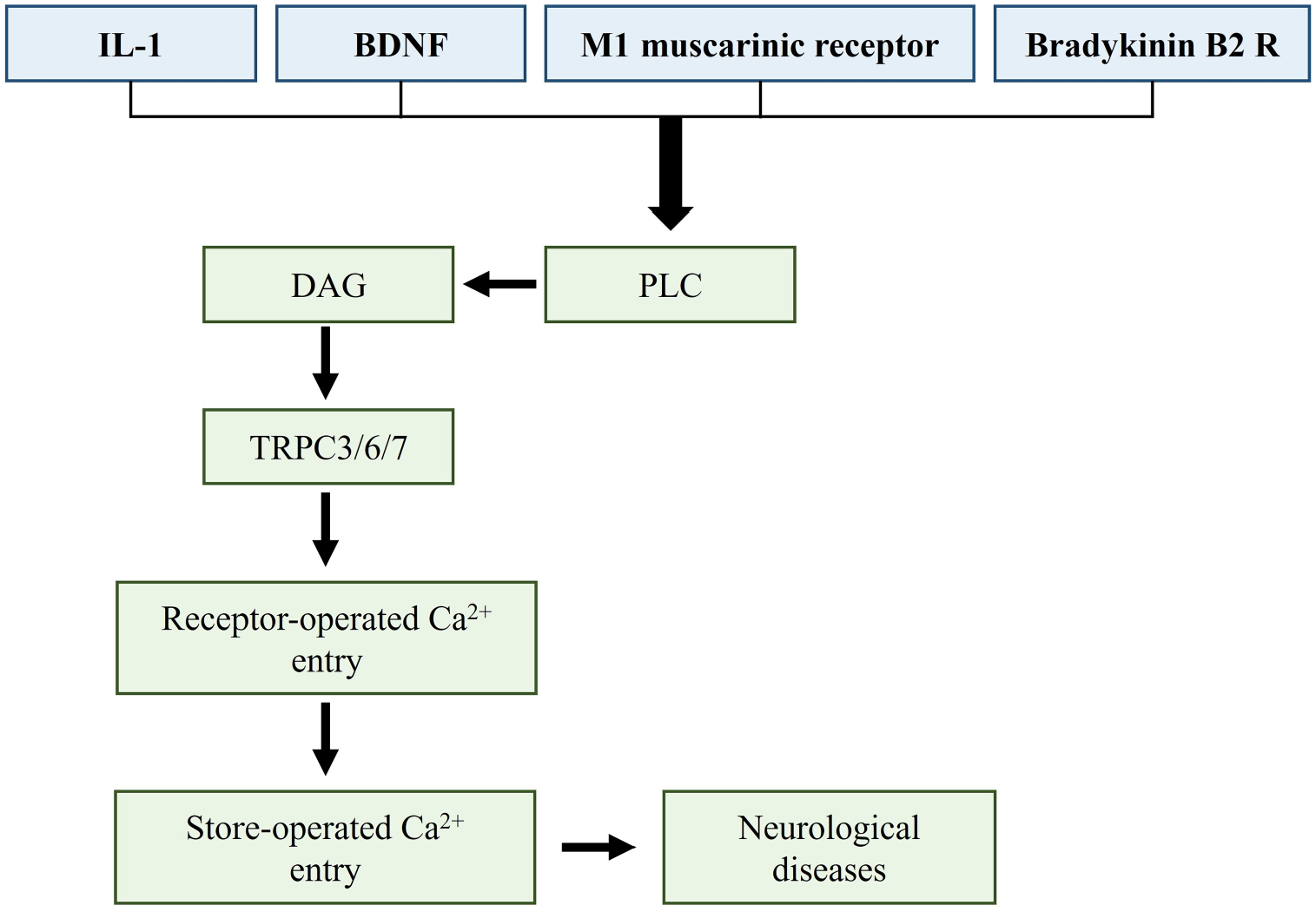
Mechanism of the TRPC3/6/7 activation in the brain. Several mechanisms involving IL-1, BDNF, M1 muscarinic receptor, and bradykinin B2 R and other receptors that activate TRPC3/6/7 may induce neurological diseases. The image has been prepared based on the image in Duitama et al. 76 Abbreviations: DAG, diacylglycerol; TRPC3/6/7, transient receptor potential canonical 3/6/7; IL-1, interleukin-1; BDNF, brain-derived neutrophic factor; PLC, phospholipase C. 76
TRPCs are expressed in the brain from the embryonic period to adulthood apart from TRPC2.79,80 For this reason, TRPC2 is not an important role in the NDs, and it is not considered a therapeutic target in these disorders.
TRPC1 and TRPC4 are expressed and involved in the early development and proliferation of neurons and in neuronal cell death.81–83
TRPC3 channels are present in CNS and PNS (Fig. 13), but is mainly identified in structures in CNS such as cerebellum, caudate nucleus, putamen, and striatum.84,85 These channels are necessary for the physiology of Purkinje’s cells in the cerebellum. 86 Moreover, the changes in their expression or mutations in their genes have dramatic consequences on motor neurons as it has been demonstrated in Moonwalker mice. 87 These mice show many problems in the coordination of movement; TRPC3 upregulation impairs the development of dendrites ameliorating motor functions. 88 TRPC3 likely interacts with the brain-derived neutrophic factor (BDNF), which is an the important regulator of striatal neuron survival, differentiation, and plasticity. 89 Then, BDNF stimulates the factor-tropomyosin receptor kinase B (TrkB), and this network leads to the activation of TRPC3, which is fundamental for spine remodeling.90,91 TRPC3 activity deregulation may induce structural alterations in the cerebellar ataxia by modulating dendrite development. Furthermore, TRPC3 can form functional channels by interactions with non-selective Ca2+ entry channels; in this way, TRPC3 has other important roles in regulating basic neuronal processes. 92
The TRPC1/4/5 channels have been demonstrated in the parietal and frontal lobes of the brain and in the hippocampus.80,84,93 In particular, TRPC5 determines neurite length, and axonal outgrowth during neural differentiation.85,94,95,96 Moreover, TRPC5 promotes cell death induced by oxidative stress; based on this effect, it is known that TRPC5 has a role in both growth and death of neurons. 85
TRPC6 is widely expressed in CNS 84 ; it is expressed in excitatory post-synaptic neurons, and it is responsible for the number of spines and spatial learning and memory.85,97–99
TRPC7 share the functions with TRPC3/4 as mentioned earlier.
The TRPV subfamily comprises of six members located mostly in the plasma membrane, and they are divided in two subgroups: TRPV1–4 and TRPV5/6.100,101 TRPV1–4 are also called “thermo-sensitive” TRPVs because they are activated by heat. 101 TRPV5/6 have different features compared with the other TRPV channels and they are not expressed in CNS 102 (Table 2).
TRPV1 is the family member originally described, and it is the most studied; it was first described as an ion channel activated by vanilloid capsaicin as well as high temperature (>42C), and ROS.103,104 TRPV1 is identified in sensory neurons (Fig. 12) with a role in pain modulation, brain cortex, hippocampus, cerebellum, substantia nigra, hypothalamus, midbrain, olfactory bulb, and astrocytes.105–108 It has many other functions being expressed in several dynamic structures of neurons such as the growth cone, neurite, and axonal filopodia. Its activation induces retraction of growth cones in sensory neurons 109 ; it plays a role in guidance during neurite extension of spiral ganglion neurons. 110 In addition, TRPV1 activation induces caspase-3-dependent programmed cell death of cortical neurons through Ca2+ influx 111 and microglial phagocytosis in damaged neuronal cells. 112 Morover, it affects the migration of astrocytes. 113
TRPV1–4 channels are identified in a variety of cell types including neuronal cells as mentioned in Table 2.114,115 It is important to underline that in aged ECs TRPV1/2/4 share with the TRPC4 their role in the neurogliovascular unit (NGVU) and the BBB disruption and increased permeability, whereas TRPV3 and TRPV4 are involved with the other TRP channels in the neurovascular coupling as reported by Negri et al. 115
Downregulation of TRPV2 channel in TRPV2 heterozygous rats, a non-diabetic model of impaired myogenic reactivity and blood flow autoregulation in the retina, induces a loss of retinal blood flow autoregulation and microvascular lesions resembling those observed in diabetic retinopathy (DR). 116 The retina in these animals also shows inflammatory, gliosis, and neurodegenerative changes like those observed in DR. The authors showed that TRPV2 is expressed in the retinal vascular smooth cells of the retinal arterioles as well as pericytes of retinal capillaries. These findings support that targeting of TRPV2 dysfunction may well be important to evaluate its potential to prevent the onset and progression of DR. 116
Only one member of the TRPA channels is known, TRPA1. Its function is well known, and its expression has been found in astrocytes; in these cells, TRPA1 plays a role in their hyperactivation and their synaptic dysfunction. 117
The TRPM channels consist of eight members and showed common structural characteristics with other TRP channels (Fig. 11, Table 2). They are involved in the regulation of Ca2+ and magnesium (Mg2+) homeostasis; TRPM2–8 are mainly expressed in the CNS. 64
TRPM1/2/3/7 channels may be critical in regulating BBB permeability under normal and pathophysiological conditions. These channels can be activated by a number of noxious stimuli and they are important for regulating BBB permeability and functions in normal and pathophysiological disorders. 2
TRPM2 is stimulated by oxidative stress and depletion of glutathione and induces the influx of Ca2+ and neuronal death. It is expressed in young EC inducing the Ca2+ influx, endothelial nitric oxide synthase (eNOS) stimulation, and increased levels of nitric oxide (NO) 115 ; it is also present in both astrocytes and microglia, which are responsible for gliosis and immune cell function. 80 Furthermore, it impairs in neurovascular coupling in aged EC together with TRPV3/4, TRPA1, and TRPC1. 115
TRPM7 maintains Mg2+ permeability and the disbalance in the homeostasis of this ion has been involved in the BBB permeability and function.2,85,118 Increased levels of Mg2+ mediated by TRPM7 stimulates the proliferation of ECs and enhances the integrity of the endothelial barrier in the brain. This is due to its effects on the cytoskeletal organization and on the expression of some proteins present in the TJs. 119 In addition, it is expressed in both astrocytes and microglia where it plays a role in the neuronal migration and proliferation. 120
The expression of TRPM8 and TRPM2 has been evaluated in the dorsal root ganglia as reported in Fig. 13. TRPM8 is a non-selective cation channel; it can act as a Ca2+ channel regulating mitochondrial activity that is involved in the modulation of cerebral vascular tone.85,121
The TRPML subfamily has a predominant role in Ca2+-permeability channels although other permeabilities have been described to be important for their functions as well.122,123 They are associated with cellular processes such as vesicle trafficking and endolysosomal-dependent degradation pathways. 124
TRPML1 modulates several processes in membrane trafficking and, in particular, biogenesis and specific functions of lysosomes. 125
Gate Mechanisms and Biological Functions of TRP Channels
As mentioned above, TRP channels are polymodal receptors that are activated by various chemical and physical stimuli such as mechanical variations (e.g., laminar shear stress, membrane stretch, temperature alterations) and incorrect lifestyle. 115 Furthermore, some TRP channels have specific ion selectivity and regulatory pathways that integrate multiple signaling mechanisms.126,127
TRPs are activated by depletion of endoplasmic reticulum (ER) Ca2+ stores; this is related to the networks with of stromal-interaction molecule 1 and calcium release-activated calcium channel protein 1 (Orai1). This process is known as store-operated Ca2+ entry (SOCE; Fig. 15).80,126,127
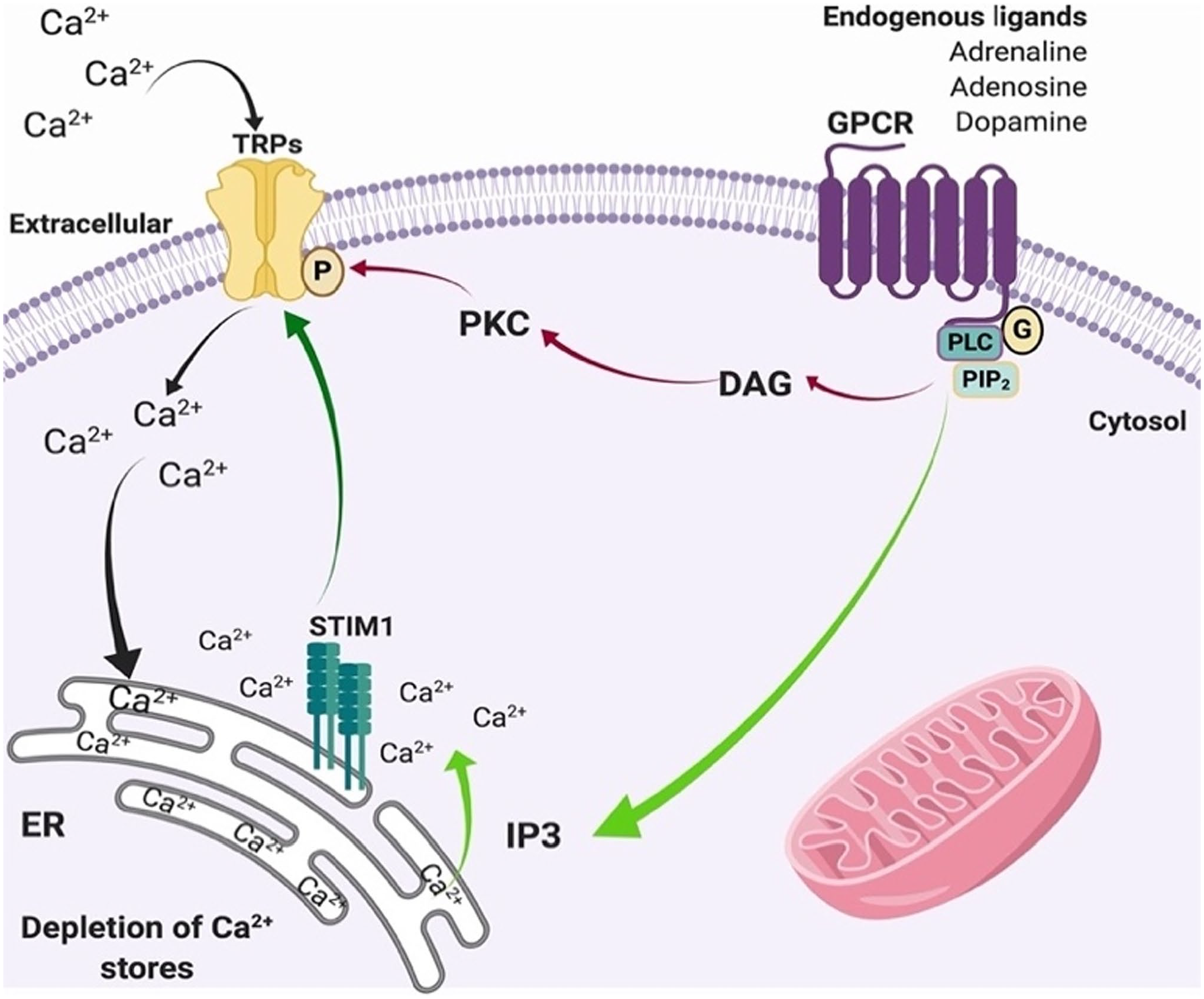
Protein storing Ca2+ entry and TRP channels (TRP channels). The activation of GPCRs stimulates PLC, which induces the hydrolysis of PIP2 and induces DAG (red arrows). This mechanism activates PKC, which in turn phosphorylates TRP channels. At the same time, the generation of IP3 (PKC, green arrows) promotes the release of Ca2+ from the ER. The depletion of intracellular Ca2+ stores from the ER is sensed by stromal-interaction molecule 1, which also activates Ca2+ channels in the plasma membrane such as TRPs (dark green arrow). These channels induce the entry of Ca2+ into the cytosol (black arrows) and the store of Ca2+ ER is modified. The image has been published previously in Duitama et al. 76 Abbreviations: GPCR, G protein-coupled receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; IP3, phosphoinositol 3; STIM1, stromal-interaction molecule 1; ER, endoplasmic reticulum; TRP, transient receptor potential; DAG, diacylglycerol; PKC, phospholipase K.
Ca2+ plays a crucial role in regulating several neuronal functions by acting as second messengers in various processes. 128 Activity of neurons regulated by the influx of Ca2+ from the extracellular environment or its release from the stores in the ER. Normally, the free intracellular concentration of Ca2+ is very low, but when the cells are in an activated stage, its intracellular concentration is increased. 129
Upregulation of intracellular Ca2+ levels and high pH intracellular levels are associated with several NDs and age-related disorders76,80,130; based on these findings, we present a scheme in which Duitama et al. 76 indicated how the alterations in Ca2+ homeostasis induce the modifications of various cellular processes 80 (Fig. 16).
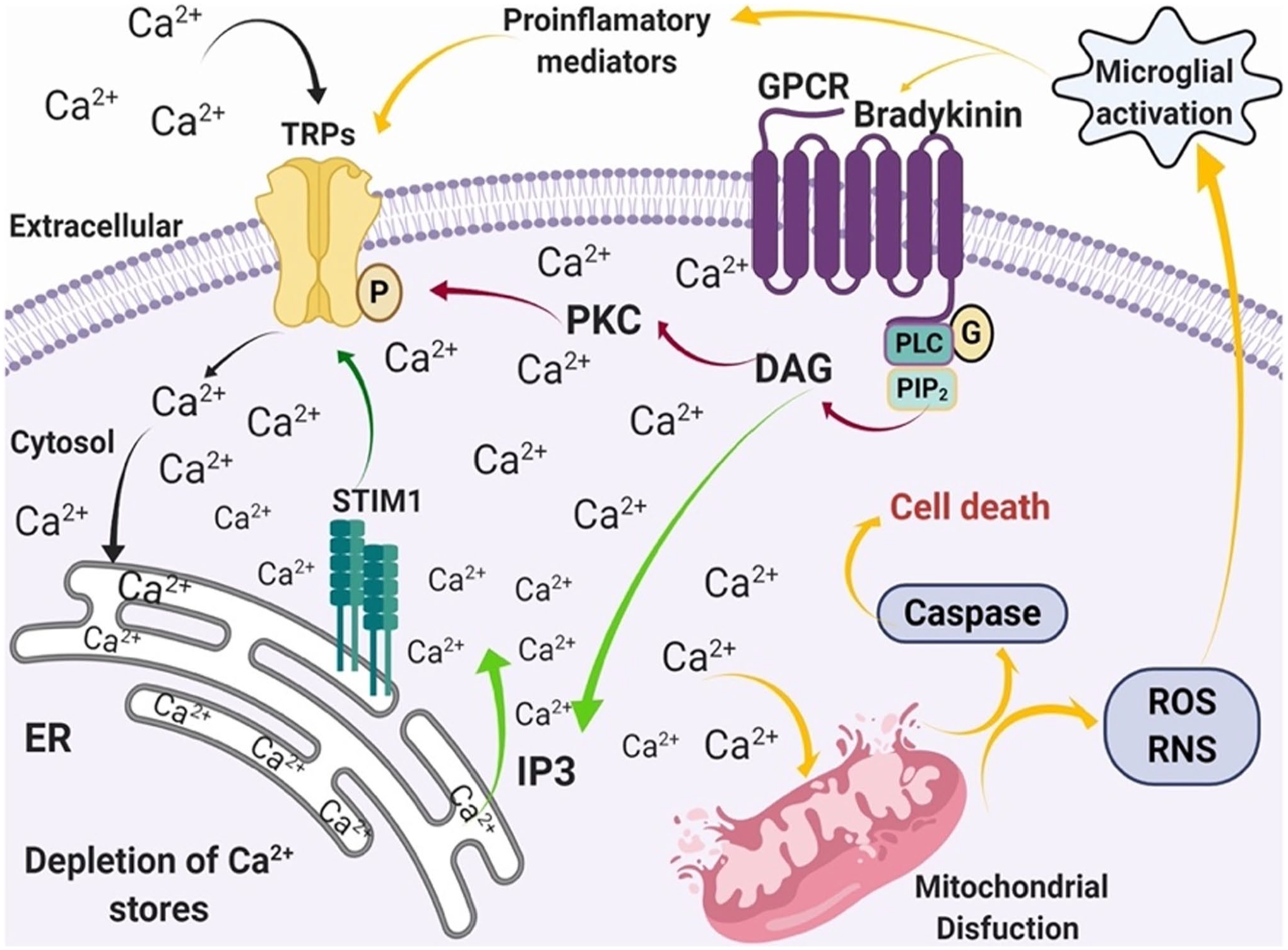
Calcium homeostasis and its role in pathological conditions mediated by a store-operated Ca2+ entry process. Activation of GPCRs by pro-inflammatory mediators induces the release of Ca2+ from the ER as is regulated by PLC pathway (green arrows), followed by an influx of Ca2+ through Ca2+-permeable channels such as TRP channels (black arrows). The increase in Ca2+ induces cellular alterations (e.g., mitochondrial dysfunction, caspase activation, microglia activation, ROS and RNS production, and production of pro-inflammatory mediators; yellow arrows). The image has been published previously in Duitama et al. 76 Abbreviations: TRP, transient receptor potential; GPCR, G protein-coupled receptors; PLC, phospholipase C; PKC, phospholipase K; DAG, diacylglycerol; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3, phosphoinositol 3; STIM1, stromal-interaction molecule 1; ER, endoplasmic reticulum; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Physiological Pathways of the Transient Receptor Potential Channels in Modulating BBB
As mentioned above, BBB integrity is regulated by EC calcium signals, which may be mediated by TRP channels.115,126
It is known that NGVU is impaired in age-related neurodegenerative disorders131,132; the integrity of the unit depends on various vasoactive molecules (e.g., NO and prostacyclin), and endothelial TRPC channels have an important role in these pathways. 115
The mechanisms by which thermo-sensitive TRPVs modulate BBB permeability are complex and not well understood. Herein, we focus on the possible action mechanisms of TRPV1 and TRPV4 in ischemic stroke.
The TRPV1 action on BBB permeability is summarized in Fig. 17.
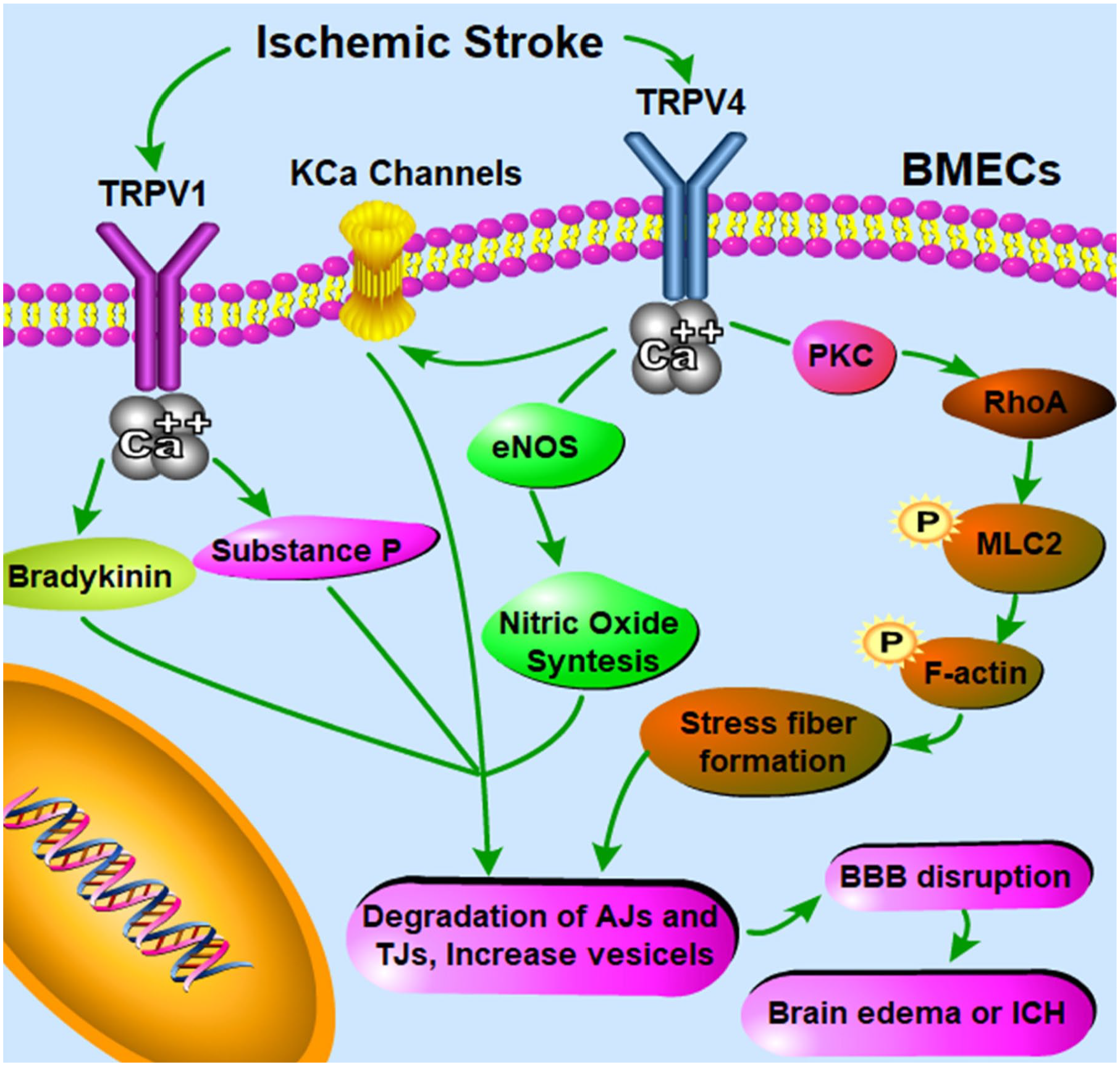
Scheme of TRPV1/4 and BBB disruption in ischemic stroke. TRPV1 activation induces Ca2+ influx and activation of bradykinin and substance P release and BBB disruption. Furthermore, TRPV4 is involved in the modulation of the BBB and determines its alterations. First, BBB disruption is induced by a Ca2+ influx and activation of KCa channels. Second, the eNOS phosphorylation and its activation produce high levels of nitric oxide and high concentrations of cyclic guanosine monophosphate, the number of pinocytic vesicles containing caveolin-1 and caveolin-5 increases, which induces BBB disruption. Third, TRPV4 activation induces the formation of actin stress fibers through PKC and phosphorylation of myosin light chain 2 and the consequence BBB disruption. The image has been published previously in Luo et al. 100 Abbreviations: BBB, blood–brain barrier; BMECs, brain microvascular endothelial cells; TRPV1/4, transient receptor potential vanilloid 1/4; eNOS, endothelial nitric oxide synthase; ICH, intracerebral hemorrhage; RhoA, Ras homolog gene family member A; MLC2, myosin light chain 2; TJ, tight junction; AJ, adherens junction; PKC, phospholipase K; KCa, Ca2+-sensitive K+.
Several mechanisms of TRPV4 activation are responsible for BBB disruption. First, the activation of TRPV4 induces an increase in Ca2+ influx that stimulates Ca2+-sensitive K+ channels 133 causing disruption of the BBB.100,134 Second, the increased Ca2+ levels via TRPV4 activation can also determine eNOS phosphorylation and the release of NO activation of high activating guanosine monophosphate (cGMP) levels. The high levels of cGMP lead to increased numbers of pinocytic vesicles containing caveolin-1 and caveolin-2 and alteration in BBB permeability by its disruption. 135 Third, the formation of actin stress fibers mediated by TRPV4 activation induces the alterations of EC permeability, the phosphorylation of myosin light chain 2, and subsequently BBB disruption.136,137
Other TRP channels such as TRPV3 may have similar effect, but the exact mechanisms of other TRPV channels that may be involved have not yet been identified. 127
Physiological Functions of TRP Channels and Their Involvement in Neurological Diseases
As mentioned above, TRP channels at high or low expression levels have been demonstrated to be involved in in a variety of biological functions of different tissues.66,138 TRP channels are known to have functional roles in NDs related to the disruption in Ca2+ homeostasis.139–145
Therefore, we discuss here on the latest findings related with involvement of TRPCs in a number of NDs such as AD, PD, ALS, and MS. These NDs all show disruption of the BBB.
TRP Channels in AD
A correlation between the pathological markers of AD and perturbed cellular Ca2+ homeostasis has been reported to occur in in vivo and in vitro studies of AD patients.40,41,42,146
To date, TPRA1, TRPC1, TRPC3, TRPC6, TRPM2, TRPM7, TRPV1, TRPV4, TRPA1, and TRPML1 have been shown to be involved in AD (Fig. 18).
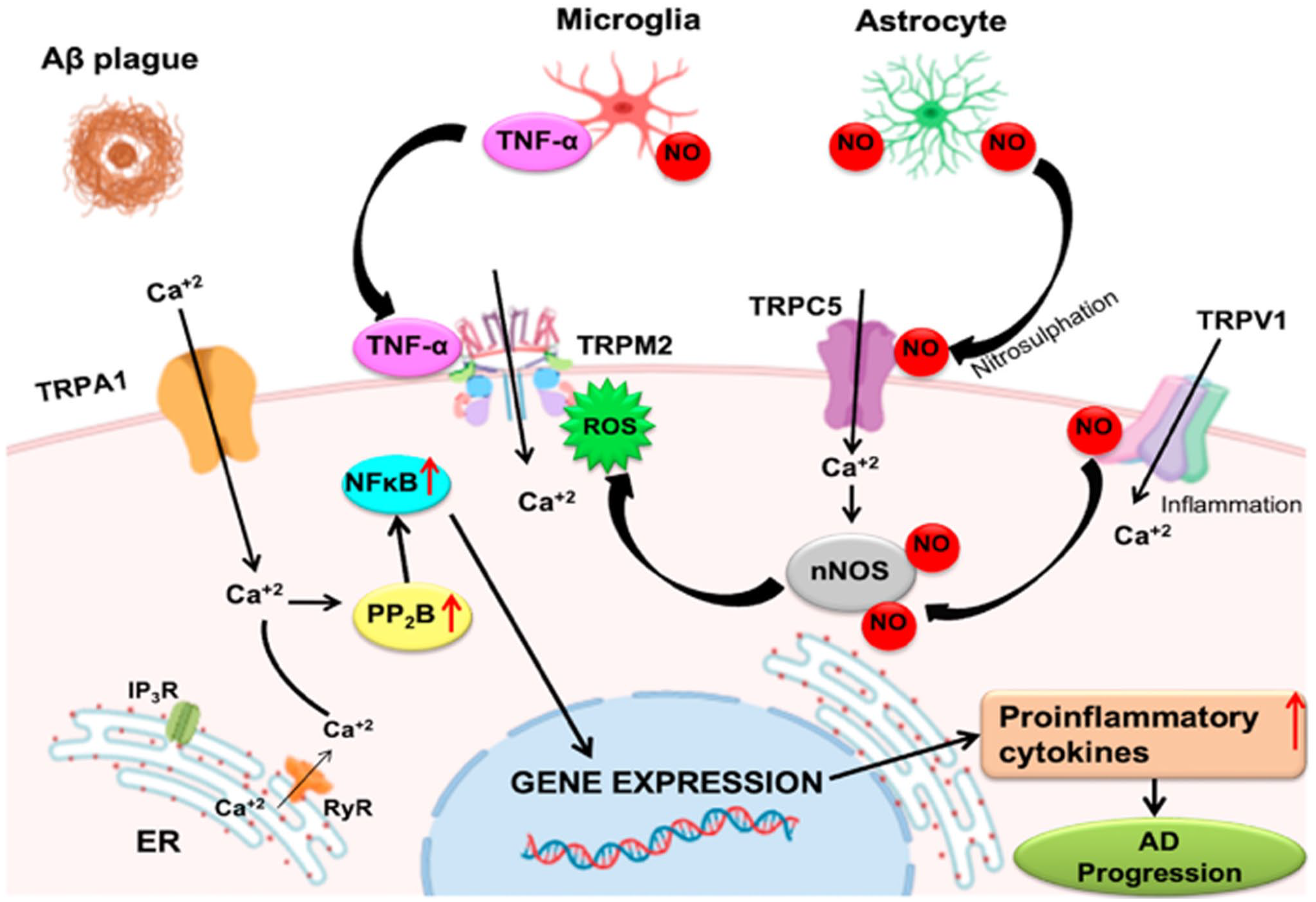
Roles of the major transient receptor potential (TRP) channels in AD. The role of TRPM2, TRPC5, and TRPV1 channels is shown in neuronal damage. All the biological mechanisms in which TRP channels are involved lead to AD and its progression. The image has been published previously in Rather et al. 128 Abbreviations: Aβ, amyloid β; TNF-α, tumor necrosis factor-α; NO, nitric oxide; TRPA1, transient receptor potential ankyrin 1; TRPC5, transient receptor potential canonical 5; TRPM2, transient receptor potential melastatin 2; TRPV1, transient receptor potential vanilloid 1; NF-κB, nuclear factor kappa B; ROS, reactive oxygen species; nNOS, neuronal nitric oxide synthase; AD, Alzheimer’s disease; ER, endoplasmic reticulum; IP3R, inositol-1,4,5-triphosphate receptor; RyR, ryanodine receptor; PP2B, protein phosphatase-2B.
TRPC1 is the most prevalent member of TRPCs in the brain; TRPC1 is linked to the SOCE process and its downregulation in astrocytes has been demonstrated to reduce this pathway in amyloid precursor protein (APP) knockout (KO) mice. 147
Alterations in BDNF-TrkB-TRPC3 signaling pathway induce hyperphosphorylation of tau protein via modulation of Ca2+ levels. 148 Another link between APP and TRPC3 is a major component of plasma membrane caveolae (caveolin-1) which interacts with APP as reported by Ikezu et al. 149
Moreover, TRPC5 is involved in AD progression by Ca2+ influx and production of NO by neuronal nitric oxide synthase (nNOS). 150
Pharmacological studies demonstrated that TRPC6 has a significant role in reduction of Aβ accumulation, 151 increased hippocampus neurogenesis, and increased long-term spatial memory. 152 This pathway interacts with cleavage of APP inhibiting its γ-secretase and not its α- or β-secretase activity. 153
It is known that TRPM2 channel ablation decreases microglia activation, improves the expression of synaptic markers, and reduces the deficits in memory observed in aging animals. 154
Furthermore, the expression of TRPM2 in rat striatum neurons and activation by Aβ and oxidative stress are involved in driving cell death in AD. 155
Increased ROS levels are regulated by complex mechanisms that involve antioxidant enzymes; reduced levels of these enzymes occur in aging and their supplementation downregulates the expression and activity of TRPM2. Increased activity of these channels leads to excitotoxicity and apoptosis in neuronal cells. 150
TRPM7 channels are involved in neuronal cell death in the brain; they play a role in the disruption of Ca2+ and Mg2+ homeostasis and increased susceptibility to degenerative processes.80,156,157
In experimental conditions, the genetic loss of TRPA1 has been shown to restrict spatial learning and memory deficits, increase Aβ deposition, promote of the release of pro-inflammatory cytokines such as IL-1β, IL-4, IL-6, and IL-10, and inhibit the activities of transcriptional factors such as nuclear factor kappa B (NF-κB) and nuclear factor of activated T cells. 158
Finally, it has been shown that overexpression of TRPML1 is sufficient to rescue memory and cognitive deficits by diminishing neuronal apoptosis in APP-presenilin (PS)-1 transgenic (Tg) mice (APP/PS-1 mice). 159
Figure 18 shows the main TRPCs involved in AD. 128 It is important to underline that microglia and astrocytes stimulated by Aβ produce TNF-α, which induces TRPM2 activation and increases levels of ROS and damage of the neuronal cells. On the other hand, the Ca2+ influx by the activation of TRPA1 stimulates transcriptional factors such as NF-κB to promote pro-inflammatory effect.128,158,160 TRPV1 also induces pro-inflammatory processes inducing NO production by nNOS. 161
When reduced levels of antioxidants, cytokines, and Aβ accumulation in AD stimulate Ca2+ influx that induces the progression of this pathology, a better understanding of these pathways is critical for developing novel therapeutic strategies.
TRP Channels in PD
As reported above, disruption of the dopaminergic neurons in the substantia nigra, Lewy bodies, and intracellular inclusions of α-syn protein are the most important hallmarks of PD.
Several mechanisms are known to be involved in the development and the progression of this disease (e.g., oxidative stress, mitochondrial dysfunction, and alteration in Ca2+ homeostasis).
TRPC1, TRPC3, TRPM2, TRPM7, and TRPV1 have been shown to be involved in the progression of PD. The activation of TRPC1 channels has been demonstrated to induce neuroprotection against apoptosis in SH-SY5Y neuroblastoma cells158,162; these mechanisms have been evaluated using a neurotoxin, which is able to induce the translocation of TRPC1 from the cell membrane into the cytoplasm as reported by Selvaraj et al. 164 Decreased levels of TRPC1 in the membranes as channels have been detected in brain lysates from the pars compacta of the substantia nigra (SNpc) of PD patients. 163 Moreover, TRPC3 levels are not altered in SNpc neurons in PD patients even when their levels are increased as compensatory effect for decreased TRPC1 level in 1-methyl-4-phenyl-1,2,3,6-tetrahyrdropyridine-induced PD-like conditions. 164
On the other hand significative oxidative stress induced by 1-methyl-4-phenylpyridinium ion (MPP+) increases intracellular Ca2+ influx via the TRPM2 channel and promotes dopaminergic neuronal cell death in the SNpc. 165 TRPM7 channel regulates the Mg2+ homeostasis in cells, and increased concentrations of these ions significantly inhibit MPP+-induced neurotoxicity by reducing the number of dopaminergic neurons and ameliorating the length of dopaminergic neurites.158,166
TRPV1 activation determines intracellular Ca2+ influx and upregulation of cytochrome-c release, caspase-3 cleavage and susequently mitochondrial disruption.108,128,158,164,167 Furthermore, activation of TRPML1 induces Ca2+ release from the ER and Ca2+ influx 168 ; these effects determine upregulation of lysosomal exocytosis, thus preventing α-syn accumulation in dopaminergic neurons.158,169
The role of a number of these channels and their possible involvement in PD are summarized in Fig. 19.
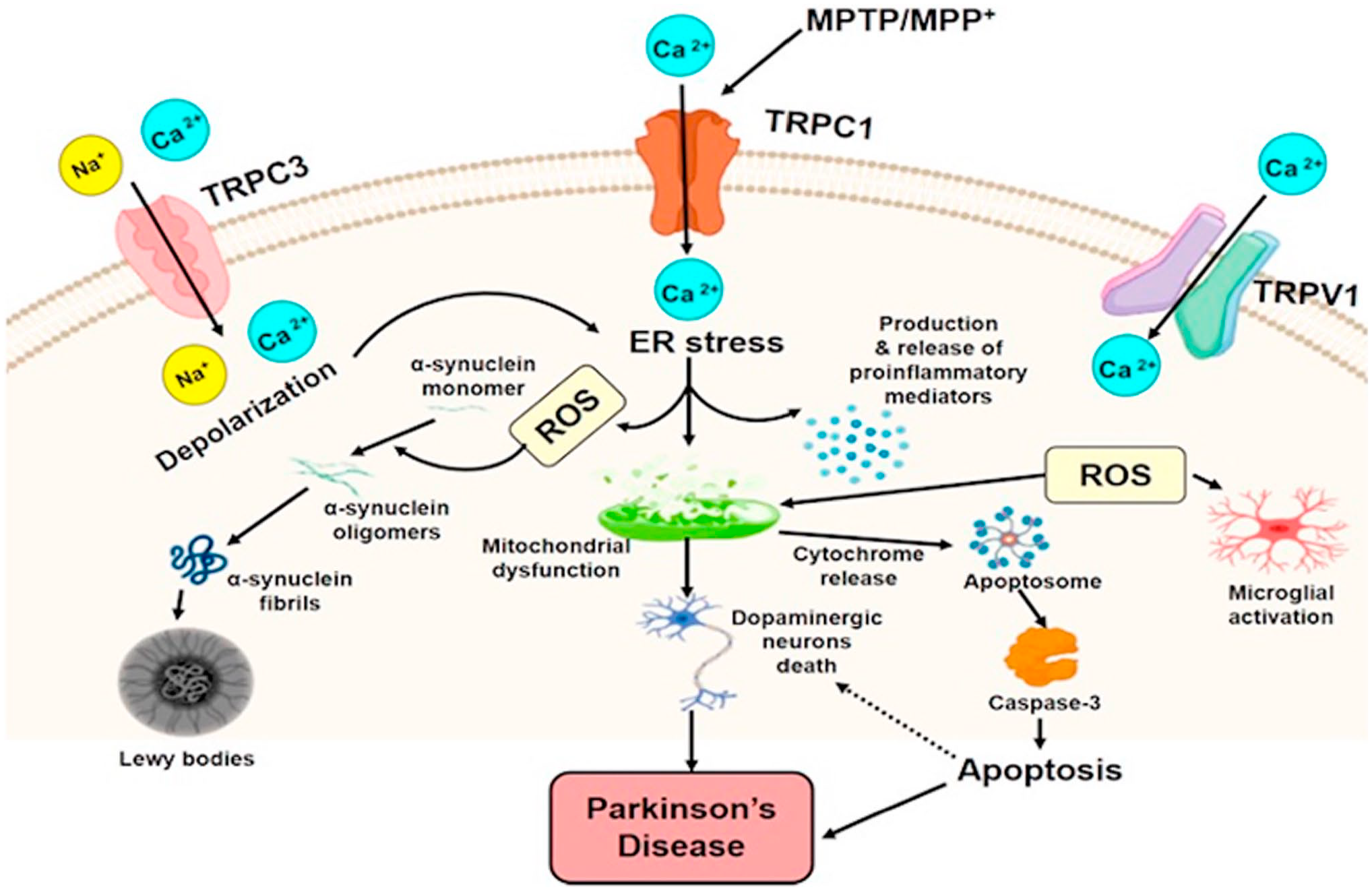
Roles of the major transient receptor potential (TRP) channels (TRPC1/3 and TRPV1) in PD. In particular, their role in the neuronal damage and their involvement in ROS formation and inflammation are shown. All the biological mechanisms of TRP channels lead to PD and its progression. The image has been published previously in Rather et al. 128 Abbreviations: TRPC1/3, transient receptor potential canonical 1/3; TRPV1, transient receptor potential vanilloid 1; ROS, reactive oxygen species; PD, Parkinson’s disease; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahyrdropyridine; MPP+, 1-methyl-4-phenylpyridinium ion; ER, endoplasmic reticulum.
TRP Channels in ALS
TRPC4, TRPM2, TRPM3, TRPM7, and TRPML1 have been shown to be involved in ALS (Fig. 20).
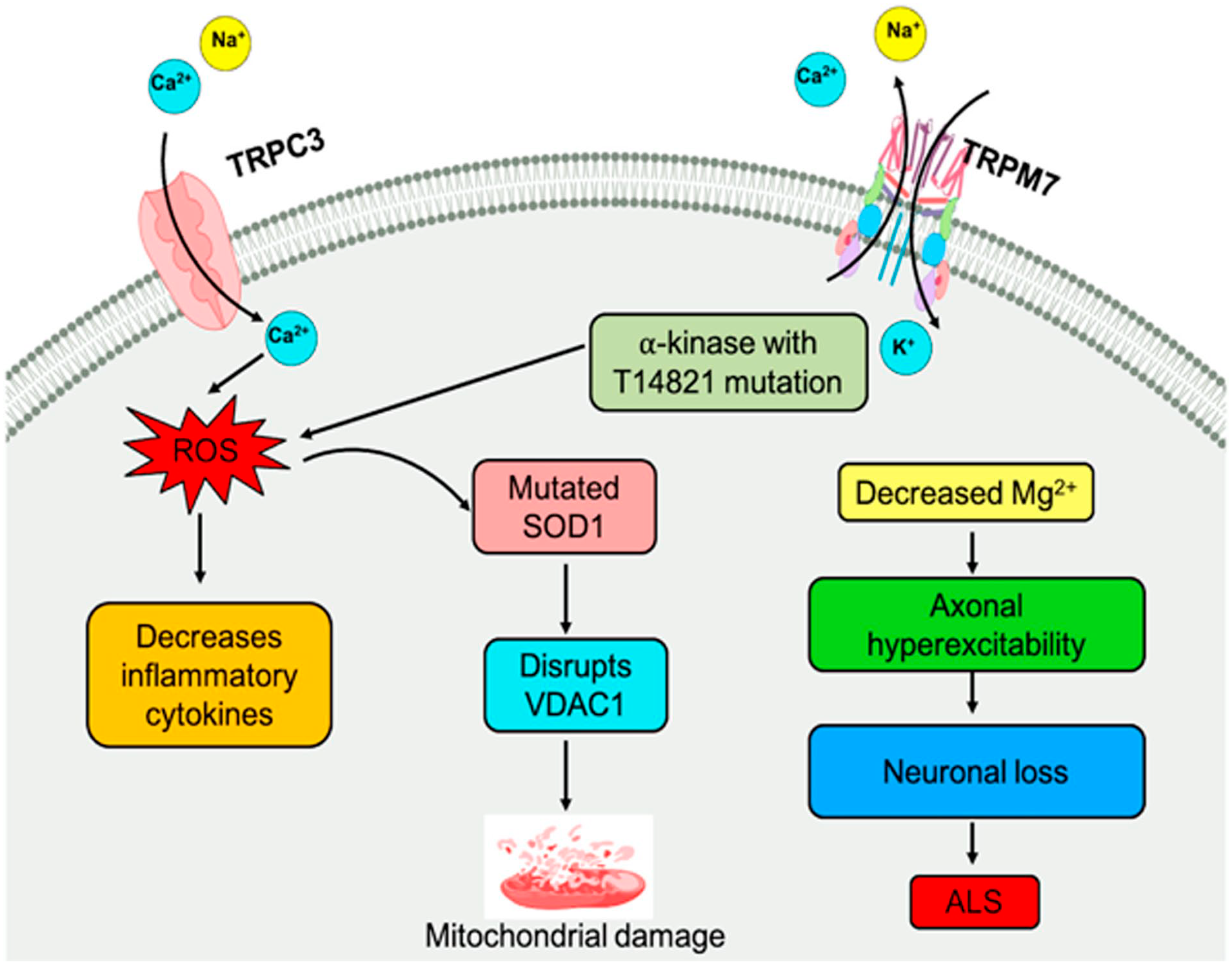
Some transient receptor potential channels involved in ALS. Activation of voltage-gated Na+ ion channels and decreased conduction of K+ ions cause hyperexcitability of axons. ROS generation causes superoxide dismutase1 dysfunction, which disrupts the VDAC1 inducing mitochondrial-dependent apoptosis, and alteration in the Mg2+ ions homeostasis contributes to the etiology of ALS. The image has been published previously in Rather et al. 128 Abbreviations: TRPC3, transient receptor potential canonical 3; TRPM7, transient receptor potential melastatin 7; ROS, reactive oxygen species; VDAC1, voltage-dependent anion channel 1; SOD1, superoxide dismutase; ALS, amyotrophic lateral sclerosis.
In particular, activation of TRPM7 channels, following the low levels of extracellular divalent cations, significantly contributes to cell death. The involved mechanisms (e.g., ROS production, α-kinase changes in its activity, superoxide dismutase1 activity and mitochondrial damage) are similar to those observed in stroke or brain in ischemia brain as well as ASL and other NDs. 141 In ALS patients, a TRPM7 channel variant, T14821, has been identified between the channel and the kinase region. Although T14821 does not induce changes in kinase activity, it decreases intracellular Mg2+ levels. Inhibition of this variant can alter Mg2+ homeostasis, lowering intracellular contents of the ion and stimulating progression of NDs.128,170
TPML1 regulates lysosome homeostasis and autophagy.124,128,171,172 Interestingly, phosphatidylinositol 3,5-biphosphate levels are significantly impaired in some forms of ALS.173,174 Moreover, in an experimental model of ALS, the expression of TRPML1 was reduced through autophagy causing ER stress and neuronal cell death. 175
TRP Channels in Huntington’s Disease (HD)
TRPC1, TRPC5, and TRPV1 have been shown to be involved in HD.
Figure 21 shows the findings published by Rather et al. on TRPC5 and TRPC1 channels in HD. 128
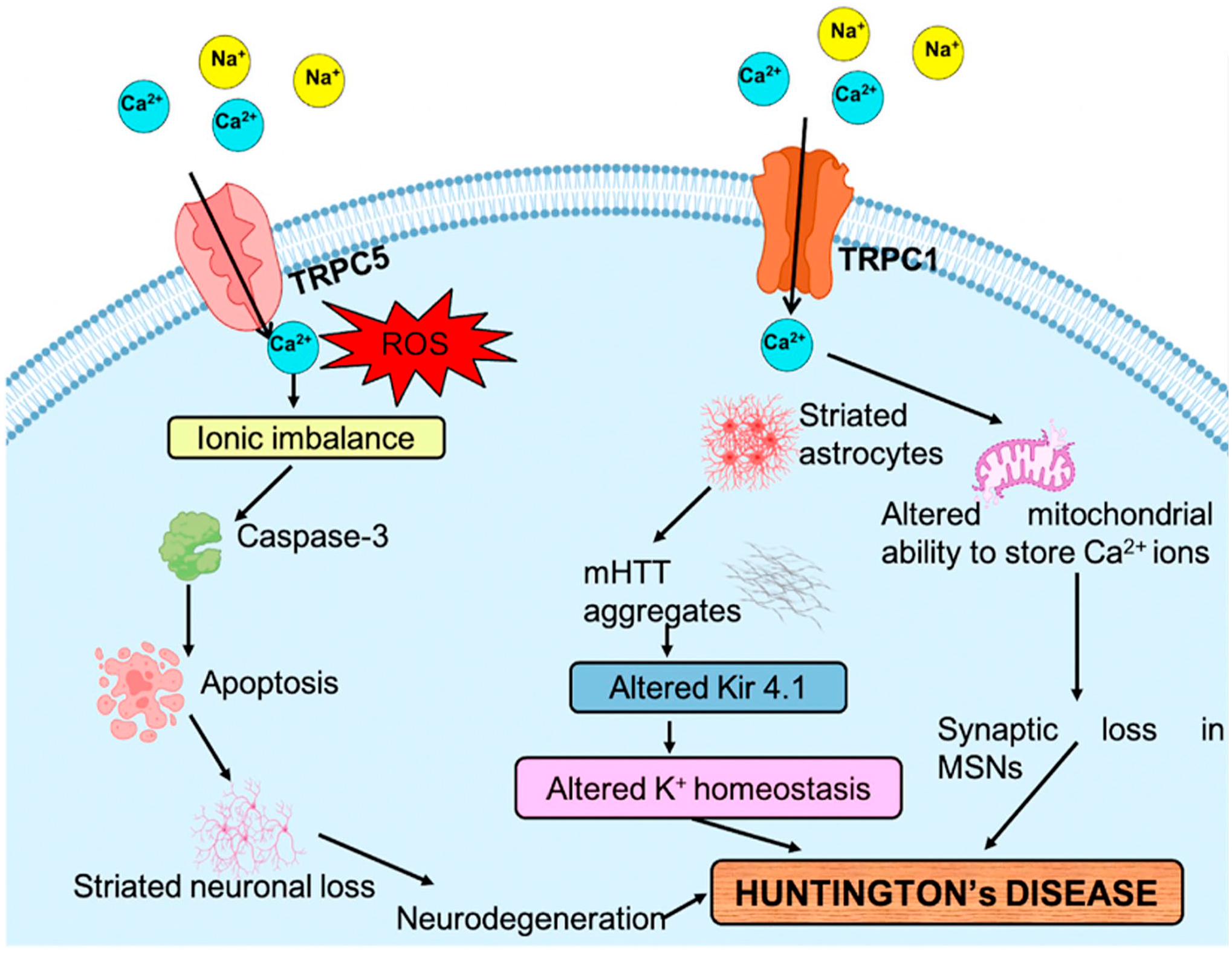
Role of transient receptor potential (TR channels in Huntington’s disease (HD). HD induces the expression of TRPC1 and TRPC5 channels and increases the generation of ROS with the entry of cations into the cell. Kir4.1 channel alters the K+ homeostasis and activates neuronal cells in mHTT protein, causing hyperexcitability. Alteration in Ca2+ homeostasis leads to mitochondrial dysfunction and loss of synapsis in MSNs. TRPC1 and TRPC5 allow the Ca2+ ions involved in the striatum causing striatal neuronal loss and then the development of HD. The image has been published previously in Rather et al. 128 Abbreviations: TRPC1/5, transient receptor potential canonical 1/5; ROS, reactive oxygen species; MSNs, medium spiny neurons; mHTT, mutant huntingtin.
Recently, it has been suggested that the expression of TRPC1 is decreased, whereas the expression of TRPC5 is increased due to activation by, oxidants that causes Ca2+-induced apoptosis in HD Tg mice.158,176
About TRPV1, it has been suggested that its activity may contribute to dysfunction in HD patients as that has been found in a rat model of HD. 177
TRP Channels in MS
As mentioned above, MS is an inflammation disease and Schattling et al. 178 demonstrate that the deficit of TRPM4 reduces the disease severity in TRPM4 KO mice resulting in the reduced axonal and neuronal degeneration without altering the immune function. 178 Moreover, it has been shown that the expression of TRPM4 is higher in patients with MS as compared with healthy controls. These findings suggest that the high expression of TRPM4 is involved in the toxic effects of glutamate levels, which is an important problem for MS. 178
Recently, Rosenkranz et al. 179 showed that TRPV4 inhibition increases endothelial resistance of BBB under homeostatic conditions; but this effect was lost during inflammation. 179 Furthermore, TRP4 activation causes disruption of TJs. 180 The findings are sometimes contradictory and the mechanisms and the conditions which lead to the inhibition of TRPV4 regulating the BBB permeability during inflammation need to be better evaluated. 179 In this case, the findings of Rosenkranz et al. 179 do not support therapeutic TRPV4 channel inhibition in MS or ischemic stroke.
The involvement of TRPV1 in MS is still not clear whether its activation has positive or negative effects. It is also unknown whether TRPV1 affects neuroinflammatory responses and whether TRPV1 can re-gulate Nod-like receptor protein 3 (NLRP3) inflammasome activation in the CNS. The findings reported by Zhang et al. 181 revealed that TRPV1-mediated NLRP3 inflammasome activation in microglia and its inhibition reduce neuroinflammation. The authors suggested that therapeutic targeting of TRPV1 channel may be a promising strategy to suppress NLRP3-driven neuroinflammation and subsequently MS.
Concluding Remarks
Research in the field of BBB disruption and NDs is challenging; it may be important to identify new targets for therapy. The BBB has a strong capacity for adaptation and plasticity over the life span 182 ; moreover, there is a significant variation in brain EC properties at different locations in the brain with respect to protein expression patterns along the cerebrovascular tree indicating that various proteins regulate the transfer molecules between blood and brain that mediate brain homeostasis. In the last decades, our understanding of regulated exchange of molecules over the various interfaces between blood and neural diseases has gratly expanded. This regulated transport between blood and neural tissue is very.
Similar to what countries, such as Switzerland and EU countries, do with import and export regulations to support their economic activity despite the existence of borders between countries as was demostrated by Badaut et al. 182 that shows this concept with characteristic impact (Fig. 22).

Schematic figure showing the trade between Switzerland and European Union with changes of adaptations depending on the needs of the partners. This change is controlled and selective with taxes fixed (before) by the partners. The image has been published previously in Badaut et al. 182
Furthermore, the role of TRP channels in the physiology and pathology should not be underestimated. As far as we know, it seems that the relationship among the homeostasis of the bivalent ions (e.g., Ca2+ and Mg2+), the BBB breakdown, and the development of NDs may be well due to increased expression of TRP channels. Thus, the changes in the activity mutations or posttranslational modifications of TRP channels may well result in diseases that affect human.
As a consequence, modulation of expression of TRPs may be important to identify new therapeu-tic strategies and possible benefits to treat NDs. Therefore, it needs to be investigated which mechanisms are responsible for changes in the BBB permeability and what roles the various TRO channels have in these changes. The present review underlines the role of BBB integrity in health and its al-terations in several NDs and the role of TRP ch-annels in NDs therapeuic as potential targets in these diseases.
Footnotes
Acknowledgements
The authors sincerely thank Dr Giorgia Cominelli (Ana-tomy and Physiopathology Division, Department of Cli-nical and Experimental Sciences, University of Brescia, Brescia, Italy) for her assistance during the revised ma-nuscript editing.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have contributed to this article as follows: conceptualization (RR, CJFvN, FR), writing of manuscript draft (RR, GF, MG, DP, ML, CJFvN, FR), drawing of figures and tables (GF, MG, DP, ML), supervision and improvisation of the manuscript content (RR, CJFvN, FR), and all authors have read and agreed to the published version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CJFvN is supported by the Slovenian Research Agency (Research project J3-2526).