
Abstract
Pressure ulcers represent a crucial clinical problem, especially in hospitalized patients. Ischemia–reperfusion (I-R) is an important cause of these lesions. Natural killer (NK), invariant NK T (iNKT), and dendritic epidermal T-cells, which express the natural killer group 2, member D (NKG2D) receptor, have been reported to have physiological roles in skin tissue repair and wound healing. However, a role for NKG2D–NKG2D ligand interactions in I-R-induced skin injury has not been determined. Using a murine pressure ulcer model, we demonstrated that I-R-induced ulcers in NKG2D-deficient mice were larger than those in wild-type or T-cell receptor δ knockout mice. Histopathological evaluation revealed that accumulation of macrophages and neutrophils at the peripheral deep dermis and subcutaneous tissue of the ulcers was enhanced in NKG2D-deficient mice. Rae-1 mRNA, which encodes an NKG2D ligand, was induced, and RAE-1 protein was detected immunohistochemically in fibroblasts and inflammatory cells in the dermis after reperfusion. RAE-1 expression was also increased in primary mouse fibroblasts treated with sodium arsenite. These results suggested that NKG2D ligand expression was induced by oxidative stress after I-R injury and support a putative role for this ligand in wound repair. Furthermore, the influx of NKG2D-positive cells at I-R sites may mitigate pressure ulcers via NKG2D–NKG2D ligand interactions.
Introduction
Pressure ulcers, also known as pressure sores or bedsores, are common in patients who require long-term institutional care and in those with sensory dysfunction and/or immobility. Geriatric medicine and improvements in nursing care and wound treatment techniques have reduced the incidence of pressure ulcers. Nevertheless, pressure ulcers remain a substantial clinical problem, especially when considering the continuing increase in elderly populations with limited mobility or underlying disease. 1
Tissue ischemia has long been regarded as important in pressure ulcer pathogenesis. However, the reperfusion following ischemia has been shown to be a major etiology of pressure ulcers. 2 Migration of polymorphonuclear leukocytes, mainly neutrophils, into the lesion is a histopathological characteristic of pressure ulcers induced by ischemia–reperfusion (I-R). 3 Oxygen-derived free radicals produced in the reperfused tissue accelerate cell damage. 4 Reactive oxygen species (ROS) are generated in the tissue under hypoxic conditions. Neutrophils also produce high levels of ROS, which are essential for bacterial digestion in the phagosome.
In addition to neutrophils and macrophages, various other inflammatory cells, including resident dendritic cells, are involved in pressure ulcer development. 5 Studies have shown that natural killer (NK) and invariant NK T (iNKT) cells play a key role in wound healing.6,7 These cells express natural killer group 2, member D (NKG2D), which is one of the major activating NK cell receptors. NKG2D is also expressed in CD8+ αβ T-cells and subsets of γδ T-cells. 8 Multiple ligands for this receptor have been identified, including major histocompatibility complex class I chain-related A and B and UL16-binding protein in humans and retinoic acid early transcript 1 (RAE-1) α–ε, histocompatibility 60 (H60) a–c, and mouse UL16-binding protein 1 (MULT1) in mice. Ligands for NKG2D are expressed on the surface of cells that are stressed by DNA damage, heat shock, infection, and tumorigenesis.9,10 Ligand expression is varied depending on the tissue-specific distribution and/or cause of cell stress. 11 Studies of NKG2D ligands have mainly focused on viral infection, malignant cells, autoimmune diseases, and post-transplantation disorders. Cell damage caused by I-R injury is a putative stimulator of NKG2D ligand expression.12,13
Although NKG2D ligand expression related to wound repair has been shown in several reports, 14 expression in pressure ulcers and its significance have not been fully elucidated. In this study, we used an experimental murine I-R model consisting of NKG2D-deficient mice to elucidate the involvement of NKG2D–NKG2D ligand interactions in the pathogenesis of pressure ulcers.
Materials and Methods
Animals
C57BL/6J mice were purchased from Sankyo Laboratory Service (Tokyo, Japan). B6.Cg-Klrk1tm1Dhr/J (NKG2D-deficient) (stock no. 022733) and B6.129P2-TcrdtmlMom/J [T-cell receptor δ knockout mice (TCRδ KO)] (stock no. 002120) were purchased from The Jackson Laboratory (Bar Harbor, ME), and KEAP1-dependent oxidative stress detector-luciferase (OKD-LUC) mice were obtained from TRANS GENIC INC. (Fukuoka, Japan). 15 Male and female mice (10–16 weeks old) were used for the experiments. All mice were bred and housed under specific pathogen-free conditions in our animal facility. All experiments using animals were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals at Hokkaido University Graduate School of Medicine and approved by the Institutional Review Committee of Hokkaido University (approval number 20-0037).
Antibodies
Antibodies and their sources were as follows: mouse RAE-1 pan-specific antibody and anti-mRae-1 (pan-specific) phycoerythrin (PE)-conjugated rat IgG (FAB17582P; R&D Systems, Minneapolis, MN), allophycocyanin (APC)-conjugated anti-mouse CD314 (NKG2D, clone CX5, cat. no. 130211; BioLegend, San Diego, CA), Brilliant Violet 510–conjugated anti-mouse NK-1.1 (clone PK136, cat. no. 108737; BioLegend), APC-conjugated anti-mouse Ly-6G/Ly-6C (Gr-1, clone RB6-8C5, cat no. 108411; BioLegend), PE/Cy7-conjugated anti-mouse CD68 (clone FA-11, cat. no. 137023; BioLegend), fluorescein isothiocyanate (FITC)-conjugated hamster anti-mouse CD3e (clone 145-2C11, cat. no. 553062; BD Biosciences; Franklin Lakes, NJ), PE-conjugated rat anti-mouse CD45 (clone 30-F11, cat no. 561087; BD Biosciences), and horseradish peroxidase–conjugated rabbit anti-mouse secondary antibody (AB_2340063; Jackson ImmunoResearch, West Grove, PA)
I-R Cycles
Experiments using the murine skin I-R model were performed as previously described.2,16,17 Briefly, backs of the mice were shaved for in vivo imaging experiments under anesthesia. The dorsal skin (comprising the epidermis, dermis, subcutaneous adipose tissue, and loose connective tissue layer and excluding the muscle layer) was gently pulled up and pinched between a pair of round ferrite magnetic disks that were 12 mm in diameter (113 mm2) and 5 mm thick; each disk had an average weight of 2.82 g and 1176 G magnetic force (Magfine; Sendai, Japan). This process created a compressive pressure of 50 mmHg between the two magnetic disks and was sufficient to reduce blood flow by 80% and induce skin ulceration and necrosis. For NKG2D ligand expression studies, flow cytometric analysis, in vivo imaging analysis, and evaluation of inflammation, the dorsal skin was pinched for 12 hr (ischemic period) followed by removal of the disks for the reperfusion period (Supplemental Fig. S1A). For I-R wound size comparisons, I-R was repeated four times with a 5-hr ischemia followed by 2- or 12-hr reperfusion to simulate clinical bedsores (Supplemental Fig. S1B). Mice were not immobilized or anesthetized during the I-R cycles. Each mouse developed a separate, round ulcer on the left and right sides of the dorsal skin. Wound healing of each pressure ulcer was photographed and analyzed digitally using Image J (NIH; Bethesda, MD).
Real-time PCR
Total RNA was extracted from the skin tissues using ISOGEN (NIPPON GENE; Tokyo, Japan) and treated with DNase I (Invitrogen; Carlsbad, CA) to remove genomic DNA in accordance with the manufacturers’ protocols. Total RNA was converted to cDNA with GoScript Reverse Transcriptase and an oligo(dT)15 primer (Promega; Madison, WI). Quantitative PCR was performed with the GoTaq 2-Step RT-qPCR System (Promega) using StepOnePlus (Applied Biosystems; Foster City, CA) following the manufacturers’ instructions. Each reaction was performed in triplicate. The relative expression of PCR products was calculated using the ΔΔCT method and normalized to Hprt. The list of primer pairs is shown in Supplemental Table 1.
Histological Examination and Immunohistochemistry
The I-R mice were euthanized, the dorsal skin tissue was collected, and the tissues were fixed in 10% neutralized formalin. The magnet-compressed, round injured areas were cut into halves and embedded in paraffin, and 4-μm-thick sections were prepared and stained with hematoxylin and eosin. The skin lesions were evaluated based on the histological alterations in the epidermis and hair follicles. The degree of inflammatory cell infiltration at the lesion site was evaluated using a three-tiered grading scale as follows based on the number of inflammatory cells per 0.01 mm2 area: mild, <50; moderate, 50–150; and severe, >150.
For immunohistochemistry, formalin-fixed tissue sections were deparaffinized in xylene and rehydrated in graded alcohols and distilled water. Sections were treated with 3% hydrogen peroxide (Kanto Chemical Co. Ltd; Tokyo, Japan) for 5 min at room temperature to block endogenous peroxidase. Antigen retrieval was performed using EnVision FLEX (Dako by Agilent; Santa Clara, CA) at pH 6.0, 97C for 20 min. After blocking with 10% normal rabbit serum (Fuji Film-Wako; Osaka, Japan) for 1 hr at room temperature, the sections were incubated with primary antibodies overnight at 4C followed by incubation with horseradish peroxidase–conjugated secondary antibody. Immunoreactivity was visualized with 3,3′-diaminobenzidine tetrahydrochloride, and the sections were counterstained with Mayer’s hematoxylin.
Flow Cytometry
Inflammatory cell infiltrates within the skin I-R injury sites were examined by flow cytometry. Skin fragments from the I-R sites were placed in RPMI medium supplemented with 1% fetal calf serum (FCS) and minced using scalpels. The samples were digested with Liberase TL (Roche; Mannheim, Germany) and DNase I (Roche) in RPMI with 1% FCS at 37C for 1.5 hr. Digested tissues were mashed and dissolved in phosphate-buffered saline with 0.1% bovine serum albumin and stained with antibodies as described above. Flow cytometric analyses were performed using an FACSCalibur instrument and CellQuest software (BD Biosciences). Dead cells were discriminated with 7-AAD Viability Staining Solution (cat no. 420404; BioLegend).
In Vivo Imaging and Analysis
Oxidative stress induced by I-R was evaluated using the In Vivo Imaging System (IVIS Spectrum CT; PerkinElmer, Waltham, MA) as previously described. 15 Briefly, OKD-LUC transgenic mice were injected intraperitoneally with 150 mg/kg luciferin (Promega) and anesthetized with a combination of anesthetics: 0.3 mg/kg medetomidine chloride, 4 mg/kg midazolam, and 5 mg/kg butorphanol tartrate. Fifteen min after luciferin injection, photon emissions from the dorsal skin of I-R mice were measured for 2 min using the IVIS and quantified using Living Image software. Luciferase activity was calculated as photons/sec/cm2.
Cell Culture (Rae-1 Expression in Primary Fibroblast by Oxidative Stress)
Primary fibroblast cells were established from 10- to 16-week-old C57BL/6 mice in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco; Thermo Fisher Scientific; Waltham, MA) supplemented with 10% FCS, 1% penicillin/streptomycin as previously described, and incubated at 37C in 5% CO2.18,19 Primary fibroblasts express RAE-1 on their surface. Therefore, the fibroblasts were cultured in serum-free DMEM for 3 days to lower RAE-1 expression. 20 Then, the culture medium was replaced with one of the following solutions: serum-free DMEM, 10% FCS/DMEM, or 10% FCS/DMEM with sodium arsenite (ASN) (Honeywell Fluka; Thermo Fisher Scientific) at a final concentration of 5 or 10 mmol/l. Flow cytometric analyses were performed to measure RAE-1 expression on the cell surface 1 day after the addition of ASN.
Statistical Analysis
Statistical analyses included two-tailed unpaired Student’s t-test, Fisher’s exact test, one-way ANOVA, and multiple Mann–Whitney tests, which were performed with Prism 9 (GraphPad; San Diego, CA). A p value of <0.05 was considered statistically significant.
Results
NKG2D Ligand Expression Is Induced at the I-R Site
To examine whether NKG2D–NKG2D ligand interactions were involved in cutaneous I-R, we used a murine I-R model to measure NKG2D ligand expression and the accumulation of NKG2D-positive cells at the injury site. A single 12-hr ischemia followed by reperfusion was conducted using wild-type mice in the following experiments to avoid the effects of repeated mechanical stimulation (Supplemental Fig. S1A).
Quantitative PCR revealed that reperfusion after ischemia markedly increased Rae-1 mRNA expression approximately 3.5-fold in the skin compared with that in the group without reperfusion (p<0.05) (Fig. 1A). Persistent ischemia alone had a mild influence on Rae-1 expression when the ischemia period exceeded 12 hr (p<0.05). Upregulation of Mult1 mRNA expression was detected in the skin only after 36 hr of reperfusion (p<0.05) (Fig. 1B). Long reperfusion time allows the injury site to develop ulcers and become infected, which may lead to the expression of NKG2D ligands. Therefore, Rae-1 mRNA induction after 12 hr of ischemia followed by 12 hr of reperfusion was worth further consideration. H60c, one of the NKG2D ligands that is expressed mainly during epidermal injury, was not induced either by ischemia or by reperfusion (data not shown). These results suggested that RAE-1 is the major NKG2D ligand upregulated by the I-R process.
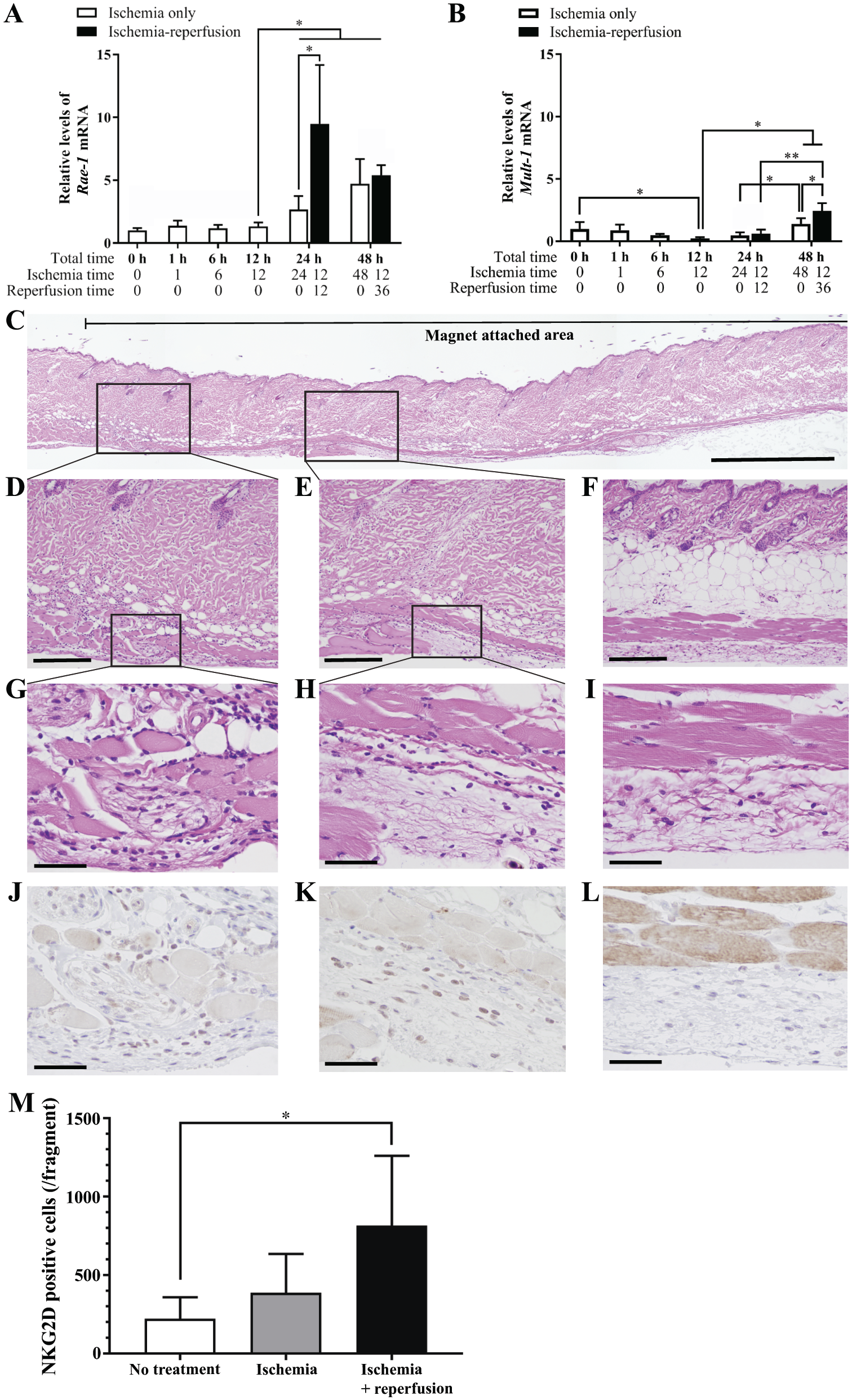
Expression of natural killer group 2, member D (NKG2D) ligands and infiltration of NKG2D-positive cells after cutaneous ischemia–reperfusion (I-R) in C57BL/6J mice. Relative levels of (A) Rae1 and (B) Mult1 mRNA expression in the cutaneous I-R lesions. Each sample was harvested after ischemia with or without reperfusion for the indicated times over a total period of 24 or 48 hr following the I-R schedule A (shown in Supplemental Fig. S1A). Black and white bars represent I-R and ischemia-only samples, respectively. Relative mRNA expression was quantified by real-time PCR. n=4–6 for each time point in each condition. All values represent means ± SD. *p<0.05 and **p<0.01 were determined by Student’s t-test. (C–L) Expression and distribution of RAE-1 protein in cutaneous I-R lesions. Skin sections were collected after 12-hr ischemia followed by 12-hr reperfusion and stained with (C–I) hematoxylin and eosin or (J–L) anti-RAE-1 antibody. (C) Low-magnification image. The area of the magnet disk attachment is indicated by the thin black line on the top. (D, E) Fibrosis and inflammatory cell infiltration were observed at the periphery and in the deep dermis of the lesion (indicated by black boxes in Fig. 1C). (G, H) High magnification of the areas of inflammation (indicated by black boxes in Fig. 1D and E, respectively). (J, K) RAE-1 immunohistochemistry. Dorsal skins from control mice were stained with (F, I) hematoxylin and eosin or (L) anti-RAE-1 antibody. The scale bars represent (C) 1 mm, (D–F) 200 µm, and (G–L) 50 µm. (M) The number of NKG2D-positive cells in cutaneous I-R tissue fragments after 12 hr of ischemia and 12 hr of reperfusion were counted using flow cytometry. No treatment (n=4), ischemia (n=6), and I-R (n=6). *p<0.05 was determined by Student’s t-test.
Next, we determined the expression of RAE-1 protein in tissue sections from the murine pressure ulcer model. Inflammatory reactions with fibrosis and leukocyte infiltration were observed at the periphery and deep dermis, subcutaneous tissue, and muscular layer of the I-R sites, although the damage of the epidermis was not apparent (Fig. 1C to I). Immunohistochemical staining demonstrated the presence of RAE-1-positive cells. Morphological examination indicated that spindle-shaped fibroblasts and infiltrating mononuclear leukocytes were positive for RAE-1 (Fig. 1J to L). These data suggested that the NKG2D–NKG2D ligand interaction may regulate wound healing after cutaneous I-R injury.
Reperfusion Increased the Influx of NKG2D-Positive Cells at the I-R Site
Induction of RAE-1 expression at the I-R site suggested the accumulation of NKG2D-positive effector cells. We performed flow cytometric analysis to compare the number of NKG2D-positive cells in the I-R lesions using wild-type C57BL/6J mice (Supplemental Fig. S2). NKG2D-positive cells were significantly increased at the I-R site after 12-hr ischemia/12-hr reperfusion (p<0.05) (Fig. 1M). These data revealed that NKG2D-positive cells were recruited to the I-R site.
NKG2D-deficient Mice Exhibited Impaired Wound Healing of Cutaneous I-R Injuries
Our results suggested a role for NKG2D–NKG2D ligand interactions at I-R sites. We used NKG2D-deficient mice to investigate the importance of this interaction in vivo. Four cycles of 5-hr ischemia followed by 2- or 12-hr reperfusion were conducted to simulate clinical pressure ulcers (Supplemental Fig. S1B). A series of digital images of the I-R lesions were captured daily until wound closure, and comparisons were made between NKG2D-deficient mice and wild-type mice. NKG2D-deficient mice developed larger wounds than wild-type mice at 24–72 hr after the final ischemic period (Supplemental Fig. 2A).
Histological examination and evaluation were performed to clarify the cause of enlarged wounds after the I-R cycles. The lesions were categorized into four groups based on the alterations in the epidermis and hair follicles (Fig. 2B): no change (normal, Fig. 2C), reactive acanthosis (hypertrophy, Fig. 2D), thinning of epidermis and atrophic hair follicles (atrophy, Fig. 2E), and loss of epidermis and hair follicles (Fig. 2F), and the widths of the wounds were measured. Areas of atrophy and loss of epidermis were located in the center of the skin lesion, while hypertrophic epidermis was observed in the periphery of the I-R site. Wound widths, which consisted of atrophied tissue and loss of epidermis, were not significantly different between NKG2D-deficient and wild-type mice at 12 hr after the last ischemic period. Wounds in the NKG2D-deficient mice remained wide and were significantly larger than those in wild-type and TCRδ KO mice (p<0.05 and p<0.01, respectively) at 60 hr after the final ischemic period (Fig. 2G). These data suggested that NKG2D-positive cells play a role in promoting wound healing after cutaneous I-R injury.
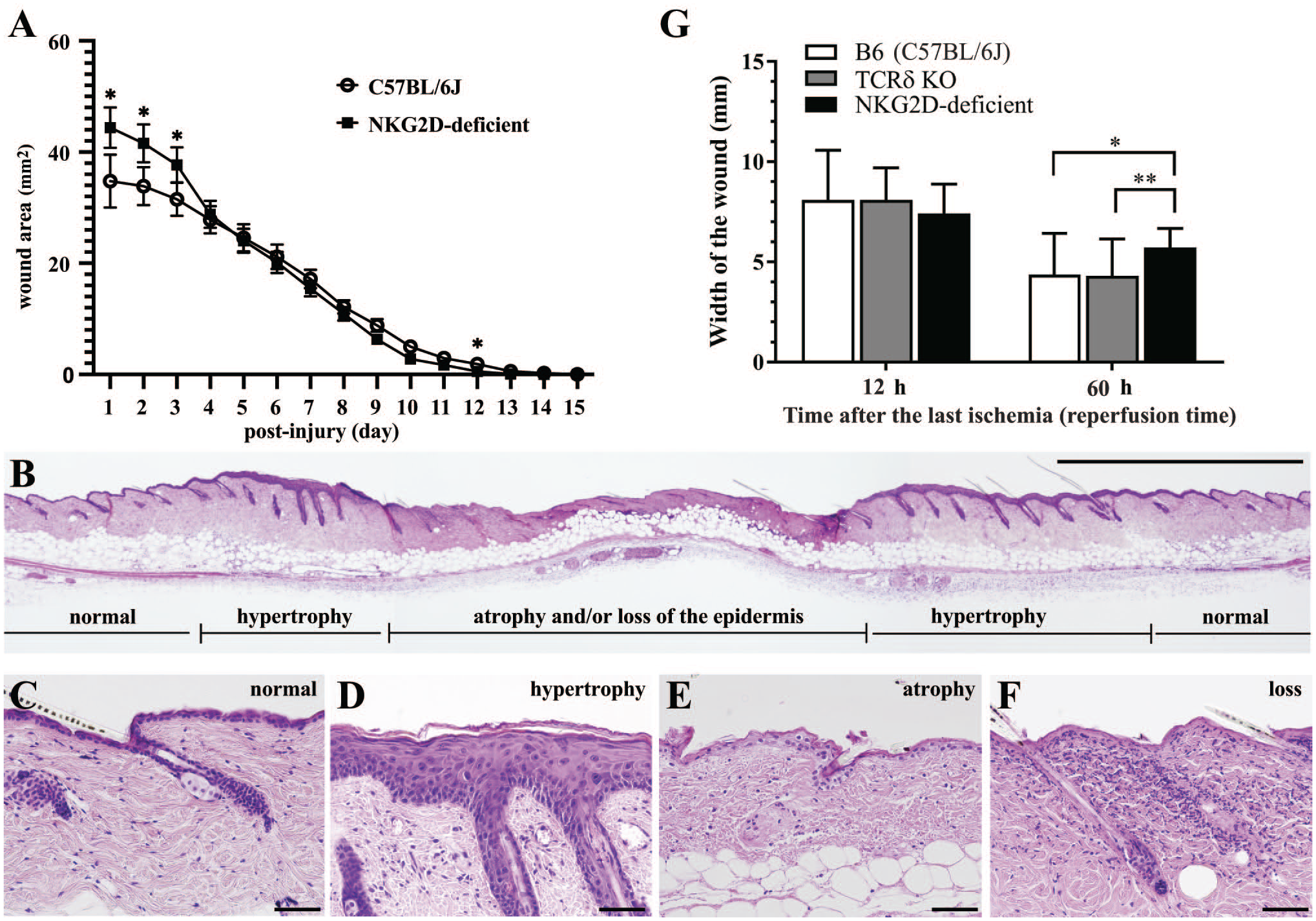
Histological examination of cutaneous ischemia–reperfusion (I-R) injuries. (A) The wound areas after I-R injury were measured in natural killer group 2, member D (NKG2D)-deficient (n=7) and control C57BL/6J mice (n=7) for 15 days. I-R was performed as described in schedule B (Supplementary Fig. S1B). *p<0.05 was determined by multiple Mann–Whitney tests. (B–F) Histological changes of the epidermis and subcutis at the I-R site in a control C57BL/6J mouse. I-R was performed as in schedule B. (B) A histological image of the entire I-R injury site. Low magnification, hematoxylin and eosin (H&E). (C) There were no histological changes in the epidermis outside of the magnet disk. (D) Epidermal reactive acanthosis (hypertrophic change) was observed adjacent to the edge of the magnet disk attachment. Atrophic thin epidermis (E) and loss of epidermis (F) were observed at the center of the magnet disk in the cutaneous I-R site (H&E staining). The scale bars represent 2 mm and 50 µm in (B) and (C–F), respectively. (G) The width of the wounds (atrophy and/or lack of epidermis) after cutaneous I-R. The diameters of the wounds were measured using microscopy at 48 hr (12-hr reperfusion) and 96 hr (60-hr reperfusion) as shown in schedule B (Supplemental Fig. S1B). All values are presented as means ± SD of 10–21 wounds at each time point for each mouse genotype; the white bars represent control C57BL/6J mice (n=6, 12-hr reperfusion; n=12, 60-hr reperfusion), gray bars represent T-cell receptor δ (TCRδ) knockout mice (n=5 and 9 for 12 and 60 hr, respectively), and black bars represent NKG2D-deficient mice (n=5 and 9 for 12 and 60 hr, respectively). *p<0.05 and **p<0.01 were determined by Student’s t-test.
Inflammation Is Prolonged at the I-R Lesion in NKG2D-deficient Mice
We performed histological analysis to evaluate the degree of inflammation at the I-R sites and explore the cause of delayed wound healing in NKG2D-deficient mice. Increased infiltration of macrophages at the I-R site was confirmed in NKG2D-deficient mice by flow cytometry compared with that in wild-type mice (p=0.006) (Fig. 3A). Neutrophil numbers at the I-R sites were not significantly different between the mouse groups by flow cytometry (Fig. 3B). However, accumulation of neutrophils within the subcutaneous tissue was observed in the periphery of the wounds by histological analysis (Fig. 3C). We employed a three-grade evaluation system to assess neutrophilic infiltration: mild (Fig. 3D), moderate (Fig. 3E), and severe (Fig. 3F). Neutrophilic infiltration was severe in 9 out of 36 lesions from NKG2D-deficient mice and only 2 out of 40 lesions from wild-type B6 mice (p<0.05) (Fig. 3G). These results suggested that the healing of I-R pressure wounds is impaired by NKG2D deficiency because of inflammatory cell accumulation within the deep dermis at the periphery of the I-R wound site.
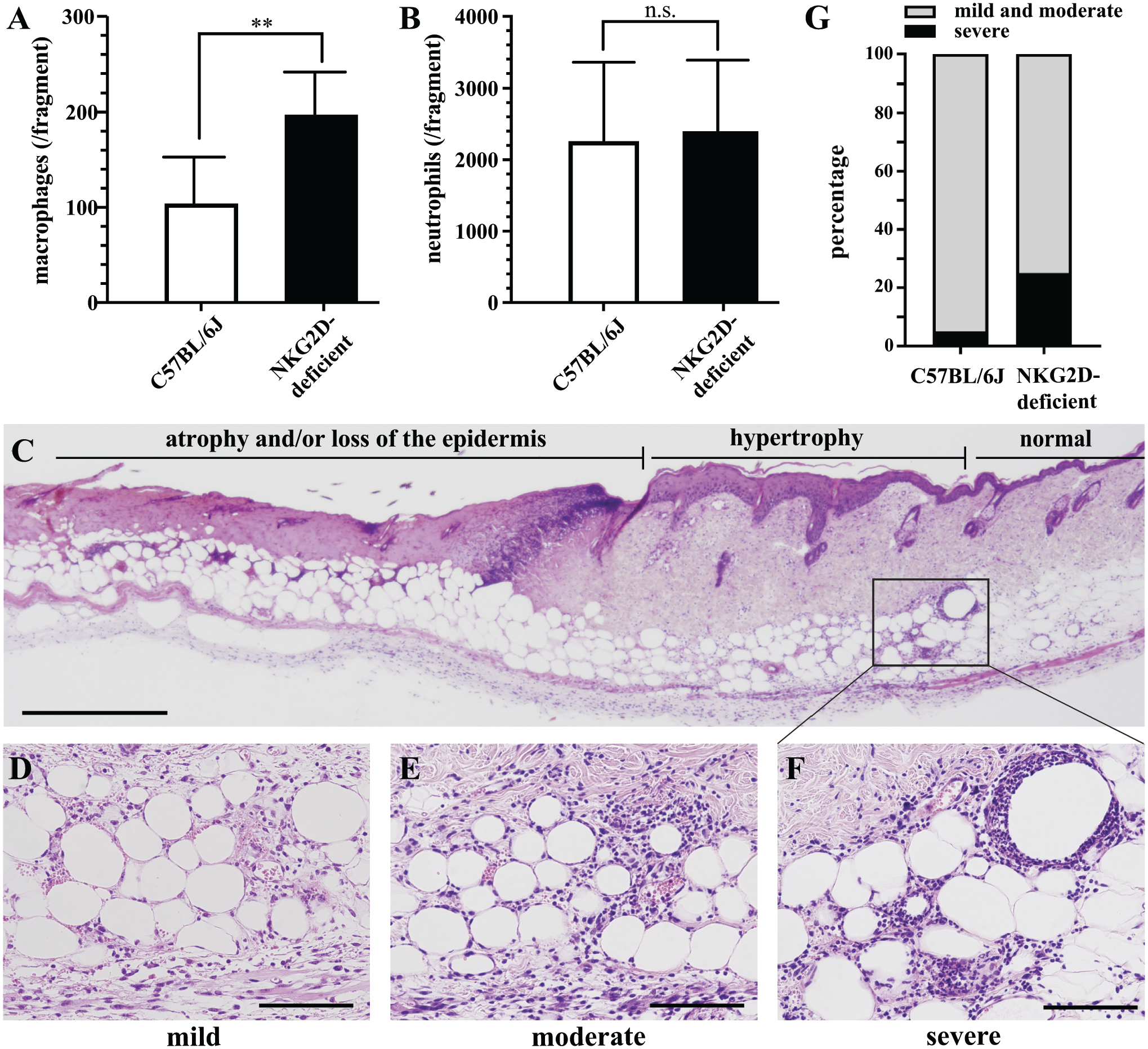
Infiltration of neutrophils in cutaneous ischemia–reperfusion (I-R) lesions. The number of macrophages (A) and neutrophils (B) in cutaneous I-R tissue fragments at 12 hr after ischemia were counted using flow cytometry. Control C57BL/6J mice (n=6) and natural killer group 2, member D (NKG2D)-deficient mice (n=6). **p<0.01 was determined by Student’s t-test. (C) Accumulation of inflammatory cells was observed mainly within the peripheral area of the wounds. The degree of neutrophil infiltration was scored using a three-tiered scale: (D) mild, 50 inflammatory cells; (E) moderate, 50–150 inflammatory cells; and (F) severe, >150 inflammatory cells per 0.01 mm2. Scale bars represent (C) 500 µm and (D–F) 100 µm. (G) The frequency of severe neutrophilic inflammation in the peripheral areas of the I-R sites. The cutaneous I-R lesions were collected at 96 hr as described in schedule B (Supplemental Fig. S1B). Control C57BL/6J mice (n=40) and NKG2D-deficient mice (n=36). p<0.05 was determined by Fisher’s exact test.
Oxidative Stress Enhanced RAE-1 Expression in Fibroblasts
Previous reports have indicated that oxidative stress plays an important role in cutaneous I-R injury. 16 Oxidative stress is a putative inducer of NKG2D ligands21,22; hence, we proposed that oxidative stress would induce expression of the NKG2D ligand RAE-1 in the cutaneous I-R injury site. To validate this hypothesis, we used OKD-LUC mice to confirm and visualize the oxidative stress elicited at the I-R site. OKD-LUC mice express luciferase via the KEAP1-NRF2 pathway in tissues that are susceptible to oxidative stress. 15 Administration of luciferin enabled the visualization of luminescence, which was detected using the IVIS. The cutaneous I-R site in OKD-LUC mice exhibited ring-shaped enhancement of luminescence after 15-hr ischemia followed by 9-hr reperfusion (Fig. 4A). The areas of enhanced luminescence were located at the periphery of the ischemic lesion. This distribution was consistent with the locations of immunohistochemical expression of RAE-1 and neutrophilic accumulation (Fig. 1C to L and Fig. 3C to F).
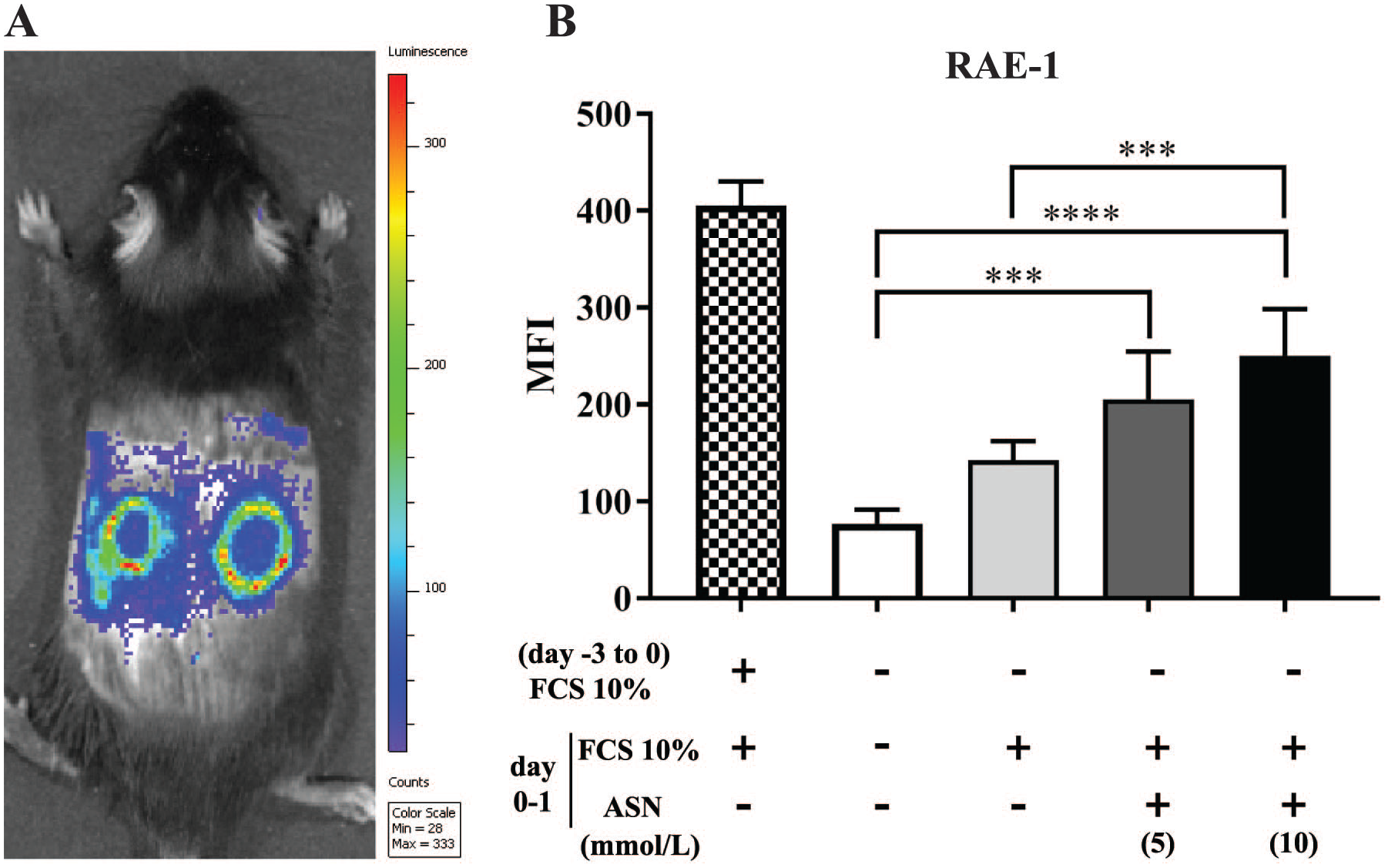
Oxidative stress in cutaneous ischemia–reperfusion (I-R) lesions. (A) The In Vivo Imaging System was used to visualize OKD-LUC (KEAP1-dependent oxidative stress detector-luciferase) mice after cutaneous I-R (15-hr ischemia followed by 9-hr reperfusion). A representative image of a pair of ring-shaped enhanced luminescence patterns at the I-R wound site on the dorsal skin is shown. (B) RAE-1 expression induced by oxidative stress in primary fibroblast cultures. Fibroblasts were cultured without serum for 3 days. Then, the culture medium was replaced with fresh medium supplemented with fetal calf serum (FCS) with or without sodium arsenite (ASN) at the indicated final concentrations. RAE-1 expression was analyzed by flow cytometry at 12 hr after retreatment. The y-axis shows the mean fluorescence intensity (MFI) values after subtraction of the isotype control antibody signal. Data represent means ± SD of five experiments per condition. ***p<0.001 and ****p<0.0001 were determined by one-way ANOVA.
To confirm that oxidative stress induced RAE-1 expression in murine skin, primary fibroblasts harvested from wild-type mice were cultured with or without the ROS inducer ASN, 23 and RAE-1 expression was assessed using flow cytometry. RAE-1 expression was induced in fibroblasts cultured with ASN (Fig. 4B). These results suggested that oxidative stress induced RAE-1 expression in fibroblasts at the cutaneous I-R injury site.
Discussion
NKG2D is a major activating receptor of NK cells, NKT cells, and dendritic epidermal T-cells (DETCs) and aids in the killing of stressed cells. Recently, a novel role for NK and NKT cells in tissue repair has been reported.6,7 A pressure ulcer is a chronic skin wound in which acute and chronic inflammatory processes disturb wound healing. 24 Reperfusion has been shown to be a main etiology of pressure ulcers. This study was conducted to elucidate the role of NKG2D–NKG2D ligand interactions in pressure ulcer development.
In this study, NKG2D-deficient mice showed significantly larger wounds than wild-type mice at 60 hr after the last ischemic period. NKG2D-deficient mice showed delayed wound healing, although DETC-deficient TCRδ KO mice did not display expanded wounds at the I-R sites. TCRδ KO mice were reported to have delayed epidermal regeneration during wound healing. 25 The lack of delayed wound closure in the TCRδ KO mice in our study supports our finding that I-R injury was initiated at the deep dermis, subcutaneous tissue, and muscle layer as shown in the histological analysis.
Tissue contraction and epidermal regeneration are the major processes of wound healing. Tissue injury in a pressure ulcer is located mainly in deep cutaneous tissue, which is susceptible to ischemia and external force. 26 DETCs reside in the epidermis and support the wound healing mainly via epidermal regeneration.14,25,27 Our results showing relatively mild injury in the epidermis and little influence of DETCs in epidermal regeneration at the I-R site were consistent with those previous reports.
Our histological findings exhibited tissue damage and inflammatory reaction caused by I-R injury located mainly in the dermis, suggesting prolonged neutrophilic infiltration inhibited wound contraction at the site. Neutrophilic accumulation in injured areas is indispensable for the tissue repair process. 28 However, prolonged and excessive retention of acute inflammatory cells, such as neutrophils and macrophages, delays wound repair.29,30 NKT cells also participate in skin wound healing. 6 NK cells have been shown to control the degree of neutrophilic accumulation in corneal abrasions. 31 Our results support a role for NKG2D-positive cells in wound healing. Although the mechanism for controlling neutrophils via NK cells remains unclear, some reports suggested that NKG2D-positive cells eliminate senile cells in the tissue or attenuate overt inflammation.32 –34 The mechanism of prolonged inflammation at cutaneous I-R sites, the determination of subsets of NKG2D-positive cells, and the details of cellular interactions with neutrophils and macrophages will require further investigation.
The expression of NKG2D ligand is upregulated under stressful conditions, including DNA damage, heat shock, infection, and tumorigenesis. Our in vitro study with primary fibroblasts suggested that reperfusion-induced oxidative stress upregulated RAE-1 expression at the I-R site. E2F transcription factors were reported to be regulators of Rae-1 expression. 20 E2F1 transcription factor plays a pivotal role for cell tolerance in oxidative stress. 35 E2F1 is a putative transcription factor inducing RAE-1 expression in I-R injury.
Taken together, we conclude that cutaneous I-R upregulates RAE-1 expression in fibroblasts at the periphery and deep dermis of the I-R site, which may be caused by reperfusion-induced oxidative stress. RAE-1 protein interacts with NKG2D-positive cells and suppresses acute inflammation at the wound site. Without NKG2D–NKG2D ligand interactions, inflammation remains and inhibits wound healing. The mechanisms underlying the murine cutaneous I-R model are summarized in Fig. 5.
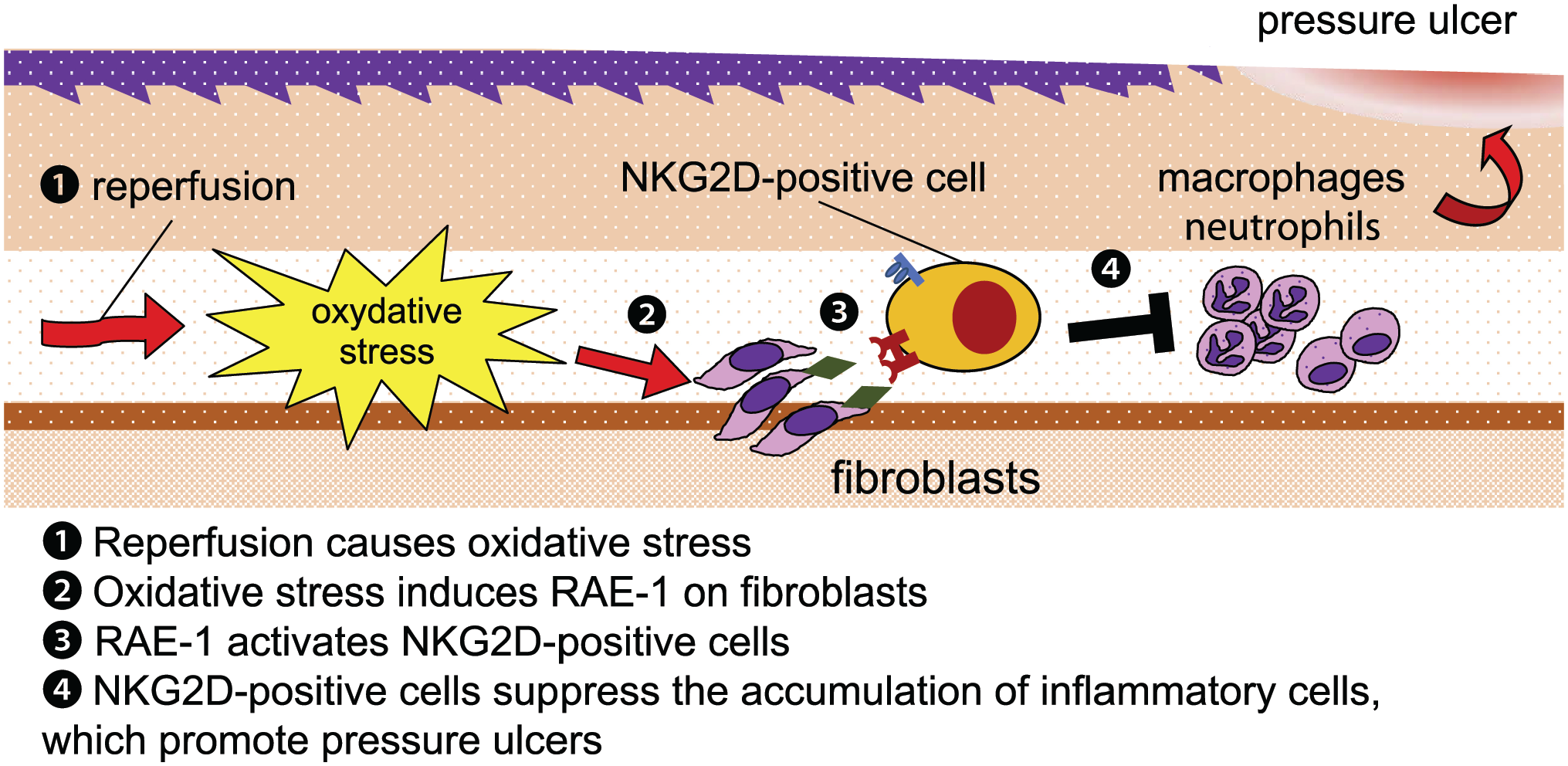
Schematic illustration of the natural killer group 2, member D (NKG2D)–NKG2D ligand interaction in an ischemia–reperfusion lesion. Reperfusion causes oxidative stress and induces the expression of the NKG2D ligand RAE-1, which modulates the function of recruited NKG2D-positive cells and suppresses inflammatory cell accumulation. This results in the promotion of wound healing.
Several studies have reported the involvement of NKG2D and NKG2D ligands in tissue I-R injuries, such as injuries after organ transplantation, coronary angioplasty, and thrombolytic therapy following cerebral infarction.12,13,36,37 These studies proposed that NK cells caused the tissue damage, and NKG2D blockade attenuated I-R injury. However, our study indicated that NKG2D-positive cells supported wound repair of I-R injured tissue. Internal organs, such as liver and kidney, are susceptible to ischemia, whereas skin is relatively resistant to ischemic damage. In addition, the cutaneous I-R model provides relatively mild ischemic conditions, considering that the reduction in blood flow rate was 80%. Therefore, it is necessary to distinguish cutaneous I-R injury from other I-R-related conditions. There is no direct evidence showing the expression of NKG2D ligands in human pressure ulcers. However, human NKG2D ligands MICA and ULBP2 are expressed in aging fibroblasts and are required for efficient recognition and removal of senescent cells, 34 suggesting human NKG2D ligands are involved in the wound repair process as demonstrated in murine studies.
Our findings suggest that NKG2D–NKG2D ligand interactions play a role in the early pathogenic phase of cutaneous I-R injury. Substantial defects in the epidermis and deep ulcerated tissue may develop if cutaneous I-R injury is prolonged. Local induction or administration of NKG2D ligands may be a therapeutic strategy to prevent the development of pressure ulcers.
To conclude, histological analysis revealed that experimental cutaneous I-R injury was more severe in NKG2D-deficient mice compared with wild-type mice. Neutrophilic infiltration at the injured site was prolonged and prevented wound healing. Overexpression of RAE-1, which is an NKG2D ligand, at the injured site was induced by the reperfusion process. Immunohistochemical and in vitro analyses indicated that oxidative stress induced RAE-1 expression in fibroblasts. Our findings suggest that NKG2D–NKG2D ligand interactions play a key role in mitigating I-R injury in the skin.
Supplemental Material
sj-docx-8-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-docx-8-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-1-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-1-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-2-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-2-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-3-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-3-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-4-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-4-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-5-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-5-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-6-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-6-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-eps-7-jhc-10.1369_00221554221147582 – Supplemental material for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury
Supplemental material, sj-eps-7-jhc-10.1369_00221554221147582 for NKG2D Ligand Expression Induced by Oxidative Stress Mitigates Cutaneous Ischemia–Reperfusion Injury by Keishi Makita, Noriyuki Otsuka, Utano Tomaru, Koji Taniguchi and Masanori Kasahara in Journal of Histochemistry & Cytochemistry
Footnotes
Acknowledgements
We thank Kayo Miyazaki and Yasuyo Udo for preparation of histological sections of murine skin tissue and supporting the immunohistochemical analyses. We also thank Eri Murata for technical advisement on the murine I-R model and IVIS analysis and Shunji Ikeshita and Yusuke Ohta for the cell preparation and gene expression studies. We are grateful to the animal facility staff at Hokkaido University School of Medicine for supporting all animal experiments and breeding. We thank Susan Zunino, PhD, from Edanz (![]() ), for editing a draft of this manuscript.
), for editing a draft of this manuscript.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have contributed to this article as follows: KM performed the animal research, histological examinations, IVIS analysis, and wrote the initial draft of the manuscript. NO designed and coordinated the studies and performed the animal research and histological analyses. NO also contributed to the interpretation of the data and wrote the manuscript. UT performed the fibroblast culture assay and analyzed the data. MK and KT contributed to interpretation of the data and assisted in the preparation of the manuscript. All authors have reviewed and approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Japanese Grant-in-Aid for Scientific Research (C) No. 16K11076.
Data Accessibility Statement
The data supporting the results of this study are available from the corresponding author upon request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.