
Abstract
Versican, a chondroitin sulfate proteoglycan, is an essential component of the extracellular matrix (ECM) in inflammatory lung disease. Versican’s potential as an immunomodulatory molecule makes it a promising therapeutic target for controlling host immune responses in the lungs. To establish changes to versican expression and accumulation during influenza A viral pneumonia, we document the temporal and spatial changes to versican mRNA and protein in concert with pulmonary inflammatory cell infiltration. These studies were performed in the lungs of wild-type C57BL6/J mice on days 3, 6, 9, and 12 post-infection with influenza A virus using immunohistochemistry, in situ hybridization, and quantitative digital pathology. Using duplex in situ hybridization, we demonstrate that type I interferon signaling contributes significantly to versican expression in lung stromal cells. Our findings show that versican is a type I interferon–stimulated gene in pulmonary fibroblasts and pericytes in the context of viral pneumonia. These data also provide a guide for future studies to determine the role of versican in the pulmonary immune response to influenza infection:
Introduction
Versican, a chondroitin sulfate proteoglycan, is an ECM molecule that is abundantly expressed in the lungs during embryonic development and is minimally expressed in the healthy lungs of adult mice and humans.1–3 There are five isoforms of versican with different glycosaminoglycan (GAG)-binding domains: V0 containing α-GAG and β-GAG domains, V1 containing only the β-GAG domain, V2 containing only the α-GAG domain, V3 containing no GAG-binding domains, and V4 which contains a portion of the β-GAG domain.1–4 Re-expression and accumulation of versican in the mature lung is a consistent finding in the study of various inflammatory lung diseases in human patients and studies using mouse models of pulmonary disease.1,5–14 Versican is increasingly recognized as a critical component of the immune response, particularly regarding the interactions of the versican-enriched ECM with immune cells as they move from circulation to sites of pulmonary inflammation.15–22 Versican interacts with immune cell surface receptors and impacts immune cell phenotype, adhesion, migration, activation, and retention within the inflamed lung.12,16,17,23–26 Versican also retains, releases, and interacts with cytokines, chemokines, and growth factors, which further inform immune cell activity and direct the host inflammatory response.27–29 Two signaling pathways, previously identified in vitro in specific cellular lineages, are responsible for versican expression. In smooth muscle cells, the canonical Wnt/β-catenin/T-cell factor (TCF) pathway controls versican promoter activity, and β-catenin signaling is required for transforming growth factor-β1–induced versican expression.30–33 Our previous work shows that Trif-dependent and type I interferon (IFN)–dependent signaling regulates versican expression, identifying versican as a type I IFN–stimulated gene in macrophages. 21 In addition, our studies show versican is necessary for type I IFN production by macrophages following exposure to toll-like receptor agonists. 21 Type I IFNs play a critical role in regulating the immune response to viral infection and establishing the host antiviral state.34,35 Taken together, type I IFN–stimulated versican likely plays an essential role in coordinating the immune response to viral pneumonia.
However, little is known about the expression and accumulation of versican in the context of viral pneumonia. In addition, the cellular specificity of the production of type I IFN–stimulated versican during lung infection is unknown. Studies using versican-deficient mice exposed by oropharyngeal instillation to polyinosinic:polycytidylic acid [poly (I:C)], a synthetic analog of double-stranded RNA, and toll-like receptor-3 (TLR3) agonist show that versican is an immunomodulatory molecule.17,21 These studies demonstrate that mice lacking versican in macrophages (LysM/Vcan−/−) have an increased recovery of inflammatory leukocytes in bronchoalveolar lavage (BAL) fluid after instillation with poly(I: C), suggesting an anti-inflammatory role for type I IFN signaling–stimulated versican expression by macrophages. In contrast, mice with a global deficiency in versican (Rosa26/Vcan−/−) treated with poly(I: C) have a decreased recovery of inflammatory leukocytes in the BAL fluid, suggesting that versican produced by stromal cells plays a critical role in initiating leukocyte migration within the lungs. The versatile activities of versican produced by different cell types in coordinating the host immune response to pulmonary infection and inflammation warrant further investigation into its roles in the host response to viral pneumonia pathogenesis.
Therefore, this study establishes the time course of changes in versican expression, versican accumulation, and leukocyte migration into the lungs during severe influenza A virus (IAV) infection. In addition, the study characterizes the role of type I IFN signaling and identifies the cellular source of type I IFN–stimulated versican during influenza infection in the lungs. The study encompasses a 12-day time frame after infection to focus on the entire disease phase (0–9 days post-infection) and a limited portion of the recovery phase (10–12 days post-infection). We report the accumulation of versican and inflammatory cells from myeloid and lymphoid lineages in all lung compartments throughout influenza infection using immunohistochemical techniques and quantitative digital pathology. 36 We investigate the role of type I IFN during influenza infection and identify another cellular source, in addition to macrophages, in which type I IFN stimulates versican expression. Day 9 is a critical time as this is when the transition from the disease to the recovery phase occurs and there is an increased accumulation of myeloid immune cell infiltration into the lungs. We use novel image analysis techniques to accomplish this work, employing artificial intelligence and machine learning to perform high-quality quantitative digital pathology (QDP). These studies show that (1) versican mRNA expression and protein accumulation peak in influenza-infected lungs on 6 days post-infection (dpi) and 9 dpi, respectively, in concert with various immune cell populations; (2) versican expression correlates closely with type I IFN expression, particularly on 3 and 6 dpi; and (3) stromal cells contribute significantly to type I IFN–stimulated versican in the transition from the disease phase to recovery phase on 9 dpi. Our findings provide the basis for the development of future mechanistic studies.
Materials and Methods
Animal Model
Eight- to 10-week-old male C57BL6/J wild-type (WT) and B6(Cg)-Ifnar1tm1.2Ees/J (Ifnar1−) mice (The Jackson Laboratory; Bar Harbor, ME) housed under standard conditions and on a 10:14 hr dark:light cycle were infected with mouse-adapted IAV. The lungs were collected for immunohistochemistry, mRNA isolation, and in situ hybridization. In all experiments, the sample size (n) is the number of mice per group. All procedures were performed as part of an approved scientific protocol in accordance with the University of Washington Institutional Animal Care and Use Committee (IACUC).
Induction of IAV Pneumonia
Mouse-adapted Influenza A/Puerto Rico/8/34 (A/PR/8/34; H1N1) was grown in the allantoic fluid of research-grade-specific pathogen-free (SPF) embryonic chicken eggs (Charles River Avian Vaccine Services; Norwich, CT), and a hemagglutination assay was performed to determine the viral titer. 37 Male mice were infected with 20 plaque-forming units (PFU) in 50 µL PBS by oropharyngeal aspiration under isoflurane anesthesia. 38 Our lab had previously found that a dose of 20 PFU caused severe influenza pneumonia in which mice approach humane euthanasia endpoint criteria but ultimately recover from infection. In addition, 20 PFU was an intermediate dose compromising 40% of the euthanasia dose 50, which was 50 PFU for C57Bl/6 male mice infected with mouse-adapted influenza A/PR/8/34 H1N1. 39 Control mice received PBS alone. Mice were sacrificed at 3, 6, 9, and 12 dpi by exsanguination under isoflurane anesthesia.
Versican, Versikine, and Leukocyte Marker Quantitative Immunohistochemistry
Lungs for immunohistochemistry experiments were inflated with 10% neutral buffered formalin at 21 cm H2O pressure and, along with the heart and mediastinal lymph nodes, immersed in 10% neutral buffered formalin for 24 hr, transferred to 70% ethanol, and embedded in paraffin.40,41 A cutting instrument with trimming blades was used to section the mouse lungs into 2 mm sagittal sections to ensure uniform random sampling of all lung lobes. 36 Five to six sagittal lung sections were embedded per paraffin block and placed on corresponding slides.
All immunohistochemistry staining procedures including deparaffinization for 30 min at 72C were performed on the Leica Bond Automated Immunostainer (Leica Biosystems; Buffalo Grove, IL). For all protocols, except versican immunohistochemistry, deparaffinization was followed by antigen retrieval at 100C and peroxide block for 5 min at room temperature before combining with the primary antibody. The versican immunohistochemistry staining protocol includes an additional digestion step before peroxide block. Primary and secondary antibodies were applied as described for the specific protocol, followed by DAB detection, hematoxylin counterstain, and mounting as directed by the manufacturer for the BOND Polymer Refine (DAB) Detection system (Leica Biosystems).
Versican immunohistochemistry was performed using an anti-β-GAG antibody with reactivity to V0 and V1 versican isoforms based on the presence of β-GAG found in each isoform (V0 α-GAG and β-GAG, V1 β-GAG only). V0 and V1 isoforms predominate in inflamed lungs. 17 All lungs were pretreated with heat-induced epitope retrieval solution-1 (HIER1), a citrate-based buffer and surfactant (Leica Biosystems), for 10 min and then with 0.2 U/ml chondroitinase ABC (cat. no. C3667; MilliporeSigma, Burlington, MA) in 18 mM Tris, 1 mM sodium acetate, and 1 mg/ml BSA pH 8.0 for 1 hr at 37C as previously described. 8 Peroxide block was performed before combining with a polyclonal rabbit anti-mouse β-GAG antibody (cat. no. AB1033, lot no. 3050782; MilliporeSigma, Burlington, MA) at 1:250 in Leica primary antibody diluent for 30 min at room temperature to detect versican accumulation.
Versikine immunohistochemistry was performed using an anti-DPEAAE antibody with reactivity to the versican V0/V1 neoepitope degradation product. All lungs were pretreated with heat-induced epitope retrieval solution-2 (HIER2), an EDTA-based buffer and surfactant (Leica Biosystems), for 10 min before incubating with a polyclonal rabbit anti-mouse DPEAAE antibody (cat. no. PA1-1748A, lot no. MB153302; Invitrogen, Carlsbad, CA) at 1:800 in Leica primary antibody diluent for 60 min at room temperature.
For Ly6b, F4/80, CD3, and CD8 immunohistochemistry of lung tissue, antigen retrieval was performed with HIER2 for 10 min with Ly6b and 20 min with F4/80, CD3, and CD8. For CD68 and CD4 protocols, antigen retrieval was performed with HIER1 at 10 and 20 min, respectively, before incubating with a primary monoclonal antibody. The following primary monoclonal antibodies were used: rat anti-mouse Ly-6b.2 alloantigen antibody, clone 7/4 (cat. no. MCA771G, lot no. 1801; Bio-Rad, Hercules, CA) at 1:10,000; rat anti-mouse CD68 antibody, clone FA-11 (cat. no. MCA1957, lot no. 1807; Bio-Rad) at 1:1500; rabbit anti-mouse F4/80 antibody, clone D2S9R (cat. no. 70076S, lot no. 5; Cell Signaling, Beverly, MA) at 1:500; rat anti-mouse CD3 antibody, clone CD3-12 (cat. no. MCA1477, lot no. 1708Bio-Rad) at 1:100; rat anti-mouse CD8 antibody, clone 4SM15 (cat. no. 14-0808-80, lot no. 2144532; eBioscience, San Diego, CA) at 1:1000; and rat anti-mouse CD4 antibody, clone 4SM95 (cat. no. 14-9766-80, lot no. 4342629; eBioscience) at 1:800. All primary antibodies were diluted in Leica Primary antibody diluent for 30 min at room temperature except for the primary F4/80 antibody, which was diluted in Signal Stain(R) Ab Diluent (cat. no .8112L, lot no. 30; Cell Signaling) for 30 min at room temperature. For Ly6B, CD68, CD3, CD4, and CD8 protocols, the primary antibody was followed by a rabbit anti-rat IgG(H+L), Mouse Adsorbed, Unconjugated secondary antibody (cat. no. AI-4001, lot no. ZF-0513; Vector, Burlingame, CA) at 1:300 in 5% normal goat serum and tris-buffered saline (TBS) for 8 min at room temperature. For F4/80, the primary antibody was followed by a goat anti-rabbit polymer-HRP-IgG in 10% animal serum and TBS for 8 min at room temperature. Negative controls were performed using purified Rabbit IgG (cat. no. AB-105-C, lot no. ER1314071; R&D Systems, Minneapolis, MN) at 1:1000 or purified Rat IgG2b Isotype (cat. no. 553986; BD Pharmingen, San Jose, CA) at 1:1000 in Leica primary antibody diluent for 30 min at room temperature.
Image analysis was performed following immunohistochemistry using whole-slide digital images and automated image analysis. Bright-field whole-slide digital images were created using a 20× objective and the Nanozoomer Digital Pathology slide scanner (Hamamatsu; Bridgewater, NJ). Analysis of the whole-slide digital images was performed using Visiopharm software (Visiopharm; Hoersholm, Denmark). Regions of interest (ROIs) were manually defined by drawing around the visible area of the lung, excluding any cardiac or mediastinal lymph node tissue present on the slide. Digital features RGB-G, RGB-B, HDAB-DAB, H&E Eosin, and Chromaticity Red were trained to label positive staining for versican or leukocyte markers and the background tissue counterstain (hematoxylin) using project-specific configurations based on a threshold of pixel values. Images were processed to generate desired outputs (area of positive staining for leukocyte markers or versican as a ratio of positive staining to total tissue area). All ROIs were sampled at 100%. Based on the output for each slide analyzed, the mean value for percentage positive staining for each day post-infection was calculated by averaging the results for mice collected on particular days post-infection.
Acute Lung Injury Scoring
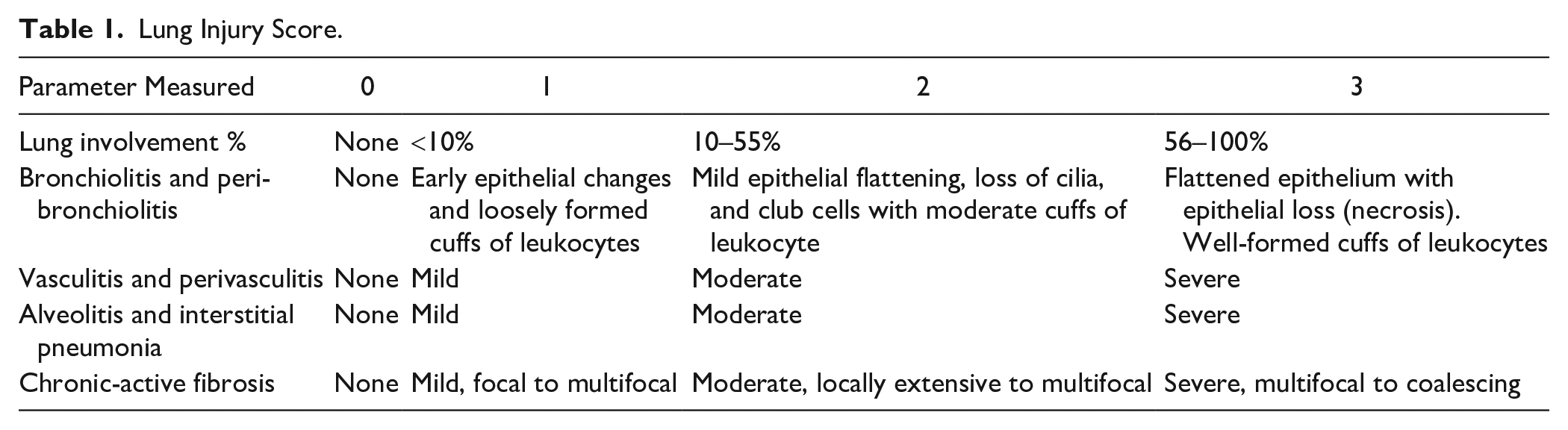
Histological assessment of lung inflammation and injury was completed using formalin-fixed paraffin-embedded lung tissue stained with hematoxylin and eosin (H&E). 42 The lung injury score was performed using a modified version of previously described semiquantitative scoring systems.43–46 The analysis was performed by a comparative pathologist (C.W.F.) who was blinded to the treatment group. Assessment of lung inflammation and injury included the extent and severity of the following parameters: (1) lung involvement (%), (2) bronchiolitis/peribronchiolitis, (3) vasculitis/perivasculitis, (4) alveolitis and interstitial pneumonia, and (4) chronic active fibrosis, including dysplastic alveolar regeneration.42,47–49 The score for each parameter was given a value from 0 to 3, with 0 being normal and 3 being the most severe. Due to the significance of lung involvement and chronic-active fibrosis to long-term health, the scores for these two parameters were weighted. The score was doubled for lung involvement and tripled for chronic-active fibrosis, resulting in the maximum lung injury score of 24. The complete scoring system used to evaluate lesions observed in H&E-stained lung tissue is described in Table 1.
Lung Injury Score.
Quantitative Real-time Reverse-transcription PCR
The left lung lobe was used for mRNA isolation and was cut into small pieces with no dimension greater than 0.5 cm before being placed in 5 ml of RNAlater (Invitrogen). RNAlater solution containing lung pieces was gently rotated at 4C overnight. RNA was extracted using RNAeasy Mini Kit with on-column DNase digestion (Qiagen; Valencia, CA). RNA quality was measured, and an RNA integrity number (RIN) was determined for each sample (Bioanalyzer RNA 6000 Nano Kit; Agilent, Wilmington, DE). All samples had an RIN between 8.2 and 9.7. cDNA was reverse-transcribed using random primers with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time reverse-transcription polymerase chain reaction (PCR) was performed on an ABI Prism 7900HT Fast Real-Time PCR System (Applied Biosystems) using PrimeTime Gene Expression Master Mix (Integrated DNA Technologies, Coralville, IA). Gene-specific TaqMan primer-probe mixes were used for quantitative real-time PCR of versican (Mm01283063_m1), interferon beta (IFN-β) (Mm00439552_s1), and TATA-box protein (Mm01277042_m1) mRNA (ThermoFisher Scientific; Grand Island, NY). The primer-probe mix spanning the junction of versican exon 3–4 was used to detect the expression of all five versican isoforms.
Versican, PDGFRβ, and CD68 Quantitative In Situ Hybridization
Lungs for in situ hybridization experiments were inflated with 10% neutral buffered formalin at 21 cm H2O pressure, immersed in 10% neutral buffered formalin for 16–24 hr, washed in molecular-grade PBS, transferred to 70% ethanol for up to 48 hr, and embedded in paraffin. A cutting instrument with trimming blades was used to section the mouse lungs into 2 mm sagittal sections to ensure uniform random sampling of all lung lobes. 36 Five to six sagittal lung sections were embedded per paraffin block and on corresponding slides. Blocks were stored at 4C with desiccant. Instruments, work surfaces, gloves, and blades associated with microtomy were treated with RNAse Away, and RNAse free water was used for the floatation bath.
All in situ hybridization staining procedures were performed on the Leica Bond Automated Immunostainer (Leica Biosystems) with ready-to-use (RTU) 2.5 LS duplex target probes for mouse platelet–derived growth factor receptor beta (PDGFRβ) (cat. no. 411388, lot no. 19142A), CD68 (cat. no. 316618, lot no. 19142A), and versican (cat. no. 486238-C2, lot no. 19142A) at 1:50 in PDGFRβ RTU LS Probe or CD68 RTU LS Probe, control probes PPIB(C2) (cat. no. 313918-C2, lot no. 1914A) at 1:50 in POLR2A(C1) (cat. no. 320768, lot no. 19036A) RTU, negative control bacterial gene DapB-C2 (cat. no. 312038-C2, lot no. 19142A) at 1:50 in Duplex Negative Control Probe C1 (cat. no. 320758, lot no. 18344A) RTU, and the 2.5 LS duplex reagent kit (cat. no. 322440) as directed by the manufacturer (ACDBio; Newark, CA). Briefly, slides were baked and deparaffinized on the Leica Bond Automated Immunostainer, pretreated with HIER2 for 15 min at 95C, and underwent protease III digestion for 15 min at room temperature before combining with positive, negative, or test probes for 120 min at room temperature. In situ hybridization amplification steps specific to the protocol were followed by peroxide block, Leica bond mixed red detection (cat. no. DS9390, lot no. 64002), staining with ACD C1 green stain, counterstaining with hematoxylin, and baking at 60C for 30 min according to the manufacturer’s instructions.
Image analysis was performed following immunohistochemistry using whole-slide digital images and automated image analysis. All slides were scanned in bright field with a 20× objective using a Nanozoomer Digital Pathology slide scanner (Hamamatsu). Whole-slide digital images were imported for analysis into Visiopharm software (Visiopharm). ROIs were manually defined by drawing around the visible area of the lung. The digital feature contrast red-green was used to label positive staining for versican (red) or cell markers CD68 and PDGFRβ (green), and the digital feature RGB-R with a mean filter was used to label background tissue counterstain (hematoxylin) using project-specific configurations based on a threshold of pixel values. Images were processed to generate desired outputs (total pixels of positive staining for PDGFRβ, CD68, and versican as a ratio of positive staining to total tissue area). All ROIs were sampled at 100%. Next, a digital feature to identify nuclei was created using the Deep Learning module in the Visiopharm APP Author software. This module was given examples of manually segmented nuclei and trained for 24,373 iterations to develop the algorithms used to identify nuclei. The machine learning algorithms were then used to identify and label nuclei in image ROIs randomly sampled at 20%. The labeled nuclei were then processed using a range of digital features and postprocessing steps to generate desired outputs, which included the number of nuclei that were single-positive for versican mRNA and double-positive for PDGFRβ/versican and CD68/versican mRNA, and the percentage of PDGFRβ nuclei that were double-positive for PDGFRβ/versican mRNA. Based on the output for each slide analyzed, the mean values were calculated and reported by mouse strain or cell marker of interest.
Stromal Cell Culture
Mouse lung fibroblasts were isolated from whole lung explants by digestion as previously described.
50
Lungs were aseptically removed from the thorax, minced, and transferred into a 15-ml conical tube with 2.5 ml digestion solution consisting of Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) high-glucose medium, 1000 U/ml Liberase LT, and 1000 U/ml DNase I. Lung digests were incubated in a shaking water bath for 60 min at 37C. Then digest was filtered through a 100-µm cell strainer with DMEM/F-12 containing 10% fetal bovine serum (FBS), and the red blood cells were lysed by suspending the cells in sterile water for 30 sec. The fibroblasts were then washed and plated in DMEM/F-12 with 20% FBS, 2 mM
Statistics
Statistics and images were generated using GraphPad Prism (La Jolla, CA). For PCR analyses of gene expression, normalized mRNA levels were expressed as -fold of levels in untreated or vehicle-treated controls using the comparative cycle threshold (Ct) method compared with the designated housekeeping gene.8,51 Quantitative PCR (qPCR) analyses were performed with two technical replicates. Comparisons of qPCR and quantitative immunohistochemistry and in situ hybridization data were analyzed by one-way ANOVA followed by Bonferroni’s posttest for multiple comparisons, with the mean of every influenza-treated group compared with the mean of vehicle-treated groups. Comparisons for acute lung injury scoring were analyzed by Kruskal–Wallis followed by Dunn’s posttest for multiple comparisons, with the mean of every influenza-treated group compared with the mean of vehicle-treated groups. 52 In vitro qPCR data were analyzed by one-way ANOVA followed by Bonferroni’s posttest with all possible comparisons performed. Statistical results with a value of p<0.05 were considered statistically significant.
Results
Versican β-GAG Accumulation and Acute Lung Injury Scoring Correlated During IAV Infection
Disease progression was monitored daily in mice infected with IAV by measuring changes to body weight. Mice infected with IAV had significant weight loss as measured by percentage of initial weight beginning on 3 dpi (98.3% ± 0.3%) and reached their maximum weight loss (73.0% ± 0.9%) at 10 dpi (Fig. 1A). After 10 dpi, mice regained weight but maintained a significant percentage of weight loss compared with PBS-treated controls (Fig. 1A).
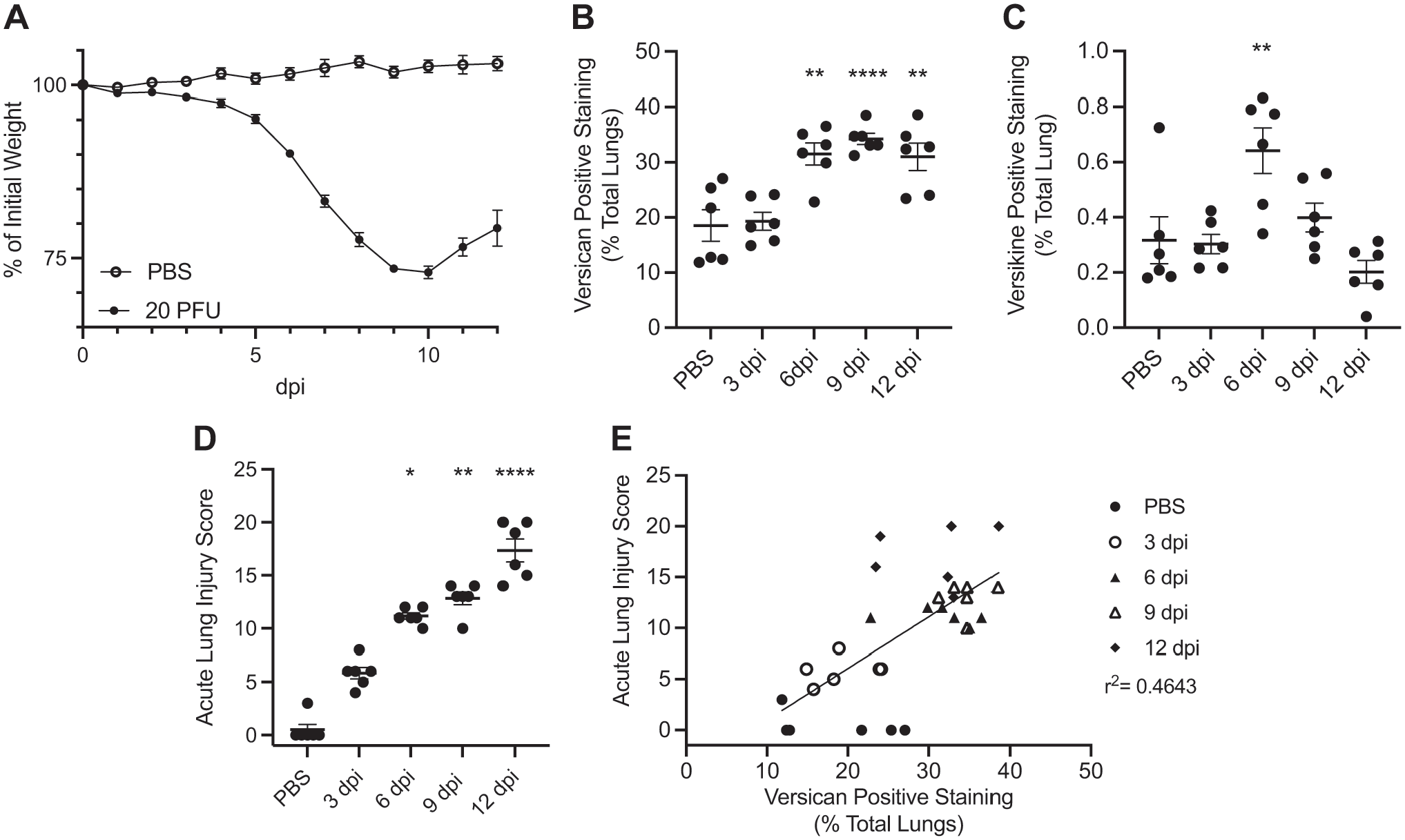
Weight loss, versican and versikine accumulation, and acute lung injury scores during influenza A virus infection. (A) Mouse percentage of initial body weight following infection with IAV over 12 days post-infection (dpi). Infected mice reached their maximum weight loss by 10 dpi. Values are mean ± SEM with n=24–6 per time. (B) Percentage of versican protein accumulation and (C) percentage of versikine accumulation in the lungs during IAV infection shown on days 3, 6, 9, and 12 after infection and in vehicle (PBS)-treated controls. Accumulation of versican and versikine was determined using immunohistochemistry and quantitative digital pathology. Values are mean ± SEM with n=6 per time. Asterisks show groups that are statistically significantly different (**p≤0.005; ****p≤0.0001) from PBS-treated controls using a one-way ANOVA with Bonferroni’s multiple comparison test. (D) Acute lung injury scores during IAV infection shown on days 3, 6, 9, and 12 after infection and in vehicle (PBS)-treated controls. Values are mean ± SEM with n=6 per time. Asterisks show groups that are statistically significantly different (*p≤0.05; **p≤0.005; ****p≤0.0001) from PBS-treated controls using a Kruskal–Wallis followed by Dunn’s posttest for multiple comparisons. (E) Percentage of versican protein accumulation correlation with acute lung injury score. Mice collected on 12 dpi demonstrated variable lung injury and versican accumulation as demonstrated by poor clustering of ◆ data points. Abbreviations: IAV, influenza A virus.
The maximum percentage of versican protein accumulation coincided closely with the maximum weight loss time point and was observed on 9 dpi (34.3% ± 1.0%). In addition, versican accumulation was significantly increased on 6 dpi (31.5% ± 2.0%) and 12 dpi (31.0% ± 2.5%) in IAV-infected mice compared with PBS controls (Fig. 1B).
The accumulation of versikine, a degradation product of versican after cleavage by A-Disintegrin-like and Metalloproteinase with Thrombospondin-1 motifs (ADAMTS) proteases, as measured by the detection of the anti-DPEAAE neoepitope was significantly increased on 6 dpi (0.64% ± 0.08%) in IAV-infected mice compared with PBS controls (Fig. 1C).
Acute lung injury scores were increased similarly to trends observed in versican accumulation. Maximum lung injury scores were observed on 12 dpi (17.3 ± 1.1) and were significantly increased on 6 dpi (11.2 ± 0.3) and 9 dpi (12.8 ± 0.6) in IAV-infected mice compared with PBS controls (Fig. 1D). Using a simple linear regression model, the correlation between versican accumulation and acute lung injury scoring was significant through 12 dpi (r2 = 0.4643, p<0.0001; Fig. 1E).
Versican β-GAG Accumulation Corresponded With Inflammatory Cell Infiltration in the Lungs of Mice During IAV Infection
To measure changes in versican accumulation in lungs infected with IAV, we performed immunohistochemistry using a polyclonal antibody developed to the β-GAG domain of versican. The accumulation of versican was measured using image analysis of whole-slide digital images. 36 Versican staining was observed in bronchiolar epithelial cells in the PBS-treated control mice. Alveolar septa in PBS-treated mice showed minimal immunoreactivity (Fig. 2A and B). Throughout IAV infection, versican immunopositivity transitioned from being primarily associated with bronchiolar epithelial cells in controls to accumulations in peribronchiolar, perivascular, and alveolar septal regions in mice infected with IAV. Significant increases in total accumulation were observed beginning on 6 dpi when compared with PBS-treated mice (Figs. 1B and 2F, H, and J).
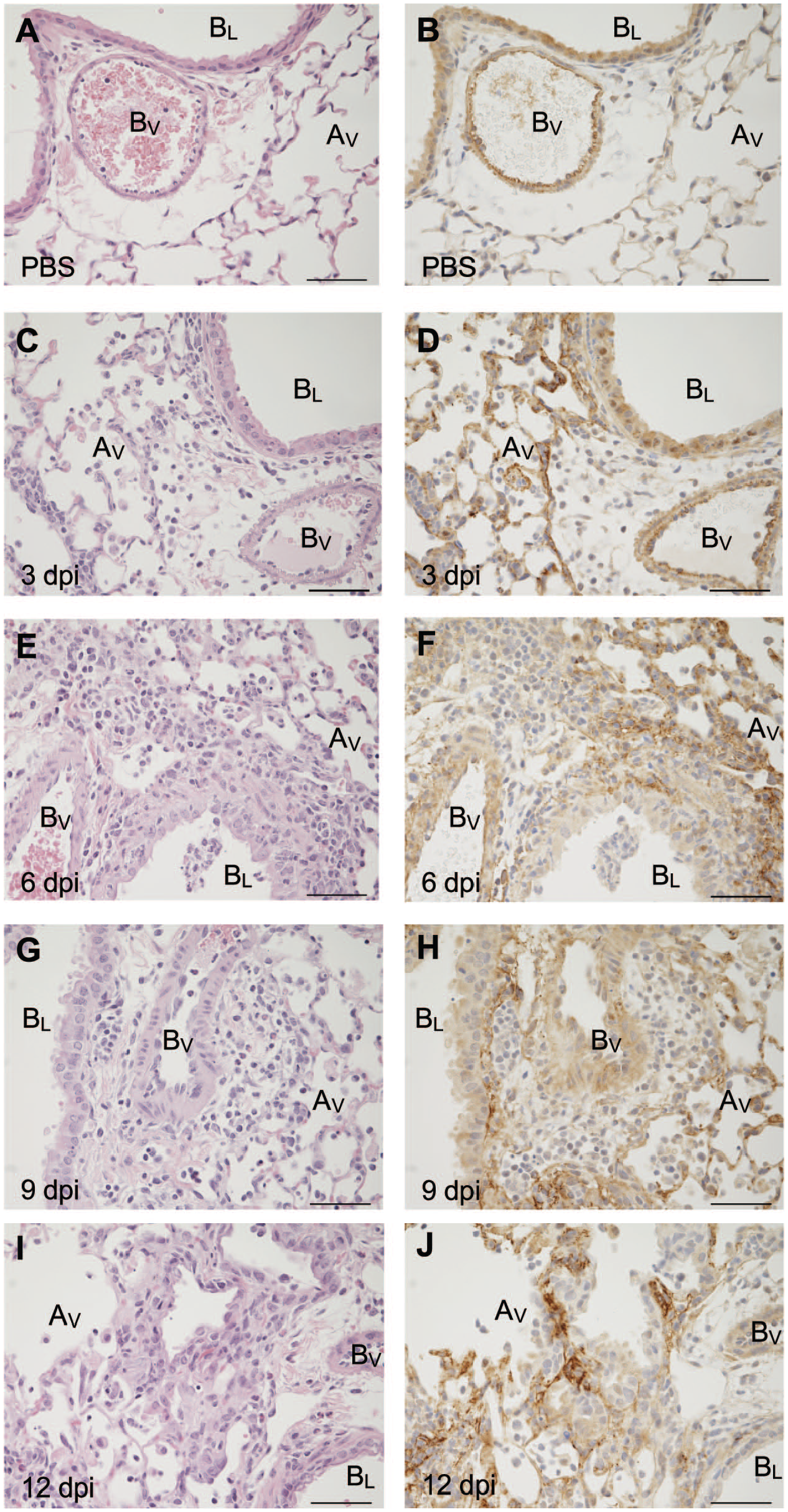
Versican β-GAG accumulation in the lungs of mice during IAV infection. Hematoxylin and eosin staining (A, C, E, G, I) and versican β-GAG accumulation (B, D, F, H, J) in PBS-treated mice at 3, 6, 9, and 12 dpi with IAV-infected lungs. For immunohistochemistry images, brown indicates positive staining for versican β-GAG domain; blue, hematoxylin counterstain. Scale (A–J) = 25 μm. Abbreviation: Av = alveolus, BL = bronchiole lumen, BV = blood vessel; GAG = glycosaminoglycan; IAV, influenza A virus; dpi = days post-infection.
At 3 dpi, there was intense positive staining for versican in the peribronchiolar spaces with a mild decrease in intensity of the positive staining of bronchiole epithelial cells compared with PBS controls. Staining of the perivascular areas was also strongly positive at 3 dpi. Multifocal areas of increased positive staining for versican in alveolar septa were seen at 3 dpi (Fig. 2D) compared with the minimal immunoreactivity of alveolar septa observed in PBS controls. Increased versican accumulation in peribronchiolar, perivascular, and alveolar septal regions was associated with mild increases in inflammatory cell infiltrates on 3 dpi. Compared with PBS-instilled controls, there was no change in total versican accumulation measured in the lungs of mice 3 dpi (Fig. 1B). A review of the tissues showed the location of intense versican staining shifted from being primarily in airway epithelial cells and smooth muscle cells of major vessels in PBS-treated mice to increased immunostaining staining for versican in the alveolar septa of influenza-infected mice 3 dpi.
On 6 dpi, increased intensity and expansion of the areas of positive staining for versican around bronchioles and pulmonary vessels contributed to statistically significant increases in versican accumulation as a percentage of total lung measured by QDP (Figs. 1B and 2F). Densely packed and markedly increased numbers of inflammatory cells expanded peribronchiolar and perivascular spaces. Versican accumulation in the alveolar septal was multifocal and coincided with areas of inflammation. Compared with that observed on day 3 after infection, there was an increased intensity and moderate thickening of the alveolar septa in regions positively stained for versican.
On 9 dpi, the overall intensity of the staining of the endothelial basement membrane of the pulmonary vessels was reduced compared with 3 and 6 dpi (Fig. 2G and H). Perivascular, peribronchiolar, and interstitial areas remain markedly expanded with inflammatory cells, with cells less densely packed into these spaces compared with 3 and 6 dpi. Positive staining of alveolar septa for versican on 9 dpi remained multifocal with thickened septa, closely associated with increased inflammatory cell infiltrates.
The alveolar spaces surrounded by positive versican staining appeared markedly expanded on 12 dpi (Fig. 2J). The positive staining for versican surrounding expanded alveoli was closely associated with loosely packed inflammatory cell infiltrates. In addition, in comparison with earlier times, the versican staining adjacent to pulmonary blood vessels and bronchioles was of lower intensity. Versican accumulation in alveolar septa remained multifocal and associated with inflammatory cell infiltrates on 12 dpi. The changes in the accumulation of versican from being predominantly epithelial in PBS controls to developing strong immunopositivity in perivascular, peribronchiolar, and alveolar septal regions throughout influenza infection culminating in expanded alveoli surrounded by versican reflect the dynamic and fluid nature of the versican-enriched ECM.
Accumulation of Myeloid and Lymphoid Lineage Leukocytes During IAV Infection
The close association and proximity of inflammatory cell infiltrates with positive staining for versican observed in IAV-infected lungs led to further characterization of changes to select populations of inflammatory cells throughout IAV infection using immunohistochemistry and QDP. 36
Ly6b is a differentiation antigen associated with mature and immature neutrophils and immature monocytes expressed in C57BL/6 mouse strains. The antigen is lost as monocytes differentiate into mature tissue-resident macrophages. 53 Positive staining for Ly6b demonstrated the relatively early influx of neutrophils into the lungs, compared with other leukocytes, during IAV infection. Mice infected with IAV had significant increases in Ly6b-positive staining beginning on 3 dpi (2.9% ± 0.5%). When compared with mice treated with PBS, the maximum percentage of positive staining for Ly6b occurred on 6 dpi (10.8% ± 0.9%) with significant increases on 9 dpi (2.5% ± 0.4%) but not on day 12 after infection (Fig. 3A).
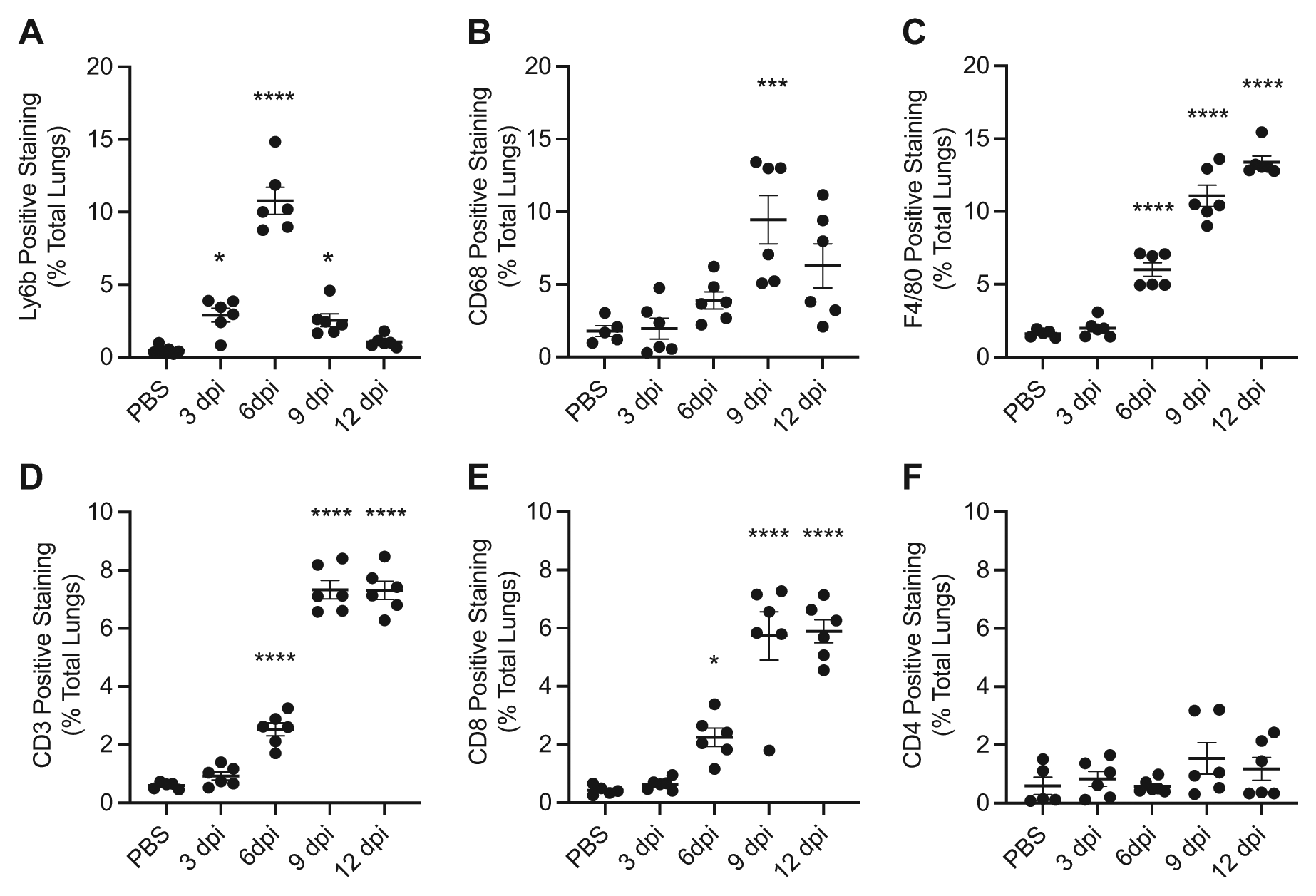
Accumulation of leukocytes during IAV infection. (A) Ly6b, (B) CD68, (C) F4/80, (D) CD3, (E) CD8, and (F) CD4 accumulation in the lungs during IAV infection shown on days 3, 6, 9, and 12 after infection and in vehicle (PBS)-treated controls. Accumulation was determined using immunohistochemistry and quantitative digital pathology. Values are mean ± SEM with n=5–6 per time. Asterisks show groups that are statistically significantly different (*p≤0.05; ***p≤0.0005; ****p≤0.0001) from PBS-treated lungs using a one-way ANOVA with Bonferroni’s multiple comparison test. Abbreviations: IAV, influenza A virus.
CD68 antigen is a glycoprotein associated with lysosomes in the cytoplasm of most cells from the mononuclear phagocyte lineage. Positive staining for CD68 captured the presence of macrophages and dendritic cells in the lung during IAV infection. Mice infected with IAV had significant increases in CD68-positive staining on 9 dpi (9.5% ± 1.7%) with no significant increases appreciated on any other days (Fig. 3B).
F4/80 antigen is a glycoprotein with surface membrane and cytoplasmic expression by resident tissue macrophages, including alveolar and interstitial macrophages, and it is also present on a subset of recruited monocytes. Significant increases in F4/80-positive staining were seen on 6 dpi (6.0% ± 0.5%), 9 dpi (11.1% ± 0.7%), and 12 dpi (13.4% ± 0.4%) in IAV-infected mice compared with PBS-treated mice (Fig. 3C). Recruited monocytes likely account for the continued increase in F4/80-positive staining even as CD68-positive staining declines on 12 dpi.
The CD3 antigen is a pan-T-cell marker present on the surface of mature T-lymphocytes and in the cytoplasm of immature T-lymphocytes. CD3-positive staining that was significantly increased compared with PBS-treated animals was seen on 6 dpi (2.5% ± 0.2%), 9 dpi (7.3% ± 0.3%), and 12 dpi (7.3% ± 0.3%) in IAV-infected mice (Fig. 3E). The increases in CD3-positive staining were consistent with the combined percentage of positive staining of CD4 and CD8 antigens, which were measured to characterize the type of T-lymphocytes recruited into the lungs. CD4 and CD8 antigens are surface membrane glycoproteins primarily of mutually exclusive, functionally different T-cell populations. CD4 is highly expressed on helper T-cells, whereas CD8 antigen is highly expressed on cytotoxic T-cells and subsets of natural killer cells. CD4-positive staining did not demonstrate significant increases in IAV-infected mice on any day post-infection. Significant elevations in CD8-positive staining mirrored the pattern of significant CD3-positive staining with significant increases seen on 6 dpi (2.2%± 0.3%), 9 dpi (5.7% ± 0.8%), and 12 dpi (5.9% ± 0.4%) in IAV-infected mice compared with PBS-treated mice (Fig. 3F). These results document the temporal changes in subpopulations of inflammatory cell infiltration into the lungs in response to influenza infection. These findings suggest increased versican accumulation in concert with inflammatory cell infiltration into the lungs is a feature of acute inflammatory response to IAV, spanning both the disease and recovery phases of our model of IAV infection.
Versican Expression Correlates With IFN-β Expression During IAV Infection
Type I IFNs are key contributors to effective antiviral responses, particularly in the acute response to viral infection. Versican is an IFN-stimulated gene in macrophages, and versican is necessary for type I IFN production by macrophages. 21 Therefore, we compared changes to versican and IFN-β mRNA expression in the lungs of mice infected with IAV by qPCR. Maximum versican expression was observed on 6 dpi (8.5 ± 0.6). Versican expression was also significantly increased on 3 dpi (5.2 ± 0.8), 9 dpi (4.2 ± 0.4), and 12 dpi (3.7 ± 0.3) in IAV-infected mice compared with PBS-treated controls (Fig. 4A).
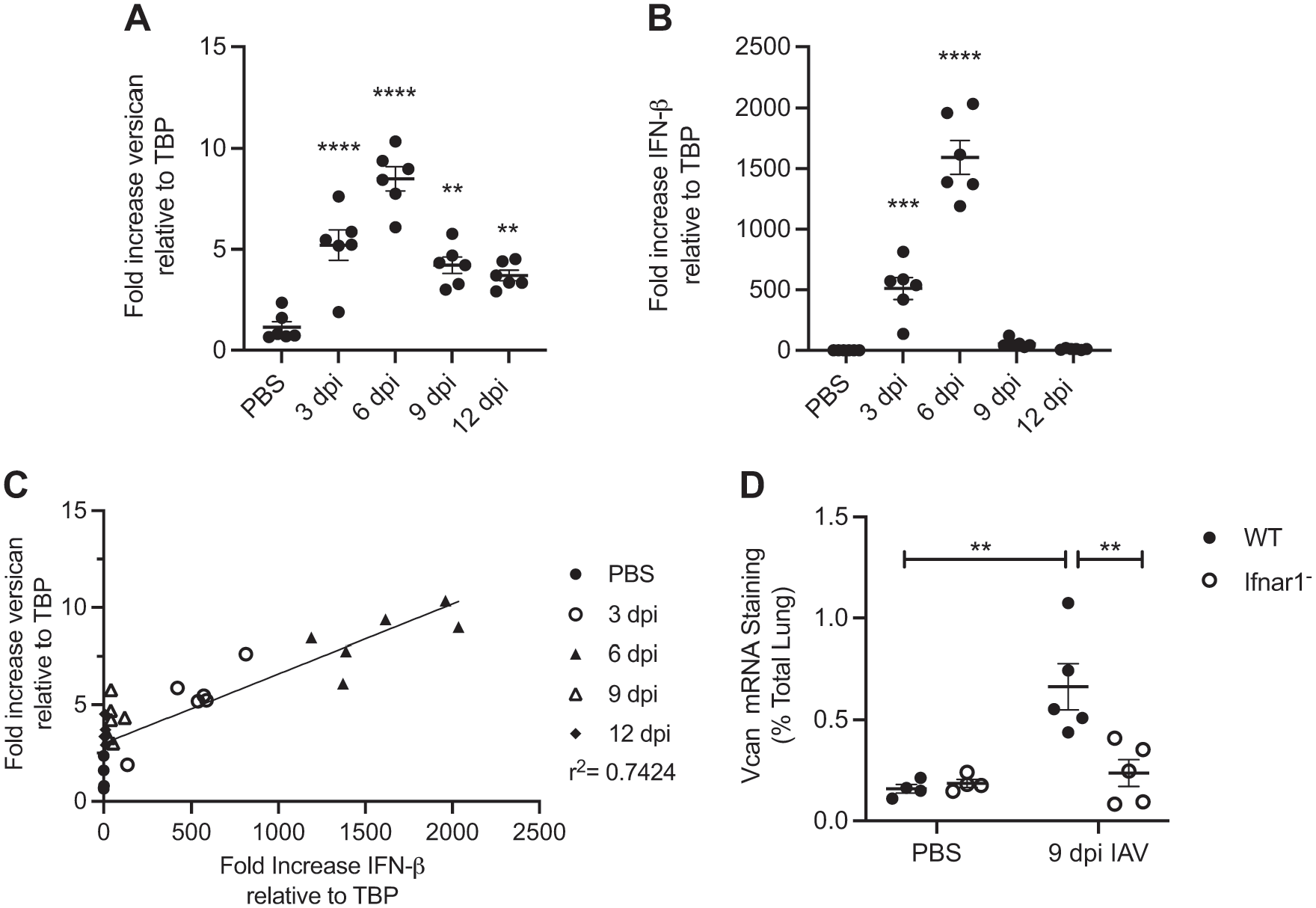
Versican expression correlation with IFN-β expression during influenza A virus (IAV) infection. (A) Total Vcan expression in the lungs by days post-infection (dpi) with influenza A virus shown on days 3, 6, 9, and 12 after infection and in vehicle (PBS)-treated controls. (B) Total IFN-β expression in the lungs by dpi with IAV shown on days 3, 6, 9, and 12 after infection and in PBS-treated controls. Change in the amount of mRNA was determined using mRNA collected from lung homogenates and quantitative real-time PCR. Values are mean ± SEM with n=6 per time. Asterisks show groups that are statistically significantly different (**p≤0.005; ***p≤0.0005; ****p≤0.0001) from PBS-treated lungs using a one-way ANOVA with Bonferroni’s multiple comparison test. (C) Vcan expression correlation with IFN-β expression. (D) Vcan expression in wild-type and Ifnar1− mice on 9 dpi with IAV and in PBS-treated controls. Vcan mRNA was detected by in situ hybridization. Values are mean ± SEM with n=4–5 per group. Asterisks show groups that are statistically significantly different (**p≤0.003) using a one-way ANOVA with Bonferroni’s multiple comparison test. Abbreviations: IAV, influenza A virus; PCR, polymerase chain reaction; IFN-β, interferon-beta.
In mice infected with IAV, the maximum expression of IFN-β mRNA was observed on day 6 after infection (1593.3 ± 139.6). In addition, IFN-β expression was significantly increased on 3 dpi (511.0 ± 91.1) in IAV-infected mice but not on any other days measured compared with PBS-treated control mice (Fig. 4B).
A simple linear regression model shows that the expression of IFN-β and versican mRNA was positively correlated up through 6 dpi (r2 = 0.8621, regression line not shown) compared with the correlation through 12 dpi (r2 = 0.7424; Fig. 4C).
Previous studies show that the activation of type I IFN receptor signaling increases versican expression in macrophages treated with poly(I: C). 21 Therefore, we investigated whether the increased pulmonary expression of versican mRNA in response to influenza depended on type I IFN receptor signaling. This was achieved by using B6(Cg)-Ifnar1tm1.2Ees/J (Ifnar1−) mice which produce type I IFNs such as IFN-β but have a non-functional type I IFN receptor. Versican expressed in response to IAV infection in the lungs of Ifnar1− mice would indicate an alternative pathway regulating versican expression. Versican mRNA was detected by in situ hybridization in wild-type and Ifnar1− mice that were 9 dpi with IAV and in PBS-treated controls. Image analysis was performed to quantify versican mRNA as a percentage of the total lung (Fig. 4D). Wild-type IAV-infected mice had significantly more versican (0.66% ± 0.11%) compared with wild-type PBS-instilled (0.16% ± 0.02%) mice. Ifnar1− IAV-infected mice had significantly less versican mRNA detected (0.24% ± 0.07%) compared with wild-type IAV-infected mice as a percentage of total lung on 9 dpi. These findings show that the temporal pattern of versican expression was closely mirrored by the pattern of IFN-β expression throughout IAV infection and that increased versican expression in response to influenza in the lungs is partially mediated by type I IFN receptor signaling.
Nuclear Colocalization of Versican mRNA and PDGFRβ mRNA by Duplex In Situ Hybridization
Previous studies have suggested that the cellular source of versican is an important factor in determining the role of versican in the context of pulmonary infection and inflammation.17,21 To address this knowledge gap, we used duplex in situ hybridization to determine important cellular sources of versican mRNA using PDGFRβ to identify a subset of versican-expressing stromal cells, including pericytes and subset of activated fibroblasts, and CD68 to identify versican-expressing macrophages and dendritic cells.54,55 PDGFRβ was selected to capture the versican expression of lung pericytes, which make up a vast network of stromal cells that are in close proximity to the lung microvascular and contribute to the vascular smooth muscle of the lung arterioles. 54 Versican expressed by PDGFRβ+ cells may play an important role in the migration of leukocytes from the vasculature and into the airspaces. CD68 was selected based on our previous work demonstrating that versican is a type I IFN–stimulated gene in macrophages. 21 However, the cellular specificity of type I IFN–stimulated versican in the context of influenza infection is unknown. Therefore, we measured whether versican and PDGFRβ mRNA colocalized in nuclei in the lungs of wild-type (Fig. 5A) vs Ifnar1− (Fig. 5C) mice 9 dpi with IAV. Nuclei were defined using a deep learning module (Visiopharm) along with single-positive versican nuclei (red), single-positive PDGFRβ nuclei (green), and double-positive versican + PDGFRβ nuclei (yellow) in both wild-type (Fig. 5B) and Ifnar1− (Fig. 5D) lungs. A similar analysis was performed in which CD68 mRNA, to identify myeloid lineage cells, and versican mRNA were detected in wild-type and Ifnar1− mice 9 dpi with IAV (image not shown).
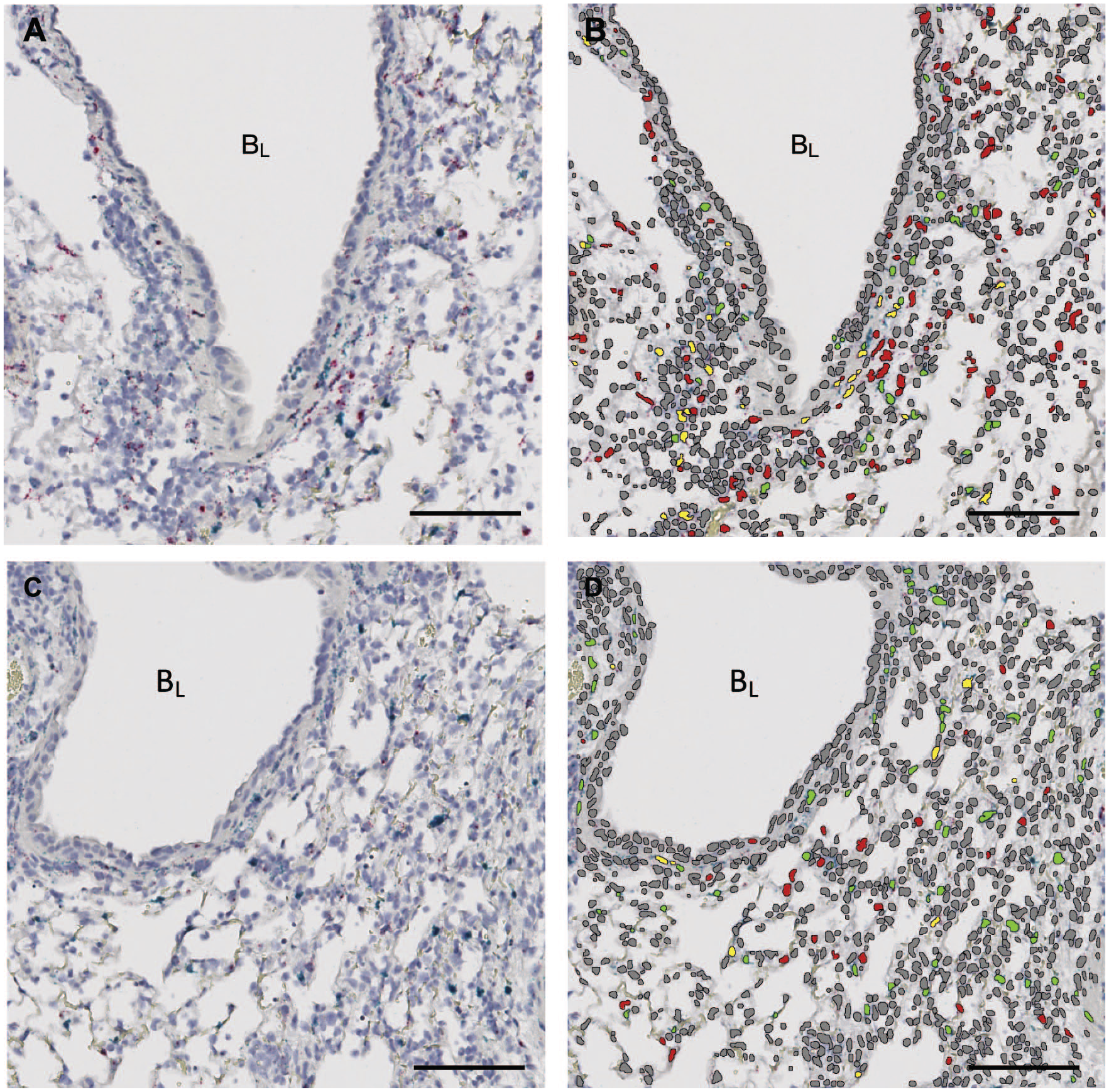
Nuclear colocalization of versican and PDGFRβ mRNA in IAV-infected mice. Digital images of wild-type (A) and Ifnar1− (C) mouse lung tissue sections stained for versican (red) and PDGFRβ (green) mRNA by in situ hybridization. Digital images of wild-type (B) and Ifnar1− (D) mouse lung tissue labeled with Visiopharm deep learning module overlay used to identify nuclei (gray) for nuclear segmentation and identify nuclei that were single-positive for versican mRNA (red), single-positive for PDGFRβ mRNA (green), and double-positive for versican and PDGFRβ mRNA (yellow). Scale (A–D) = 100 μm. Abbreviations: IAV, influenza A virus; PDGFRβ, platelet-derived growth factor receptor beta.
Type I IFN–stimulated Versican Expression in Fibroblasts During IAV Infection
In consensus with the quantification of total versican mRNA (Fig. 4D), we found that the total versican mRNA-positive nuclei were also significantly elevated in wild-type IAV-infected mice (5672 ± 967) compared with wild-type PBS-instilled mice (563 ± 69) and that there were significantly less versican mRNA-positive nuclei in the lungs of Ifnar1− mice (1627 ± 546) compared with wild-type mice 9 dpi with IAV (Fig. 6A). No statistically significant difference was observed between PBS-instilled and IAV-infected Ifnar1− lungs (Figs. 4D and 6A). These findings provide further evidence that increased versican expression in response to influenza in the lungs is partially mediated by type I IFN receptor signaling and that constitutive versican expression is mediated by signaling that is not type I IFN receptor–dependent.
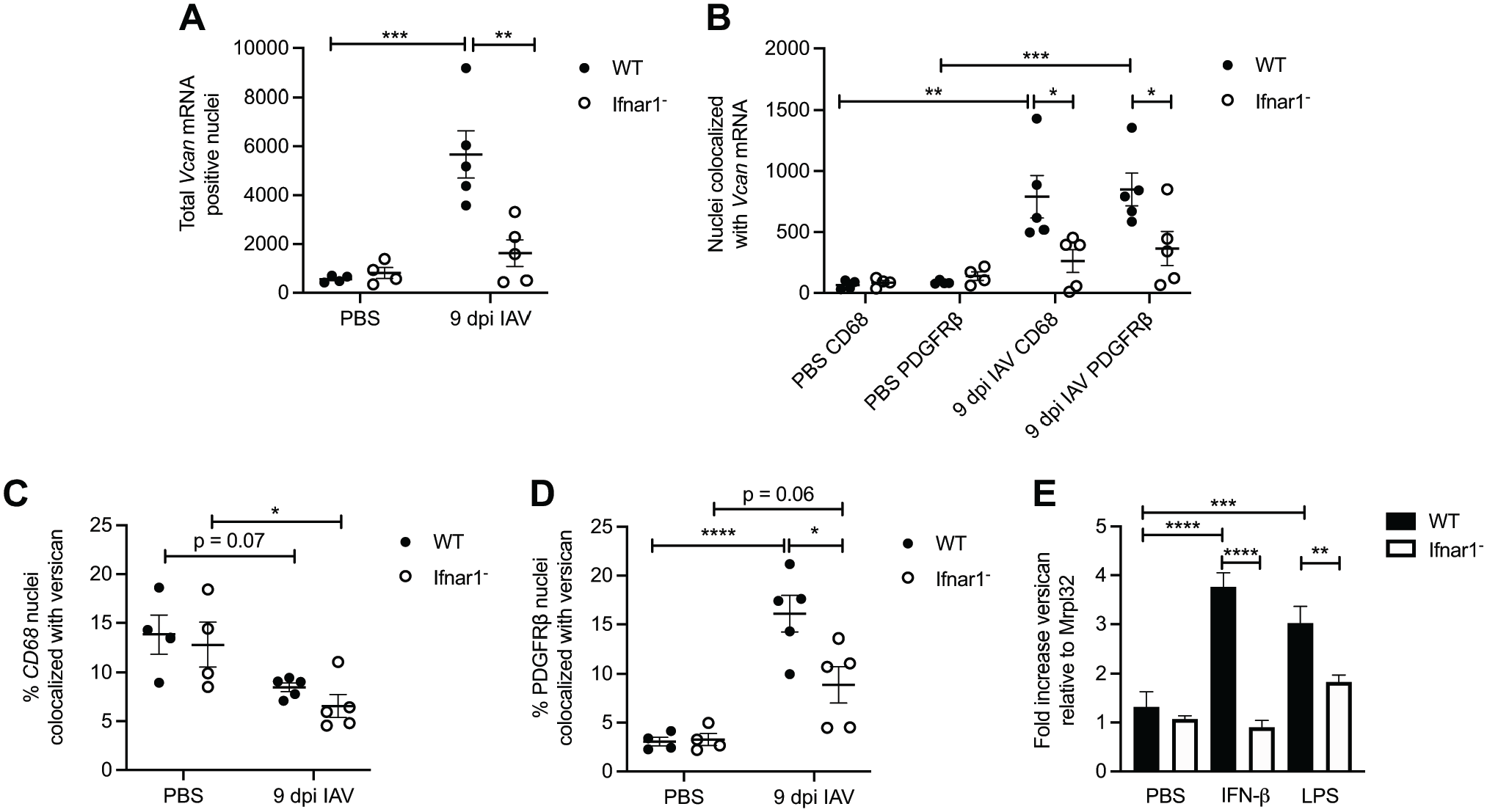
Type I IFN–stimulated versican expression in fibroblasts during IAV infection. (A) Total Vcan mRNA-positive nuclei in wild-type and Ifnar1− mice treated with PBS and 9 dpi with IAV. (B) Total nuclei with colocalization of Vcan mRNA and either CD68 or PDGFRβ mRNA in wild-type and Ifnar1− mice treated with PBS and 9 dpi with IAV. (C) Percentage of CD68 nuclei that colocalized with versican in wild-type and Ifnar1− mice treated with PBS and 9 dpi with IAV. (D) Percentage of PDGFRβ nuclei that colocalized with versican in wild-type and Ifnar1− mice treated with PBS and 9 dpi with IAV. (E) Total versican expression in wild-type and Ifnar1− primary lung fibroblasts 4 hr after exposure to PBS, IFN-β, or LPS. Values are mean ± SEM with n=3–6 per time. Asterisks show groups that are statistically significantly different (*p≤0.03; **p≤0.005; ***p≤0.0007; ****p≤0.0001) from PBS-treated cells or mice using a one-way ANOVA with Bonferroni’s multiple comparison test. Abbreviations: IAV, influenza A virus; IFN, interferon; LPS, lipopolysaccharide; PDGFRβ, platelet-derived growth factor receptor beta.
Previously, we described the role of type I IFNs and the type I IFN receptor in regulating versican expression in macrophages. 21 Therefore, we were interested in this relationship during IAV infection and whether a similar relationship existed in stromal cells. To investigate this further, we performed duplex in situ hybridization on lung tissue from both wild-type and Ifnar1− mice at 9 dpi with IAV for colocalization of versican/CD68 and versican/PDGFRβ. In Ifnar1− mice 9 dpi with IAV, there were significantly fewer versican/CD68 (262 ± 94) and versican/ PDGFRβ (365 ± 140) mRNA colocalized nuclei compared with versican/CD68 (790 ± 174) and versican/PDGFRβ (850 ± 134) mRNA colocalized nuclei in wild-type mice (Fig. 6B). It is important to point out that our quantification of versican mRNA-positive nuclei does not define the amount of versican expressed by each positive cell. The ability to measure the amount of versican mRNA in the nucleus of a cell using image analysis is an area for future refinement and study. However, these data indicate that both CD68- and PDGFRβ-positive cells are sources of versican and express versican partly by type I IFN receptor signaling during IAV infection. We expect CD68-positive macrophages to express versican in a type I IFN receptor signaling–dependent manner. Interestingly, versican/CD68 mRNA colocalized nuclei in IAV-infected Ifnar1− mice (262 ± 94) compared with PBS-instilled Ifnar1− mice (86 ± 18; Fig. 6B) showed no statistically significant difference. This is suggestive that some CD68-positive cells constitutively express versican in a manner that is not dependent on type I IFN receptor signaling.
In addition to quantifying the number of versican/CD68 mRNA colocalized nuclei, we assessed the percentage of total CD68-positive nuclei that had versican/CD68 mRNA colocalized nuclei (Fig. 6C). We found that in both PBS-instilled wild-type (13.8% ± 2.0%) and Ifnar1− mice (12.8% ± 2.3%), there was a greater percentage of CD68-positive nuclei that were constitutively expressing versican compared with IAV-infected wild-type (8.5% ± 0.4%, p=0.07) and Ifnar1− mice (6.6% ± 1.2%, p=0.03; Fig. 6C). Untreated mouse alveolar macrophages express very little versican. 21 Therefore, it is likely that this increased proportion of constitutive expression is coming from a population of interstitial macrophages or dendritic cells that express versican in the lungs during homeostasis. When considering that there is a relative increase in the number of CD68-positive cells (9259 ± 1822) that move into the lungs of wild-type mice 9 dpi with IAV compared with PBS-instilled controls (475 ± 96; Supplemental Fig. 1A), we conclude that many of the CD68-positive cells that migrate into the lungs during IAV are not expressing versican. While not statistically significant, this trend was also observed in Ifnar1− mice which had increased CD68-positive cells in IAV-infected (4697 ± 1882) vs PBS-instilled mice (673 ± 107; Supplemental Fig. 1A). A limitation of our study is that we are unable to define the subsets of CD68-positive cells in the lung and cannot positively identify the subsets of macrophages and myeloid cells that are contributing to versican expression during IAV. This represents an area of future study to further define the role of myeloid-derived versican during IAV infection.
Next, we assessed the percentage of total PDGFRβ-positive nuclei that had versican/PDGFRβ mRNA colocalized nuclei in both wild-type and Ifnar1− mice (Fig. 6D). We found that in both IAV-infected wild-type (16.1% ± 1.9%) and Ifnar1− mice (8.9% ± 1.8%), there was a greater percentage of PDGFRβ-positive nuclei that were expressing versican compared with PBS-instilled wild-type (3.1% ± 0.4%, p<0.0001) and Ifnar1− mice (3.3% ± 0.6%, p=0.06; Fig. 6D). There was also a statistically significant less colocalization of versican/PDGFRβ mRNA in wild-type vs Ifnar1− IAV-infected mice. Significant increases in the number of PDGFRβ−positive cells were observed with IAV infection (5315 ± 527), compared with PBS-instilled wild-type controls (2967 ± 287), but this increase was not observed in Ifnar1− mice (Supplemental Fig. 1B). Taken together, these results show that PDGFRβ+ stromal cells express a significant amount of versican through type I IFN signaling on 9 dpi with IAV. However, in the Ifnar1− PDGFRβ+ cells, versican expression was not completely abrogated, suggesting that the previously described β-catenin/TCF signaling may partially contribute to versican expression in stromal cells after influenza infection.
To confirm the role of type I IFN signaling in regulating versican expression in fibroblasts, in vitro studies were conducted in cultures of primary murine lung fibroblasts. For these studies, cells were treated with IFN-β or LPS, a TLR agonist known to activate type I IFN signaling and versican expression in macrophages. 21 These data show wild-type fibroblasts have significant increases in the expression of versican mRNA 4 hr after stimulation with IFN-β (3.8 ± 0.3) and LPS (3.0 ± 0.3) compared with wild-type PBS-treated (1.3 ± 0.3) fibroblasts. When wild-type fibroblasts are compared with Ifnar1− fibroblasts, we observed significant decreases in versican mRNA 4 hr after stimulation with IFN-β (0.9 ± 0.1) and with LPS (1.8± 0.1; Fig. 6E). These correlative data strengthen our finding that type I IFN signaling increases versican expression in lung fibroblasts of mice exposed to IAV in vivo.
Discussion
Consistent with the study objectives, we determined the time course and magnitude of versican accumulation and expression in the mouse lungs during severe influenza infection, which has not been previously reported. In addition, we document the following significant findings: (1) The temporal increase in versican accumulation in the lungs, in response to IAV, correlates with mouse acute lung injury scores and with pulmonary inflammatory cell infiltration; (2) the expression of versican is increased in both mononuclear phagocytic cells and stromal cells in the lungs in response to IAV; (3) the expression of versican in stromal cells is partially mediated by type I IFN receptor signaling; and (4) versican is increased in murine lung fibroblasts exposed in vitro to agonists that stimulate type I IFN signaling. These findings indicate that increased versican expression is a component of the acute inflammatory response to IAV in the lungs. In addition, versican is a type I IFN–stimulated gene in pulmonary stromal cells, both in vivo and in vitro. These data raise intriguing questions about the role(s) of versican in the early host response to IAV. This study does not directly address the function of versican. However, it does provide a guide for future studies trying to determine the role of versican in the immune response to lung infection caused by IAV. Several potential aspects of versican’s function are discussed.
In the context of IAV infection, versican likely has a vital role in coordinating the host immune response and is an IFN-stimulated gene in macrophages.17,21 Unexpectedly, we find that PDGFRβ+ stromal cells contribute to versican expression in response to IAV in a partially type I IFN–dependent manner. Type I IFNs directly contribute to the resolution of viral infection by creating the antiviral state in multiple cell types through the expression of IFN-stimulated genes. Type I IFNs are also known for limiting and resolving inflammation through stimulation of IL-10 production. 56 It is unknown whether stromal cell type I IFN–stimulated versican potentiates the expression of IFN-β and interleukin (IL)-10, as shown in macrophages, or how it contributes to the antiviral state through interactions with leukocytes, chemokines, cytokines, and growth factors. 10 The Wnt/β-catenin/TCF pathway, previously identified as controlling the versican promoter in stromal cells, could be the source of versican expression that is type I IFN–independent in stromal cells in our 9 dpi studies using Ifnar1− mice. This pathway has been implicated in regulating proteinases for which versican is a substrate, including secreted matrix metalloproteinases (MMPs) and members of the ADAMTS protein family.12,25,57–61 In both stimulating versican expression and proteases, which cleave versican, the Wnt/β-catenin/TCF pathway may be important for regulating the versican-enriched matrix’s turnover and enabling leukocyte migration through the lungs. Exploring the functional consequences of both type I IFN–dependent and type I IFN–independent pathways controlling stromal cell–derived versican is a crucial opportunity to understand the immunomodulatory effects of versican and identify host-directed therapies targeting the ECM and host response in the context of IAV pneumonia.
The importance of versican degradation in the immune response to influenza pneumonia is highlighted in two studies of the ADAMTS protein family of proteases. The work of McMahon et al. 18 shows that the degradation of the versican-enriched ECM in the mediastinal lymph nodes by ADAMTS5 is required for the migration of influenza-specific lymphocytes from the lymph node to the lungs of IAV-infected mice. ADAMTS5 is not increased in the lungs of mice exposed to IAV, but rather ADAMTS4 appears to be the protease responsible for versican degradation.15,18 In a recent manuscript, Boyd and colleagues showed that the deficiency of ADAMTS4 activity in the lungs of mice infected with IAV preserves intact ECM-localized versican and decreases CD3 lymphocyte migration into areas of infection. 15 Our study found the greatest CD3 and CD8 migration into the lungs on days 9 and 12 after infection. Studies of lymphocyte migration into the lungs from studies of ADAMTS4- and ADAMTS5-deficient mice were performed between days 7 and 10 after infection, suggesting that versican degradation may explain this increase in T-cell migration into the lungs.15,18 In both human lung fibroblasts in vitro and mice exposed to IAV, fibroblasts were identified as an important source of ADAMTS4, demonstrating that fibroblasts both contribute to the versican-enriched matrix and the degradation of the versican.12,15 Measurement of versikine, the degradation product of versican after cleavage by ADAMTS proteases, at our established time points provides additional insight into the degradation of the versican-enriched matrix. Interestingly, we see significant increases in versikine accumulation only on 6 dpi, coinciding with initial increases in lung injury scores and the initial wave of migration of various leukocyte populations into the lungs. The accumulation of versikine in our studies does not fully account for the expected amount of versican degradation, especially late in infection. Our data highlight the importance of versican degradation for future studies in versican-deficient mouse lines and the potential for other proteolytic enzymes and degradation products to be involved during IAV infection.
Much of the data presented have utility in informing future studies by providing a guide to the critical time points to measure leukocyte migration in the lungs within the context of the versican-enriched extracellular matrix stimulated by viral pneumonia. In particular, our data highlight the prominence of stromal cell–derived versican as both the number and proportion of versican/PDGFRβ mRNA colocalized nuclei increase during IAV infection. In contrast, many of the myeloid cells entering the lung are not expressing versican as the proportion of versican/CD68 mRNA colocalized nuclei of all CD68-positive nuclei decreases with IAV infection as total CD68-positive cells increase. A signature of decreased versican expression by human blood monocytes in response to LPS has been previously reported. 62 A limitation of our study is we do not know what subpopulations of monocytes and macrophages are entering the lungs and expressing versican in our tissues, but reduced expression of versican by newly recruited monocytes and macrophages is a possible mechanism explaining the reduction in the percentage of versican/CD68 mRNA colocalized nuclei during IAV infection.
The correlation of versican accumulation with the pulmonary recruitment of inflammatory cells suggests that versican influences leukocyte activity and orchestrates the host immune response in the lungs through direct and indirect mechanisms. Versican directly interacts with L-selectin and P-selectin, important mediators of cell adhesion and leukocyte rolling on the vascular endothelium. 23 Versican is also known to bind to P-selectin glycoprotein ligand-1 on neutrophils at the G3 domain and coordinate leukocyte aggregation. 63 Global knockdown of versican in mice exposed to poly(I:C) reduced leukocytes in the BAL fluid, suggesting a critical role for versican in promoting leukocyte migration into the lungs. 17 Versican can also provide indirect, fine control over leukocyte migration into the lungs through interactions of the negatively charged chondroitin sulfate chains on versican with chemokines.27–29,64,65 Previous studies demonstrate that versican interacts with chemokine CCL2, creating a gradient that promotes monocyte migration 17 and that globally versican-deficient animals exposed to poly(I: C) had reduced expression of chemokine CXCL2, a potent neutrophil chemoattractant. 66 Mice with epithelial-specific versican deficiency demonstrated increased recruitment of monocytes and neutrophils in BAL fluid and elevated expression levels of chemokines CCL2, CCL3, and CCL4 after infection with the respiratory syncytial virus, suggesting that epithelial versican attenuates leukocyte migration through chemokine-mediated mechanisms. 22 Future studies using cell-specific versican-deficient mouse strains could elucidate the potential mechanisms whereby versican directly or indirectly coordinates the migration and activation of leukocytes into lungs in response to bacterial and viral lung infection.
Viral pneumonia pathogenesis is heavily influenced by the host’s reaction to the virus, making modulation of the host immune response a critical target for drug intervention.5,67,68 The need to target the host response is especially true for IAV, which is equipped to undermine conventional therapies and prophylaxis through its ability to mutate rapidly and undergo genetic drift. These virulence traits enable IAV to develop resistance to antiviral medications and require the seasonal redesign of influenza vaccines. 69 In addition, IAV infection places a significant burden on global health, with estimates of 145,000 to 645,000 deaths attributed to influenza lower respiratory tract infections globally each year.70,71 In the United States, influenza is one of the top causes of hospitalization among patients with community-acquired pneumonia.72,73 Versican’s potential as an immunomodulatory molecule makes it an inviting therapeutic target for providing fine tune control over the immune response to viral pneumonia and IAV infection.
In conclusion, we present data showing coordination between versican gene expression patterns, versican accumulation, and leukocyte accumulation in the lungs during influenza infection. We further demonstrate that type I IFN signaling contributes significantly to versican expression in lung stromal cells during influenza infection. Of critical importance is the need to investigate the role of both type I IFN–dependent and type I IFN–independent pathways in modulating stromal cell–derived versican expression to target stromal cell versican expression and accumulation as a host-directed therapy for viral pneumonia. The data presented provide salient guidance for future studies to be performed in vivo in versican-deficient mouse models and expand upon previously published data that demonstrate the potential immunomodulatory activity of versican through direct and indirect interactions with leukocytes.
Supplemental Material
sj-pdf-1-jhc-10.1369_00221554211054447 – Supplemental material for Type I Interferon Signaling Increases Versican Expression and Synthesis in Lung Stromal Cells During Influenza Infection
Supplemental material, sj-pdf-1-jhc-10.1369_00221554211054447 for Type I Interferon Signaling Increases Versican Expression and Synthesis in Lung Stromal Cells During Influenza Infection by Jourdan E. Brune, Mary Y. Chang, William A. Altemeier and Charles W. Frevert in Journal of Histochemistry & Cytochemistry
Footnotes
Acknowledgements
The authors thank Brian Johnson and Sarah Lindhartsen in the Histology and Imaging Core at UW-South Lake Union for their assistance with in situ hybridization, immunohistochemistry, and image analysis.
Author Contributions
JB performed all animal infections; collected all tissues; carried out qPCR studies; contributed to technical aspects of immunohistochemistry, in situ hybridization, and image analysis methodology; and drafted the manuscript. MC contributed to study design, advised on technical aspects of qPCR methodology, and contributed to the drafting of the manuscript. WA contributed to study design, advised on technical aspects of tissue collection and processing, and contributed to the drafting of the manuscript. CWF contributed to study design; advised on technical aspects of immunohistochemistry, in situ hybridization, and image analysis methodology; and contributed to the drafting of the manuscript. All authors have read and approved the final manuscript.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH/NIAID Grant AI136468, NIH/NHLBI Grant HL 122895, and Histochemical Society, Cornerstone grant. The authors declare they have no competing interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.