
Abstract
“Multi-Omics” technologies have contributed greatly to the understanding of various diseases by enabling researchers to accurately and rapidly investigate the molecular circuitry that connects cellular systems. The tissue-engineered, three-dimensional (3D), in vitro disease model “organoid” integrates the “omics” results in a model system, elucidating the complex links between genotype and phenotype. These 3D structures have been used to model cancer, infectious disease, toxicity, and neurological disorders. Here, we describe the advantage of using the tissue microarray (TMA) technology to analyze human-induced pluripotent stem cell–derived cerebral organoids. Compared with the conventional processing of individual samples, sectioning and staining of TMA slides are faster and can be automated, decreasing labor and reagent costs. The TMA technology faithfully captures cell morphology variations and detects specific biomarkers. The use of this technology can scale up organoid research results in at least two ways: (1) in the number of specimens that can be analyzed simultaneously and (2) in the number of consecutive sections that can be produced for analysis with different probes and antibodies.
Keywords
Introduction
The three-dimensional (3D) cell model system “organoid” is an advanced tool for tissue engineering–based strategies suitable to investigate physiological, pathological, and developmental processes.1–6 Diverse organoid models have been generated from primary patient samples and from human reprogrammed somatic cell lines (human-induced pluripotent stem cells [hiPSCs]) and in conjunction with gene-editing technologies 7 have been used to recapitulate tumorogenesis 7 and to examine infectious diseases.8–15 These 3D structures are able to maintain the self-organizing and renewal properties of the stem cell nature as well as keep the morphology of the original tissue.16–18 Organoids preserve the genomic, physiological, and multicellular identity of the original organ; therefore, they can be very useful in analyzing, in a dish, patient diversity and heterogeneity 19 or in predicting individual drug action using patient-specific specimens 20 ; moreover, this technology can replace the use of animals.21–25 The application of the 3D technology is truly wide; contrary to two-dimensional (2D) cultures which are simple but limit morphofunctional characterization, 3D cultures allow cells to self-organize and polarize by creating their own extracellular matrix (ECM), interactions, adhesion, and communication patterns. 26 Great attention has been paid to investigate brain developmental disorders.27–29 The conventional method of analysis involves processing of each individual organoid on a microscope slide, which requires deep confocal imaging/sectioning and staining.30,31 However, the TMA approach can partially overcome the labor and reagent costs associated with single-slide analysis and scale up in situ protein results because it arranges a number of aggregates onto a single array, allowing the direct comparison of organoids grown in different conditions or subjected to different treatments. The direct comparison can facilitate studies on the inherent variability and heterogeneity between organoids. 1 The TMA technology is based on paraffin embedding of the samples; the long-term room temperature storage facilitates the storage of in vitro collections particularly useful when a large set of different experimental conditions require analysis on the same slide or in the same experimental condition of the entire collection. 32 This method of analysis can become even more advantageous if linked to multiplex staining where multiple antibody stains can be applied on a single collection of fixed and embedded aggregates placed on a single slide. 33 Here, we describe how we used the TMA technology to study cerebral organoids starting from hiPSC differentiated into neurons and describe an alternative approach to previously published methods.34,35 Generated organoids grown in different conditions and for different lengths of time were resuspended in low-melting agarose and arrayed using a semiautomatic tissue instrument with a proprietary software enabling sample deposition and condition tracing. The TMA technology faithfully captured cell morphology variations and detected stage-specific biomarkers.
Materials and Methods
Generation of Human Cortical Organoids From hiPSC
The hiPSC BF15#2, a generous gift from Dr. Domenico Delia (IFOM; Milan, Italy), was cultured in Matrigel (Stem Cell Technologies; Milan, Italy), maintained in feeder-free mTeSR-E8 (Stem Cell Technologies) media, and passaged with ReLeSR (Stem Cell Technologies) to generate optimally sized aggregates. E8 media, supplemented with Rho-kinase inhibitor (Ri-5uM) (Y-27632; Millipore, Milan, Italy), was used to generate embryoid bodies (EB). Colonies were detached into single cells with accutase (Sigma-Aldrich; Milan, Italy) and washed with PBS, centrifuged, and pellet resuspended in E8 with Ri. Cells 1.2 × 106 were distributed into 96-well U-bottom ultra-low attachment plates (Duotech; Milan, Italy) and incubated undisturbed for EB formation. After 48 hr, the EBs were transferred into 10 cm ultra-low attachment dish (Corning; Milan, Italy), and the E8 media was replaced with neural induction media (DMEM/F12; Life Technologies, Monza, Italy) consisting of dorsomorphin (5 µM; Sigma-Aldrich) and SB-431542 (10 µM; Bio-Techne, Milan, Italy) and kept for 5 days by daily media changes. On the sixth day, to the floating aggregates was added neurobasal (Neurobasal A; Life Technologies) supplemented with B-27 (deprived of vitamin A) serum (Life Technologies, Monza, Italy), GlutaMax (1:100 Life Technologies), penicillin/streptomycin (100 U/ml; Life Technologies), beta-mercaptoethanol (0.1 mM; Sigma-Aldrich), fibroblast growth factor 2 (FGF2) (20 ng/ml; R&D System, Milan, Italy), and endothelial grown factor (EGF) (20 ng/ml; Millipore) for 19 days with daily media changes. Sets of aggregates were moved onto an orbital shaker and maintained under slow movement. The neural progenitors were promoted into cortical neurons by replacing FGF2 and EGF with brain-derived neurotrophic factor (BDNF) (20 ng/ml; Peprotech, Milan, Italy) and NT-3 (20 ng/ml; Peprotech), and the media was changed every 4 days using neural medium deprived of growth factors. At specific times, organoids were resuspended in 2% Gibco Geltrex LDEV-free reduced growth factor basement membrane matrix diluted 1:1 in DMEM/F-12.
Formalin Fixation and Paraffin Embedding of Organoids
The TMA technology requires the embedding of the fixed samples in paraffin. Organoids were collected, washed twice in PBS, and fixed in 4% paraformaldehyde (PFA) at 4C for 2 hr for days 14–18 to overnight for day 50 organoids. Subsequently, the fixed organoids were moved into plastic cuvettes; 2% low-melting agarose (Merck Life Sciences; Milan, Italy) in PBS was added and allowed to solidify for at least 30 min in ice. Finally, the organoids were dehydrated in solutions with increasing alcohol content and placed in a cassette for paraffin embedding using the Fully Enclosed Processor Leica ASP300S (Leica Microsystem; Milan, Italy). 36 From this point onward, each cassette containing the organoid was treated as single donor block.
TMA Construction
The TMA block is made up of tissue rods or cylindrical cores (diameter size and core length can vary according to aggregate number and diameter) taken from different paraffin-fixed organoids and are arrayed on high-density TMA recipient block. A sampling needle punches the organoids from the donor block and places them in the predesigned recipient block. The semiautomatic tissue arrayer “Galileo Ck4600” (www.isenet.it) was equipped with a different hollow needle. Whole organoids were transferred into a recipient block; three organoids from the same preparation were placed on different spots of the TMA recipient block. Using a microtome, 3–5 µm sections were cut from the TMA and stained with hematoxylin and eosin (H&E) to control cell morphology. Sample tracking is based on the coordinate position of each spot in the TMA block, which is then transferred onto the TMA slides (Galileo proprietary software tracking). The sample tracking system is linked to a database containing salient information (origin of the primary lines, general cell maintenance, cryopreservation conditions) and the characteristics of the iPSCs, including cell line generation (reprogramming technology, genomic stability, passage number), growth and differentiation conditions, type of matrices, and organoid size and number, as well as the general management of the 3D culturing system and manipulations.
Immunostaining and Biomarker Analysis
Several microscopic slides were made from the TMA recipient blocks:
Immunohistochemistry H&E images. TMA slices were placed on polylysine-coated slides and allowed to dry before dewaxing in 3 xylene and rehydrated in methanol baths. H&E staining was performed with Mayer’s H&E.
Immunofluorescence
Deparaffinization of glass slides: TMA slides (3–5 µm) were deparaffinized by submerging in xylene for 5 min (Carlo Erba; Milan, Italy) and rehydrated by decreasing alcoholic scales (twice in 100% ethanol; twice in 95% ethanol, 70% ethanol, and 50% ethanol; and in tris-EDTA 0.5 mM) for 3 min in each step.
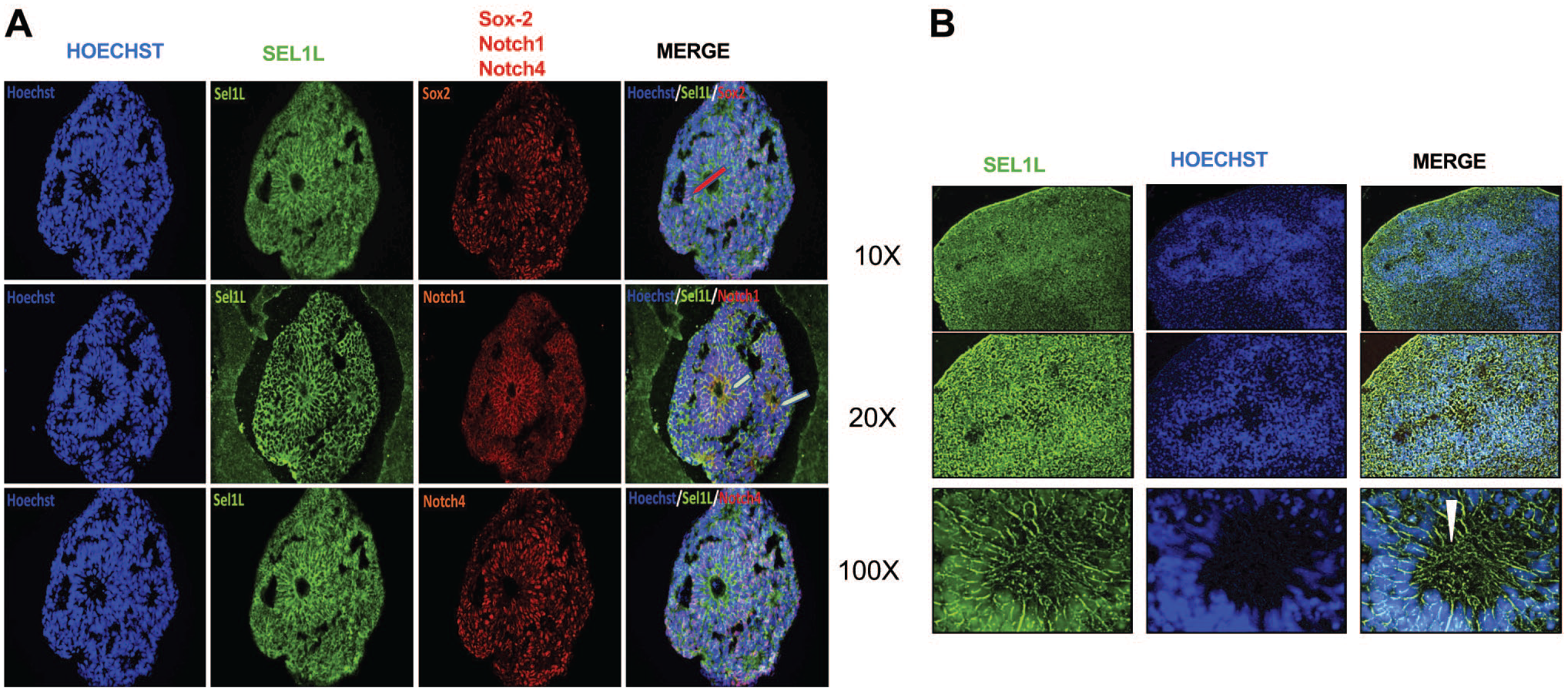
Antigen retrieval: slides were placed for 30 min in buffer (0.5 mM Tris-EDTA, pH 8.0, and 0.1 M glycine, pH 7.4), preheated at 95C, and incubated for 45 min and allowed to cool down to room temperature. Incubate slides in 1% Triton X-100 in PBS for 30 min and ulterior 30 min in blocking buffer (BB) consisting of 5% goat serum and 0.6% v/v Triton X-100 in PBS. Slides were incubated in a humid chamber with primary antibody diluted in BB (Rhodamine-Red anti-mouse IgM and anti-rabbit IgG, Alexa Fluor 488 or 568 anti-mouse IgG; 1:1000; Jackson ImmunoResearch, Milan, Italy) O/N at 4C. Slides were washed with 0.3% Triton X-100 in PBS (6× for 5 min) and incubated for 60 min with secondary antibody in BB followed by 5 min washing with PBS 6×. Nuclei were counterstained with Hoechst 33258 diluted 1:600 in PBS at room temperature. Finally, the slides were washed twice in H2O and mounted on coverslips with GelMount aqueous mounting medium (Sigma-Aldrich). Primary antibodies used were diluted accordingly to Ab specificities: Sox-2 (rabbit, 1:300; Millipore), N-cadherin (mouse, 1:100; Sigma), ZO-1 (rabbit, 1:100; Thermo Fisher Scientific, Milan, Italy), Pax-6 (rabbit, 1:500; Thermo Fisher Scientific), Tuji-1 (rabbit, 1:100; Sigma-Aldrich), Notch-1 (rabbit, 1:500; Thermo Fisher), Notch-4 (mouse, 1:1000; Thermo Fisher), and rabbit polyclonal SEL1L (rabbit, 1:50; Thermo Fisher Scientific). Antibody specificities are provided in Table 1. Images were acquired using a Leica DMI4000B inverted microscope linked to a DFC360FX camera (Leica Microsystems).
Primary Antibody Used in the Immunofluorescence Protocol.
Results
TMA Donor and Recipient Block Preparation
The preparation of TMA donor and recipient blocks is schematized in Supplemental Fig. 1. Cerebral organoids were obtained by culturing the BF15#2 line in 3D as described in MM, and the specific steps to make a TMA are shown in the different panels of the figure. Starting from an hiPSC clone (a), several embryoid bodies (EBs) of different sizes and consistency were obtained (b), which were differentiated into cerebral neurons; several aggregates (20–30) were generated (c), which were allowed to grow for days before biomarker investigation. Sets of aggregates were embedded in low-melting-agarose (d) to obtain artificial tissue cassette (s) and create several paraffin donor block(s) (e–f) ready for TMA construction. Whole organoids from each donor block were transferred and assembled into a recipient block using the Galileo TMA CK4600 (g) platform (www.isenet.it) to generate the array (h). To check for putative changes in the organoid morphology, TMA slides were monitored by H&E staining (i) and with the neuronal marker Tuji-1 (l).
Biomarker Expression of Organoids Grown in Different Culturing Conditions
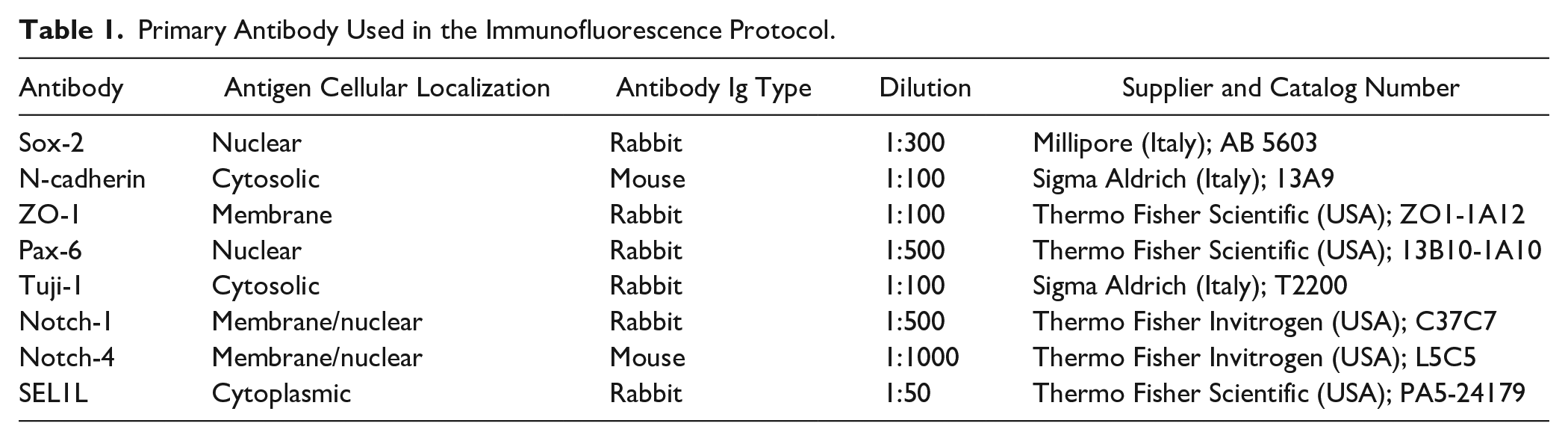
The morphology of young organoids (spheroids) and the number and size of rosettes formed during differentiation can change during growth as shown in a TMA section (Fig. 1A). The number of rosettes are indicated by 1 (a–c), 0 (b), and about 5 (d) arrows. This is useful to monitor for eventual defects in the growth of the aggregates before staining with specific biomarkers. We compared 3D TMA whole-aggregate and representative phase-contrast images at the neural induction phase (Fig. 1B–D) of an array slice (3 µm) obtained from a preparation from three different tested culture conditions. At early stages of differentiation, rosettes and cavities are formed as shown by the characteristic apical localization of the neural specific N-cadherin (red) and DAPI (blue) staining. TMA analysis indicates that the size of the cavities/lumen of rosettes and the morphological complexities of the aggregates increased in the presence of the ECM protein Geltrex. 37 Cultivation was without ECM proteins (Fig. 1B), with ECM proteins (Fig. 1C) from day 5 of differentiation onward, and with ECM protein addition from day 0 (starting from EB formation) (Fig. 1D). The addition of Geltrex from the initial phase of the differentiation increased the appearance and number of rosette-like structures (arrows), a critical morphogenetic process during neural development, whereby stem cells are enclosed in rosette niches to counterbalance proliferation and differentiation. TMAs of 3D cultures preserve cell morphology, allowing better characterization of protein expression, and visualize the subcellular localization of markers more comprehensively. Rosette identification, at day 21 of growth, is better visualized by staining with the marker zonula occludens-1 (ZO-1) (Fig.1E) in green and the neuronal differentiation marker anti-beta-III-tubulin (Tuj-1) (Fig. 1F) in red, both counterstained with Hoechst 33258 in blue at 40× magnification.
Rosette formation in organoids grown under different Geltrex conditions. Panel A shows that an H&E staining of a 3-µm TMA section (Fig. 1A) was made to monitor eventual defects in the growth of the aggregates before staining with specific biomarkers. A TMA array slice and representative phase-contrast images of live aggregates from three different culture conditions are shown in sections B to D. (B) Without Geltrex, (C) with 2% Geltrex from day 5 of differentiation onward, and (D) with 2% Geltrex from day 0. The addition of Geltrex, known to contain a high concentration of laminin 111, in the initial phase of the differentiation (D) increased the number of rosettes, a critical morphogenetic process during neural development. N-cadherin (Fig. 1B and D, red) and zonula occludens-1 (Fig.1E, green) biomarkers were used to mark the rosette structures. Neuronal differentiation was analyzed with anti-beta-III-tubulin (Tuj-1) (Fig. 1F) in red, counterstained with Hoechst 33258 in blue at 40× magnification. Scale bar, 200 µm. Abbreviations: DAPI, 4’,6-diamidino-2-phenylindole dihydrochloride; H&E, hematoxylin and eosin; TMA, Tissue microarray.
Neuroectoderm Specification Genes in Young Organoids: Day 15 and Day 21
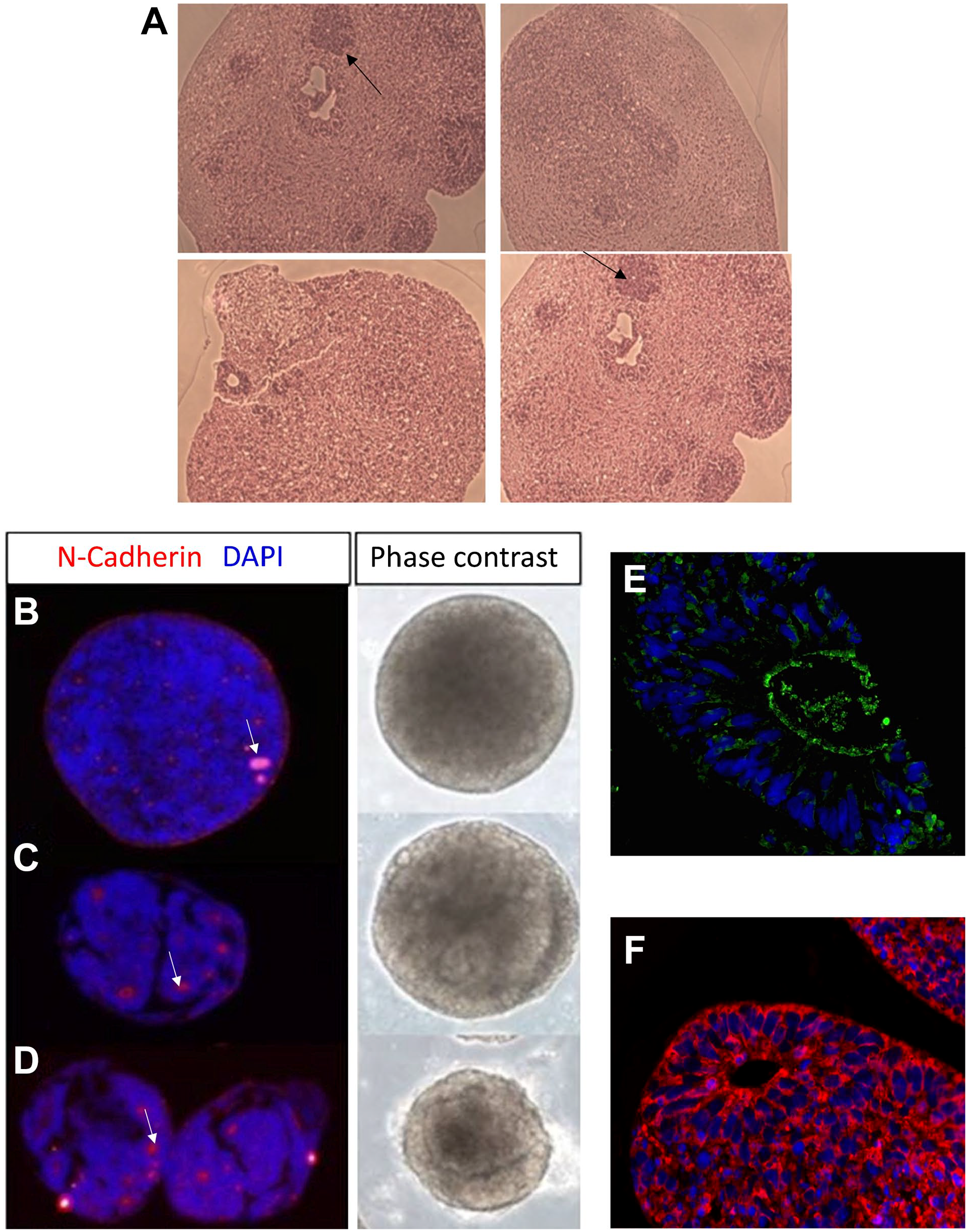
Including the organoids in agarose followed by paraffin embedding did not alter biomarker analysis as shown by analyzing several consecutive sections of the TMA block (Fig. 2). Images show a series of organoids probed with different markers: Tuj-1 (green)/Sox2 (red) (Fig. 2A), Tuji-1 (green)/Pax-6 (red) (Fig. 2B), Pax-6 (green)/N-Cad (red) (Fig. 2C), and Sox-2 (red)/N-Cad (green) (Fig. 2D). Hoechst 33258 is in blue. Arrows indicate the rosettes. Here, we show that the TMA technology does not alter the organoid morphology, protein expression and subcellular localization.
Expression of neuroectoderm specification genes in day 15 and day 21 organoids. Whole 15-day (Fig. 2A and B) and 21-day (Fig. 2C and D) organoids grown in Geltrex (as in Fig. 1D) were used to evaluate the expression of few markers and their subcellular location. Images show a series of organoids probed with Tuj-1 (green)/Sox2 (red) (A), Tuji-1 (green)/Pax-6 (red) (B), Pax-6 (green)/N-Cad (red) (C), and Sox-2 (red)/N-Cad (green) (D). N-cadherin staining shows the size and number of rosettes as indicated by arrows. Magnification: 20×. Scale bar: 100 µm.
Monitoring the Expression of SEL1L in Day 15 and Day 50 Organoids
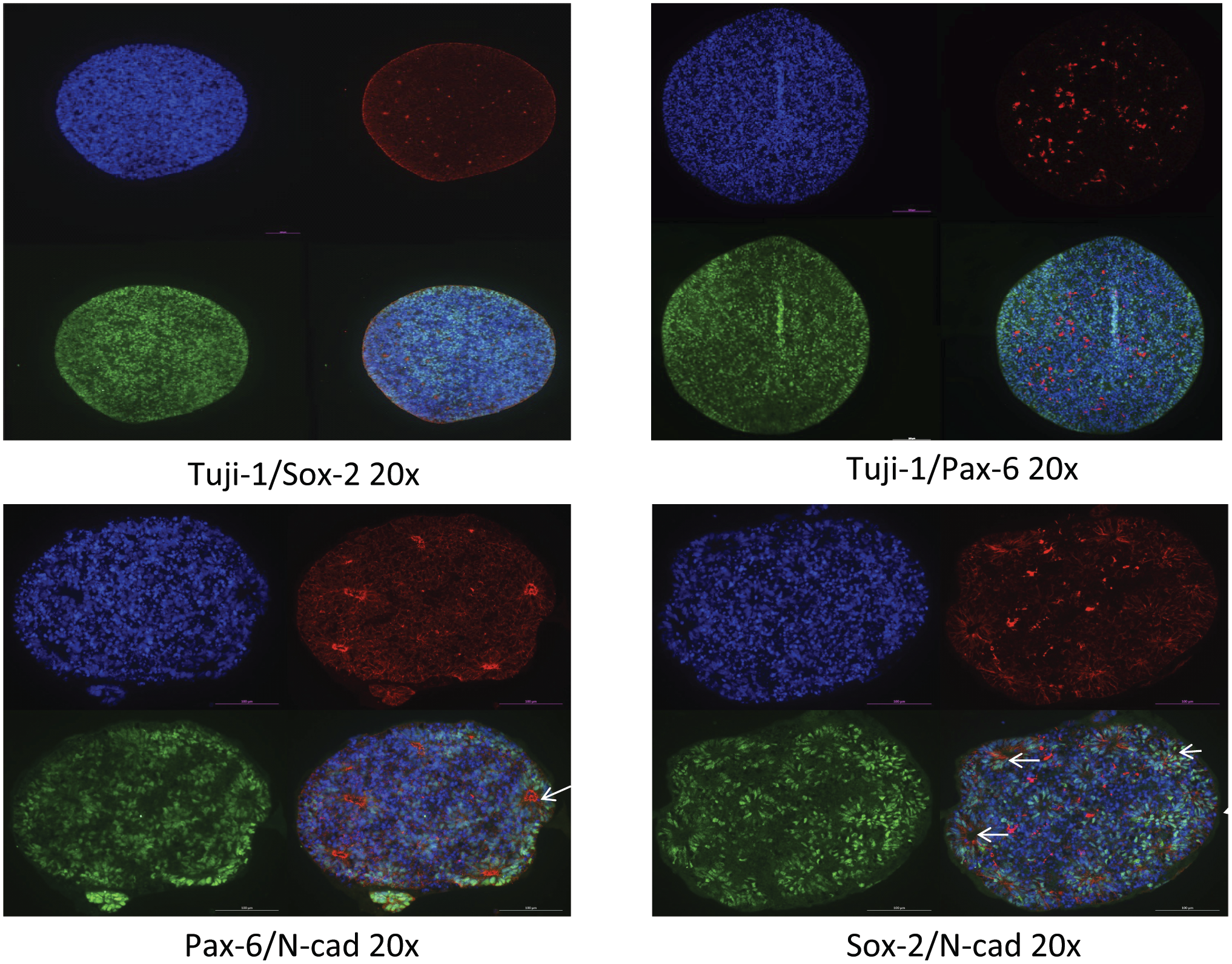
SEL1L, during murine and human embryogenesis, is an early expressing gene that was reported to be present in the neural crest and neuroepithelium.38,39 To mimic this formation, we used organoids and explored the expression of SEL1L in relation to Sox-2, Notch-1, and Notch-4 at day 15 of differentiation (Fig. 3A) and only SEL1L at day 50 (Fig. 3B). At day 15, cytoplasmic and punctuate pattern of SEL1L expression was ubiquitously present; of particular interest is its presence in cells surrounding the neo-formed rosettes representing the site where neuroprogenitors are derived (Fig. 3A). Moreover, in several cells around the germinal centers, SEL1L coexpresses with Notch-1 but not with Notch-4 nor with Sox-2. At day 50, SEL1L specifically stains neuronal progenitors, better appreciated at 100× magnification (Fig. 3B).
Monitoring the expression of SEL1L in day 15 and day 50 organoids. SEL1L expression in relation to Sox-2, Notch-1, and Notch-4 at neural induction phase (day 15) (A) and only SEL1L at day 50 (B). SEL1L is ubiquitously expressed and colocalizes with Notch-1 in several cells surrounding the rosettes (arrows) but not with Sox2 nor with Notch-4. At day 50 (B), SEL1L marks neural progenitor cells clearly seen at 100× magnification. Scale bar: 200 µm.
Discussion
The ability to generate diverse arrays of organoids from hiPSCs in conjunction with gene-editing technologies and high-throughput image screening will advance studies not only on tissue homeostasis and diseases but also as a valuable platform to test potential antiviral drugs40,41 or novel drugs for hereditary diseases. 17 The spread of organoid models over the years required technological advancements to accurately attain robust and rigorous standards to obtain ordered structures and relative functions.1,17 Furthermore, hundreds of serial sections of individual organoid would be necessary to accurately reconstruct the phenotypic information of the neo-formed 3D structure. Advances in 3D microscope methods and analytical methods have been developed42,43 but limit the analysis to a small number of markers on individual samples. The TMA technology is an unbiased high-throughput procedure that can partially resolve some problems because it arranges a number of aggregates onto a single paraffin cassette, leading to a microscope slide containing a whole collection of aggregates. The conventional method of analysis involves processing of each single spheroid/organoid on a single microscope slide; this process not only limits high-throughput analysis but increases, substantially, labor and reagent costs. Sectioning and staining of TMA slides simplify and amplify the simultaneous analysis of organoids/spheroids grown in different conditions or subjected to different treatments. Furthermore, staining comparison of specific biomarker is more reliable because all samples are processed simultaneously, thereby limiting manipulating artifacts or variables (e.g., antigen retrieval, temperature, incubation times, washing procedure, and reagent concentration/volume) that can be generated when using a single slide per sample. Depending on the organoid diameter, dozens of TMA replicates can be obtained by sampling each array block multiple times, increasing the power of the combined technologies. The number of organoids that can be placed on a TMA block depends on the size of the needle (ranging from 1.5 to 3 mm) used to punch the organoids out of the donor block and also depends on its diameter size. From a TMA block containing big aggregates (500 μm to 1 mm diameter), over 50 useful histological sections can be made and several markers can be tested on the same collection. If multiplexing is applied, as described for TMAs, 33 the number of biological questions that can be answered can drastically be expanded. Organoids are paraffin-embedded and like any other tissue sample can be stored at room temperature for long periods of time; this facilitates the generation of in vitro organoid libraries that are particularly useful in comparing the expression of multiple biomarkers and tests the effects of drugs within the concept of personalized and network medicine. The major advantage of the TMA technology is to be able to use it as a platform for the simultaneous analysis of a broad spectrum of aggregates (spheroids, organoids, tumoroids, etc.) generated from a patient’s primary cells and from pluripotent stem cells with the objective to answer a wide range of biological and pharmacological questions. However, several problems still need to be resolved before applying this technology to large-scale research. A major difficulty is to obtain uniform staining of the whole organoid because the penetration of the antibody in the complete structure at times is inefficient. It is also particularly difficult to unmask hidden or latent epitopes in paraffin-embedded aggregates and even more problematic when using antibodies directed against nuclear proteins or those that express at very levels; these parameters are also negatively influenced by fluorescence light scattering and quenching. Another technical problem encountered is the settling of the organoids in different planes during processing, causing inefficient evaluation of a histological section containing an entire collection of samples. This problem can partially be resolved using precasted agarose molds34,35 where individual spheroids are embedded in cryomolds or individual paraffin blocks, using a manual microarrayer, in such a way as to align the paraffin array block to the cutting plane of the microtome. These precasted molds are very useful, but their main limitation is that all the organoids or spheroids need to have the same dimension (size and shape), which is not always possible. In this work, we used the computer-driven instrument Galileo CK4600, a flexible platform that does not limit the number or the overall size (diameter and length) of the organoid. Using this TMA system, we accommodated different sizes of organoids and spheroids and show that the overall technical manipulations, agarose molds followed by paraffin embedding, do not alter the analysis of the organoid morphology nor the evaluation of the level and subcellular expression of representative biomarkers. We focused the attention on the formation of rosettes because these are fundamental structures to study neurogenesis, and for this purpose, we analyzed the expression of SEL1L in young (spheroids) and in older organoids. SEL1L is a ubiquitously expressed protein that colocalizes with Notch-1 in cells surrounding neo-forming rosettes, confirming previous observations38,39; in older organoids, it marks progenitor cells. Further work is necessary to establish the role of SEL1L in the processes of neurogenesis in relation to Notch-1 signaling pathway.
In summary, we indicate that the TMA technology can complement studies of organoid biology, and the benefits include the following: (1) scaling up research results by analyzing, simultaneously, a number of specimens on a single block. The number of organoids that can be placed on a TMA array depends on the aggregate diameter; from big organoids (500 µm to 1 mm diameter), over 50 useful sections can be made and several markers can be tested on the same collection; (2) more consistent expression comparisons between samples; and (3) reduction in labor and reagents cost.
Supplemental Material
sj-pdf-1-jhc-10.1369_00221554211025327 – Supplemental material for The Application of the Tissue Microarray (TMA) Technology to Analyze Cerebral Organoids
Supplemental material, sj-pdf-1-jhc-10.1369_00221554211025327 for The Application of the Tissue Microarray (TMA) Technology to Analyze Cerebral Organoids by Ida Biunno, Emanuela Paiola and Pasquale De Blasio in Journal of Histochemistry & Cytochemistry
Footnotes
Acknowledgements
The authors acknowledge Integrated Systems Engineering for having provided the Galileo CK4600 platform for the realization of the work.
Competing Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Integrated System Engineering is the manufacturing firm of the Galileo TMA platform.
Author Contributions
IB performed the differentiation protocols, standardized the embedding of the organoids protocols, wrote the manuscript, and financed the work. PDB made available the Galileo TMA platform, revised the manuscript, and financed part of the work. EP performed paraffin embedding and histology analysis.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Progetto Cariplo 2017-0886 to I.B. and by Progetto Bandiera “Interomics” to I.B.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.