
Abstract
To preserve material for future genetic studies, human B-lymphocytes from whole blood samples are routinely transformed into lymphoblastoid cell lines (LCLs) by in vitro infection with Epstein–Barr virus. To determine the rate and frequency of chromosomal changes during long-term culture, we established 10 LCLs (from eight individuals). Before transformation, these cases showed a normal karyotype (three cases), a small supernumerary marker chromosome (three cases), or an aberrant karyotype (four cases). Chromosome analyses were performed at 8-week intervals over a period of at least 1 year, up to 3 years. Surprisingly, we demonstrate that chromosomal instability is the rule, rather than the exception, during long-term culture of LCLs. The most commonly observed acquired clonal aberration was trisomy 12, which emerged in all cell lines within 21 to 49 weeks after infection. Telomeric fusions indicating telomere shortening were found after ~21 weeks. After 1 year of cultivation, the proportion of cells with the original karyotype decreased to ≤10% in 7 of the 10 cell lines. To preserve cells with aberrant genomes, we conclude the cultivation time of LCLs must be restricted to the absolute minimum time required:
Keywords
Introduction
Lymphoblastoid cell lines (LCLs), conducted through in vitro infection of human B-lymphocytic cells with Epstein–Barr virus (EBV), are a well-established tool for preserving patient material with specific genetic aberration(s) for future studies. Although reports as early as 1977 1 indicated that prolonged culture of LCLs resulted in chromosomal gains (specifically for #3, #7, #8, #9, and #12 and for the gonosomes), it continued to be widely reported that LCLs remain karyotypically stable during culture. 2 Furthermore, a 1994 publication 3 demonstrated that EBV transformation results in immortalization in only a small percentage of LCLs, that is, in those which successfully activate telomerase. 4 The majority of LCLs should therefore be considered as “EBV transformed cell lines” rather than immortalized. 5 Sugimoto et al.4,6 also refuted the idea that LCLs retain stable karyotypes during prolonged cultivation as they demonstrated that “chromosomal rearrangements and induction of strong telomerase activity are two events that take place in parallel in the process of immortalization of EBV-LCLs.” 7 Therefore, a distinction is needed between preimmortal LCLs with predominantly normal karyotypes and postimmortal lines with clonally abnormal chromosome complements. 8 In the postimmortal stage, LCLs have various degrees of tumorigenicity, ranging from benign to malignant, and have even been shown to form colonies in soft agar and initiate tumor growth in nude mice.8,9 However, only a limited proportion of preimmortal LCLs, approximately 10%, reach the final postimmortal stage characterized by the activation of telomerase, chromosomal instability (CIN), and upregulation of WRN helicase. 4 Recent reports indicate that cancer cells undergo an alternative lengthening of telomeres (ALT) 10 ; however, this has not been studied or reported in LCLs. The role of EBV for induction of chromosomal aberrations during prolonged cell culture remains unclear 11 or appears to be uncertain. 12 Interestingly, hypomethylation of repetitive satellite 2 sequences, which results in the uncoiling of heterochromatic segments of chromosomes 1 and 16, has also been observed during long-term culture of LCLs.13,14 Formation of triradials or multiradials in patients with autosomal recessive immunodeficiency, centromeric region instability, and facial anomalies syndrome is also based on hypomethylation of heterochromatic segments.15–17
In this study, we demonstrate that the clonal development of abnormal karyotypes, concurring with CIN, consistently occurs before immortalization, a seemingly late and rare event during LCL long-term culture.
Materials and Methods
Cell Lines
Derived from eight individuals, 10 independent LCLs were established by infection with EBV harvested from the B95-8 marmoset cell line. 2 These lines included three with normal karyotype (N1, N2, and N3), three with small supernumerary marker chromosomes (M1; M2 with two independent lines: M2-1 and M2-2), and four with other aberrant karyotypes (two independent lines from A1: A1-1, A1-2; A2 and A3) (see Table 1).
Characteristics of Cell Lines Investigated.

Abbreviations: Scoring: N, normal karyotype; M, marker chromosome present; A, aberrant karyotype.
Final karyotypes of A1-2:
49,XX,t(7;14)(q32;q31.1),+der(7)t(7;14)(q32;q31.1),add(8)(p23),+psu idic(9)(q12),+12 (mother).
87,XXXX,der(1),-2,-3,-4,-6, t(7;14)(q32;q31.1),+der(7)t(7;14)(q32;q31.1),add(8)(q24.3)x2,+psu idic(9)(q12)x2,+der(12)t(8;12)(q22;p13),-13,-15,-16,-18,-21 (daughter).
The cell lines for this study were either established from whole blood samples (6/10) or already present as cryopreserved cultures (4/10). In the latter case, calculation of the total cultivation time included the duration of cultivation before freezing.
The cultures were grown in RPMI 1640, supplemented with 13% fetal calf serum and penicillin/streptomycin (100 U/ml and 100 µg/ml, respectively). Half of the culture medium was replaced with fresh solution twice a week. Metaphase spreads were harvested every 8 weeks by routine procedures. However, the cumulative cultivation time at a certain harvest time point was not identical for all cell lines due to variations in the timing of the first harvest. Therefore, the duration of cultivation was divided into 5-week intervals (1–5, 6–10, 11–15, etc.). All cell lines were cultivated for at least 55 weeks. The experiments were stopped after 55 weeks (3/10), 80 weeks (3/10), or 95 weeks and more (4/10).
To observe whether aberrations would differ despite an identical background, two independent cell lines were established from a single donor in two cases (A1-1 and A1-2; M2-1 and M2-2) and two cell lines were split after 33 weeks (N2) or 62 weeks (A1-2) of culture.
Chromosome Analyses
G-banding was performed as described by Seabright. 18 Thirty GTG-banded metaphase spreads were analyzed in terms of karyotype formula, number of telomeric fusions (“dicentrics”), and breakage events (chromosome breaks, acentric fragments).
For characterization of a derivative chromosome 13 detected in a specific cell line, whole chromosome paints for #13 and #16, a region-specific probe for 13q14 (LSI13), and a centromere-specific probe for chromosome 16 were used (all probes from Abbott; Wiesbaden, Germany). Fluorescence in situ hybridization (FISH) procedures were carried out according to manufacturer’s protocols.
Quantitative FISH
For the determination of telomere length through quantitative FISH (Q-FISH) analysis, metaphase preparations were cohybridized with an all-telomere probe and a probe for the centromere of chromosome 2 (Dako; Glostrup, Denmark), the latter serving as an internal reference. The hybridization was performed according to manufacturer’s protocols. Metaphases were captured with a fluorescent microscope (Axio Imager Z2; Zeiss, Jena, Germany) and analyzed with ISIS software (MetaSystems; Altlussheim, Germany). In all, 15 to 17 metaphases per sample were reviewed. Telomere length was measured independently for each chromosome pair. Results were calculated as a percentage of internal control (test/control ratio). The detailed method was performed as described by Perner et al. 19 The telomere length was examined in three cell lines (A1-1, A2, and N1) at two independent cultivation time points.
Results
Appearance of Clonal Rearrangements
During the course of long-term observation, the cultures retained the original karyotype for only the first few months of cultivation. After this initial phase, all cell lines acquired clonal aberrations. Concordantly, the proportion of cells with the baseline karyotype continuously decreased over time. However, karyotype evolution differed between the cell lines, and even for those which originally derived from the same individual. To facilitate a more generalized review of the changes in the 10 LCLs, the median was calculated for the different characteristics which are shown in Fig. 1. The results for cell lines N1 and N2 are shown in Figs. 2 and 3, respectively. The most common aneuploidy observed was trisomy 12, which presented as a clonal change in all cell lines between 21 and 49 weeks after infection (median: 27 weeks). Over time, all cell lines reached a state where the trisomy 12 clone composed a considerable number of metaphases analyzed. Between 27 and 62 weeks of culture (median: 39.5 weeks), trisomy 12 was observed in more than 50% of cells. Cells with an additional chromosome 12 needed on average only 20.5 weeks to increase the fraction of cells with this aberration from zero to more than 50% (range: 7–32 weeks). At the end of the observation period, seven cell lines contained trisomy 12 in more than 90% of the metaphase spreads. In one cell line, the majority of metaphase spreads showed 50 chromosomes (+5, +10, +12, +12) before cell death. Another line acquired structurally aberrant chromosomes in addition to trisomy 12. Only a single line returned to its baseline karyotype after the trisomy 12 clone composed over two thirds of all cells within the culture. Three cell lines stopped proliferating during the observation period; all had acquired clonal chromosomal aberrations before entering crisis, and thus, we conclude that the presence of an aberrant karyotype is no indicator for immortalization.
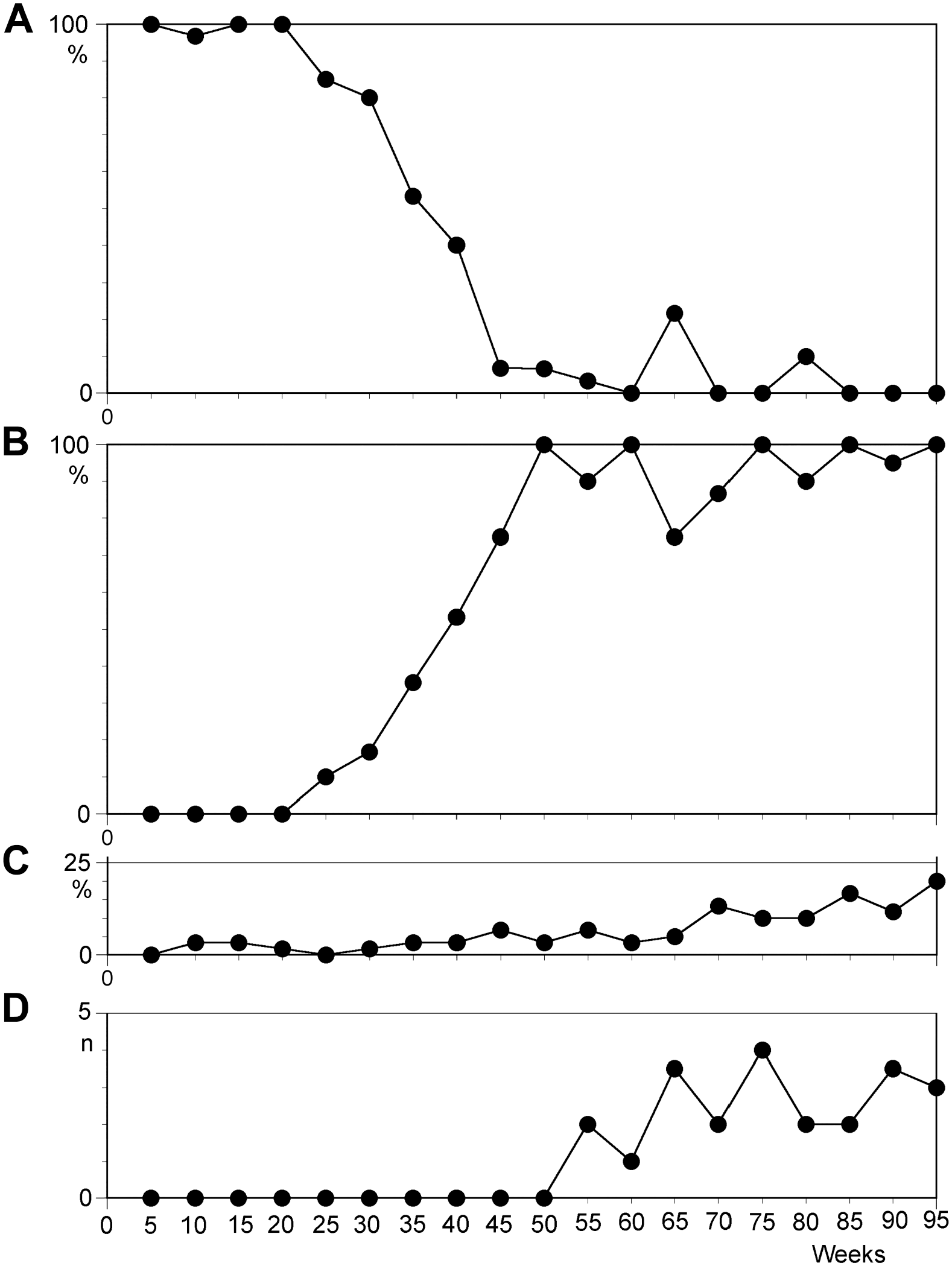
Temporal course of the median value of characteristics scored from 30 metaphase spreads per 10 lymphoblastoid cell lines as a function of culture time in weeks. Horizontal axis shows culture time divided in 5-week intervals from initiation to 5 weeks (weeks 1 to 5) to 95 weeks (weeks 91 to 95) following culture set-up. (A) Percentage of cells with original karyotype; (B) percentage of cells with trisomy 12 or derived aberrations; (C) percentage of non-clonal aberrations; and (D) number of telomeric fusions.
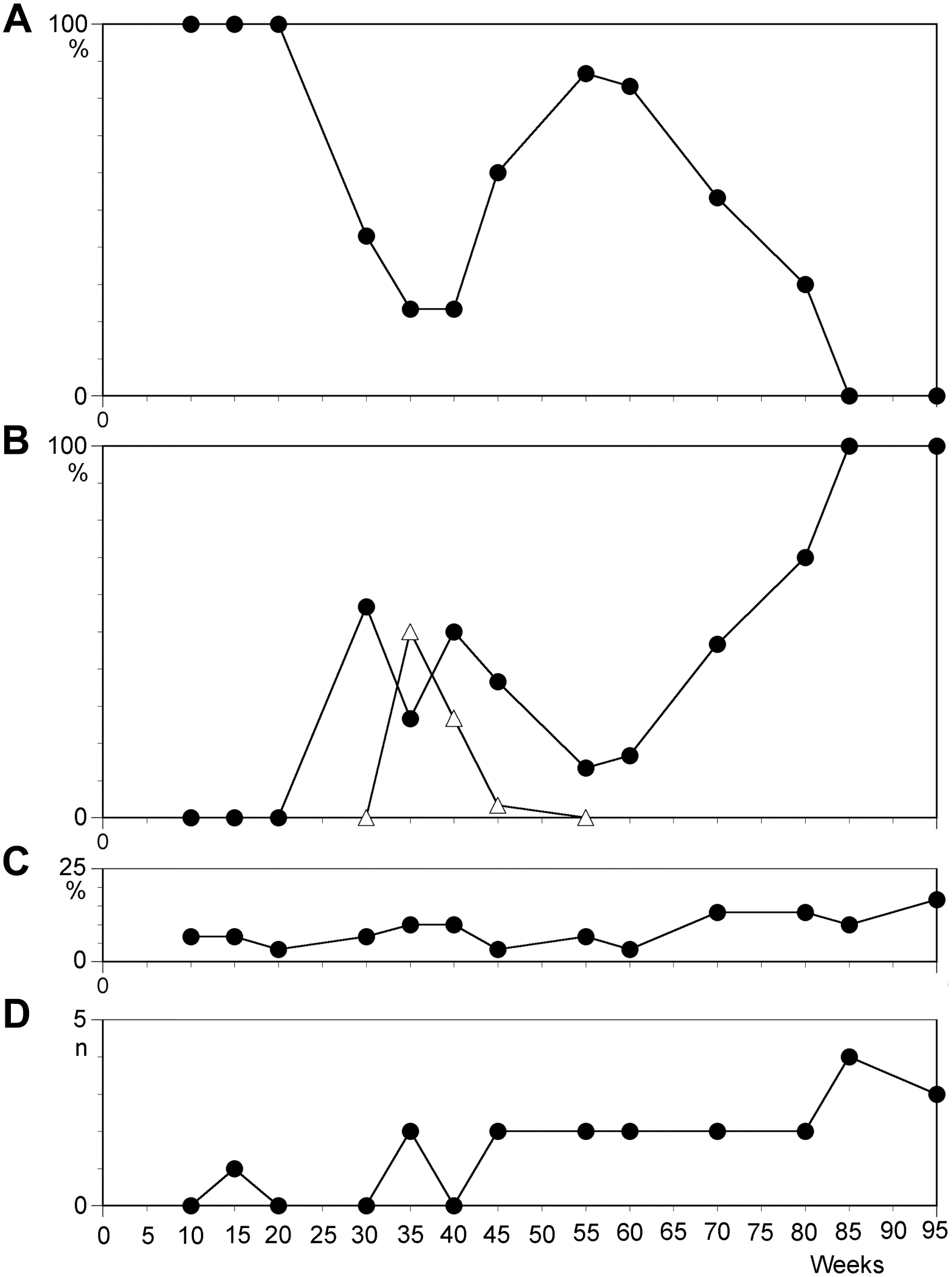
Temporal course of characteristics in cell line N1 scored from 30 metaphases. (A) Percentage of cells with karyotype 46,XX; (B) percentage of cells with 47,XX,+12 (filled squares) and 46,XX,add(11q) (triangles); (C) percentage of cells with non-clonal changes; and (D) number of telomeric associations.
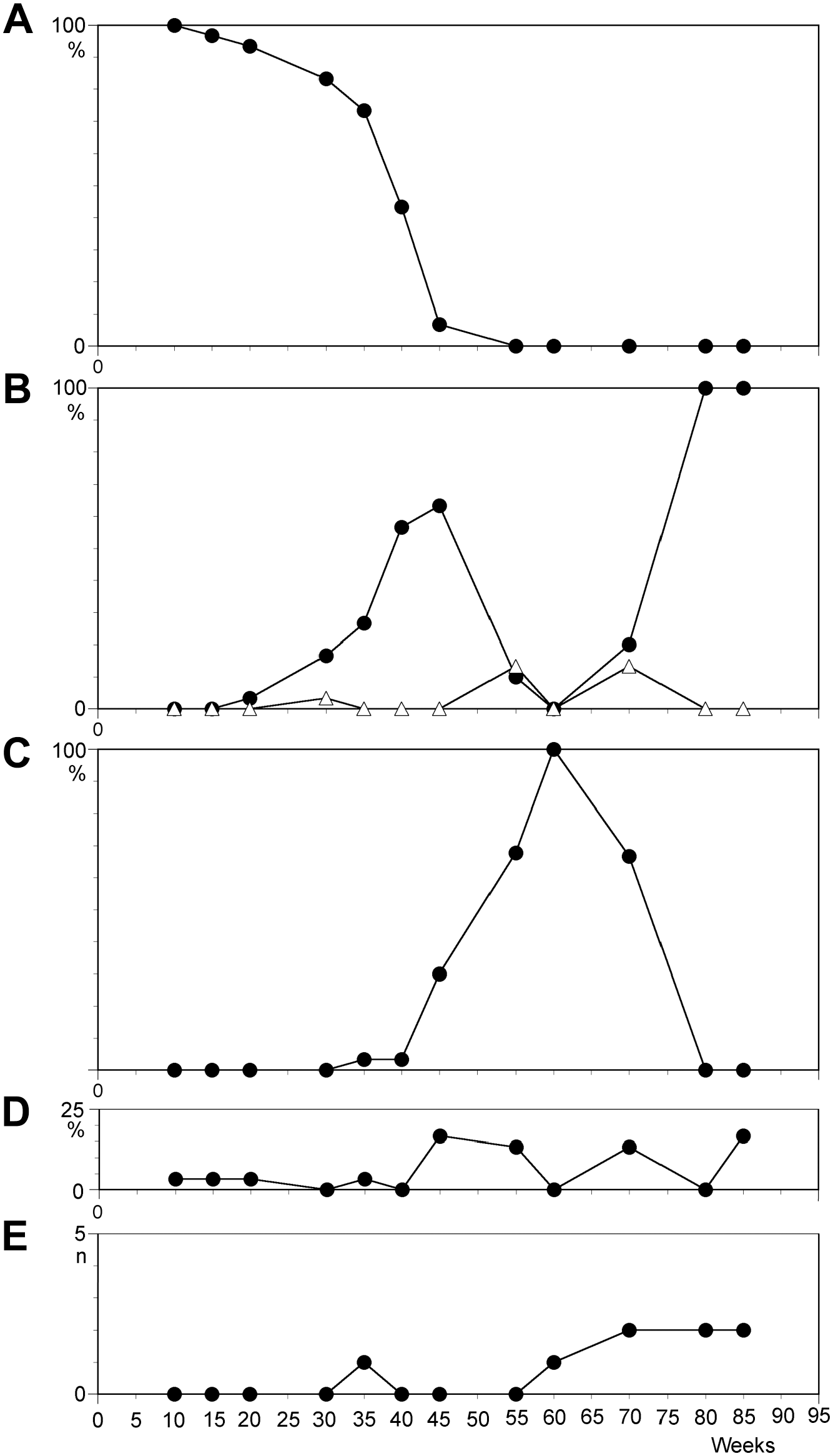
Temporal course of characteristics in cell line N2 scored from 30 metaphases. (A) Percentage of cells with karyotype 46,XX; (B) percentage of cells with 47,XX,+12 (filled squares) and 48,XX,+11,+12 (triangles); (C) percentage of cells with 50,XXX,+11,+12,+15; (D) percentage of cells with non-clonal changes; and (E) number of telomeric associations.
No Chromosomal Fragility at Early Stages
For three cell lines, the number of breakage events was scored at early stages of cultivation, designated as interval from 4 to 25 weeks after infection. Within 12 experimental observations, the number of breaks was very low in the majority (11/12), with a range from zero to three breaks in 30 analyzed metaphase spreads (0.0–0.1 breaks per cell). In a single experiment, performed after 11 weeks of cultivation, seven breakage events (0.23 breaks per cell) were observed.
Chromosomal Changes Besides Trisomy 12
Different types of rearrangements were observed during long-term culture of LCLs, and excluding trisomy 12 (as described above), trisomies and structural rearrangements dominated the cultures during chromosomal evolution. Uniquely, the two independent cultures of individual A1 showed extremely high rates of chromosomal changes. Of the eight remaining LCLs, three developed no further aberrations (excluding trisomy 12), three acquired additional trisomies, and two presented with structural changes (translocations and additions of material of unknown origin). Of the observed trisomies, only +8, +10, and +11 were found as sole aberrations. Trisomies of chromosomes 5, 10, 11, 13, 15, 20, and 21 were found in hyperdiploid clones with 48 to 51 chromosomes. However, except for cells with trisomy 12, the life span of aberrant clones was limited. Of the 12 clones with aberrant karyotypes, 7 were only transiently observed in a single experiment, and the other 5 clones were present in the cell lines for a mean duration of 25.5 weeks (median: 29; range: 8–41 weeks). Even abnormal clones that constituted up to 80% of the cells analyzed at a given time point later disappeared completely and were subsequently replaced by a cell population with a different aberration. We conclude that trisomy 12 is a state that leads to sustained clonal expansion in LCLs, whereas other observed trisomies and hyperploidy may only promote a transient clonal proliferation followed by rapid involution.
Non-clonal Chromosomal Aberrations
CIN, as defined by Heng et al., 20 was only rarely observed during prolonged periods of culture in this study, as on average only 1 to 3 metaphase spreads out of 30 (i.e., 3.3–10%) displayed non-clonal chromosomal aberrations (NCCAs). Beyond a cultivation time of 66 weeks, the levels reached 10% and greater (Figs. 1C, 2C, and 3D). Immediately before the breakdown of a cell line, NCCAs reached values ranging from 20% to 36% (observed in A1-1, A1-2, and N1). In the only immortalized LCL in this series, the daughter line of A1-2, the rate of NCCAs moderately increased in the critical phase before immortalization, but reached very high values (23.3–66.7%; median: 33%) 5 months after immortalization.
Telomeric Fusions
Telomeric associations and fusions, indicators of telomere shortening, 3 were found in only a small percentage of metaphase spreads and presented only after 24 weeks of culture. After cultivation longer than 50 weeks, most cell lines presented with one to four telomeric fusions per 30 metaphase spreads (Figs. 1D, 2D, and 3E). Shortly before breakdown of the culture or immortalization, the observed number of telomeric fusions reached the highest levels (7, 6, and 10 observed in cell lines A1-1, A1-2, and A2, respectively). Examples of telomeric fusions with the resulting dicentric chromosomes are shown in Fig. 4A.
Chromosomal modifications in lymphoblastoid cell lines: (A) Examples of telomeric fusions with non-involved homologues displayed from cell lines N1, A2 (twice), A1-2, and N3, from left to right. (B) Examples of 16qh undercondensation resulting in elongation of the region (cell lines A1-1 and N2), loss of 16q (line N2), and triradial formation (duplication of 16q; lines A1-1 and A1-2).
Low Level of Immortalization
The earliest observations of crisis and culture breakdown were made after 1.5 years of culture. Only a single cell line, the daughter cell line of A1-2, which was established from the mother line 60 weeks after infection, is believed to have reached a postimmortal stage. Similar to the mother cell line, a slightly elevated level of chromosomal instability and telomeric fusions was observed after 90 weeks of culture. Harvest after 109 weeks revealed a very low proliferation rate in both the mother and the daughter cell lines. No metaphase spreads could be obtained from the mother line after 8 weeks, whereas the daughter line returned to a good cell proliferation rate associated with a tetraploid (4n) karyotype. Approximately 5 months later, the number of telomeric fusions was very low, but CIN had reached a high level which resulted in impressive chromosomal changes (see Table 1 for final karyotype).
Undercondensation of 16qh
In 8 of 10 independent lymphoblastoid cultures, undercondensation of the 16qh heterochromatic segment and subsequent chromosomal rearrangements of chromosome 16 (see below) were observed at the earliest after 20 weeks of culture. However, in most cultures, 16qh undercondensation was a phenomenon that emerged following prolonged cultivation time, generally after 1 year. The median time of first occurrence of this aberration was 71.5 weeks. The changes in 16qh appear to correlate with increased chromosome instability (increases in non-clonal rearrangements) and telomere shortening (increases in telomeric fusions or dicentric chromosomes). As a result of the undercondensed fragile 16q11.2 region, cells with deletion of 16q were also found. In addition, triradials of chromosome 16 with two short arms, two long arms, or both were observed (Fig. 4B). Comparatively, the undercondensation of 1qh, another classical satellite 2–rich region, was a very rare event.
Occurrence of Marker Chromosomes
A dicentric marker chromosome was observed in the daughter cell line of A1-2. This cell line, established from the mother line through separation of a small number of cells after a culture period of 60 weeks, entered the postimmortal stage after 109 weeks by tetraploidization. The dicentric marker chromosome was observed for the first time after 141 weeks in culture and was then already present in 40% of metaphase plates. After composing a fraction of 93% in the culture at 169 weeks, the level reduced to less than 50% at the end of the observation period (183 weeks). The appearance of the marker occurred at a time when already one homolog of chromosomes 4, 6, and 18 was missing in the near-tetraploid karyotype. This was followed by loss of one homolog of chromosomes 2, 3, and 21. Subsequently, in combination with loss of one homolog of chromosome 15, the dicentric marker was also lost.
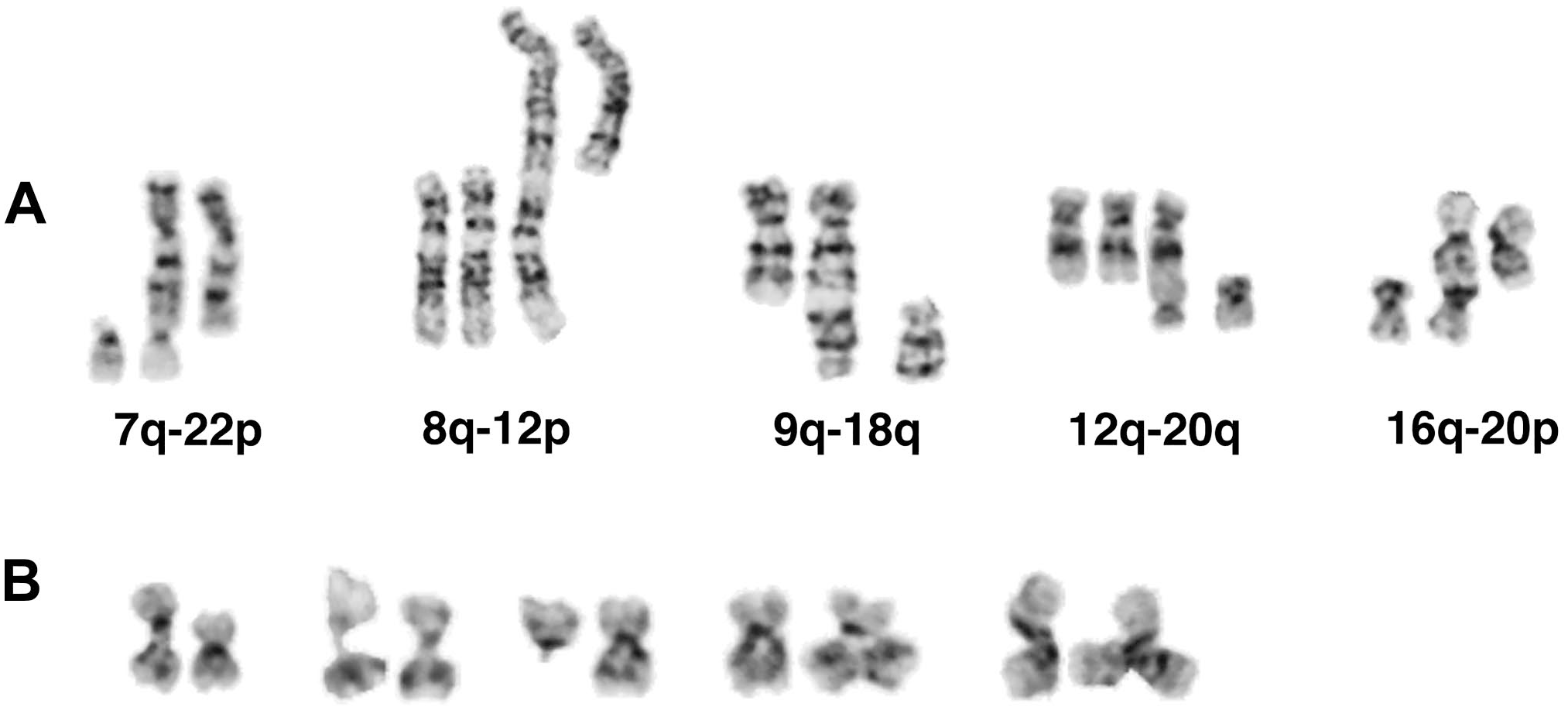
The dicentric marker was composed of a derivative chromosome 13, with deletion of the terminal band 13q34, and the short arm of chromosome 16 (Fig. 5). The centromere of chromosome 13 was active; chromosome 16 centromere displayed a constriction in one third of observed markers and was not visible in the remaining metaphase spreads. Rarely, together with the loss of one chromosome 16 homolog in the corresponding cell, a deleted chromosome 13 was observed, which was predicted to be a result of a break located at the fusion site of the marker. FISH using whole chromosome paints for chromosomes 13 and 16, and a centromere 16–specific probe confirmed this conclusion drawn from G-banded analysis (Fig. 5B and C). Possibly, it was the undercondensed heterochromatic region of 16qh that contributed to the break in 16q11.2 and subsequent fusion with chromosome 13q.
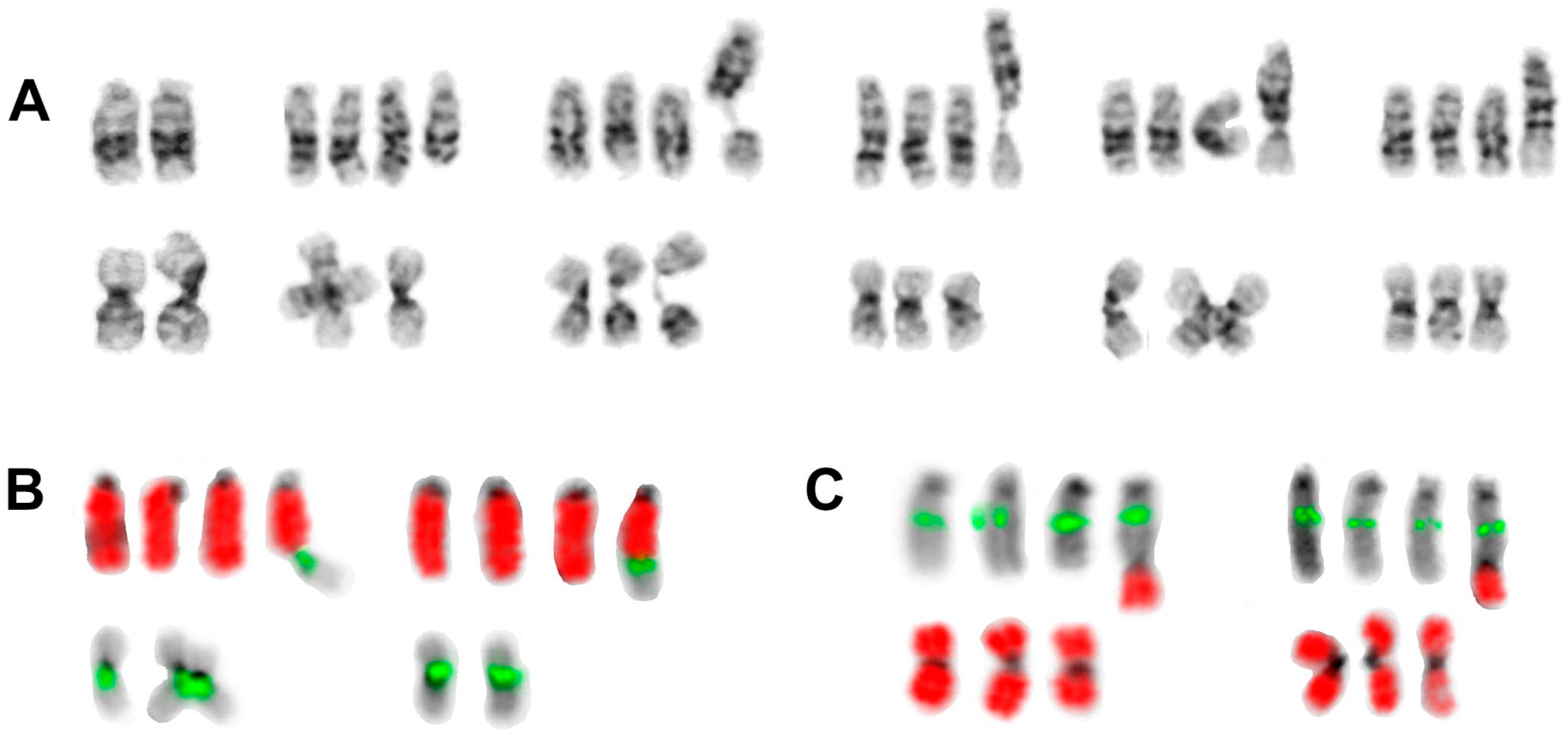
Examples of the dicentric marker dic(13;16)(q34;q11.2) from cell line A1-2 (daughter line): (A) G-banded chromosomes 13 (upper row) and 16 (lower row) from preimmortal diploid cell (first pair) and postimmortal tetraploid state; the rearranged chromosome 13 is always in the right position. After development of the dicentric marker, only three normal homologues of chromosome 16 remained in the 4n state. The second to sixth pairs: 16p deleted from marker; 16qh undercondensation (twice), second centromere of dicentric visible, and second centromere invisible by G-banding. (B) FISH with whole chromosome painting probe for chromosome 13 (wcp13, red) plus centromere-specific probe for chromosome 16 (CEP16, green) demonstrates the presence of chromosome 16 centromere in the dicentric, either with (left) or without (right, one homologue of chromosome 16 was missing in this cell) constriction. (C) FISH with locus-specific probe LSI13 (green, from 13q14) plus wcp16 (red) verifies the composition of the dicentric. Abbreviation: FISH, fluorescence in situ hybridization.
Telomere Length
Q-FISH analysis was used to determine telomere length in three cell lines at two different culture time points (15 and 75 weeks for A1, 7 and 52 weeks for N1, and 38 and 74 weeks for A2) . The mean value (and standard deviation) calculated from telomere length of all chromosomal pairs is shown in Fig. 6. The decreased telomere length co-occurred with an increase in telomeric fusions. At the first point of measurement, no telomeric fusions were observed in the three cell lines. At the second time point, after additional 45 (N1), 60 (A1-1), and 35 (A2) weeks in culture, a reduction in the average telomere length by 25% (N1), 50% (A1-1), and 10% (A2) and two, eight, and six telomeric fusions per 30 metaphase cells were observed. Thus, increases in the number of telomeric fusions can be used as an indicator for the shortening of telomeres. Heterogeneity in telomere length was especially prominent at the second time point in cell lines N1 (at 52 weeks of culture) and A2 (after 74 weeks in culture). Therefore, telomeres could be dysfunctional and, especially in N1, it is suspected that ALT mechanism could be activated. 21 Proliferation ceased at 19 and 6 weeks following the second telomere measurement in A1-1 and A2, respectively.
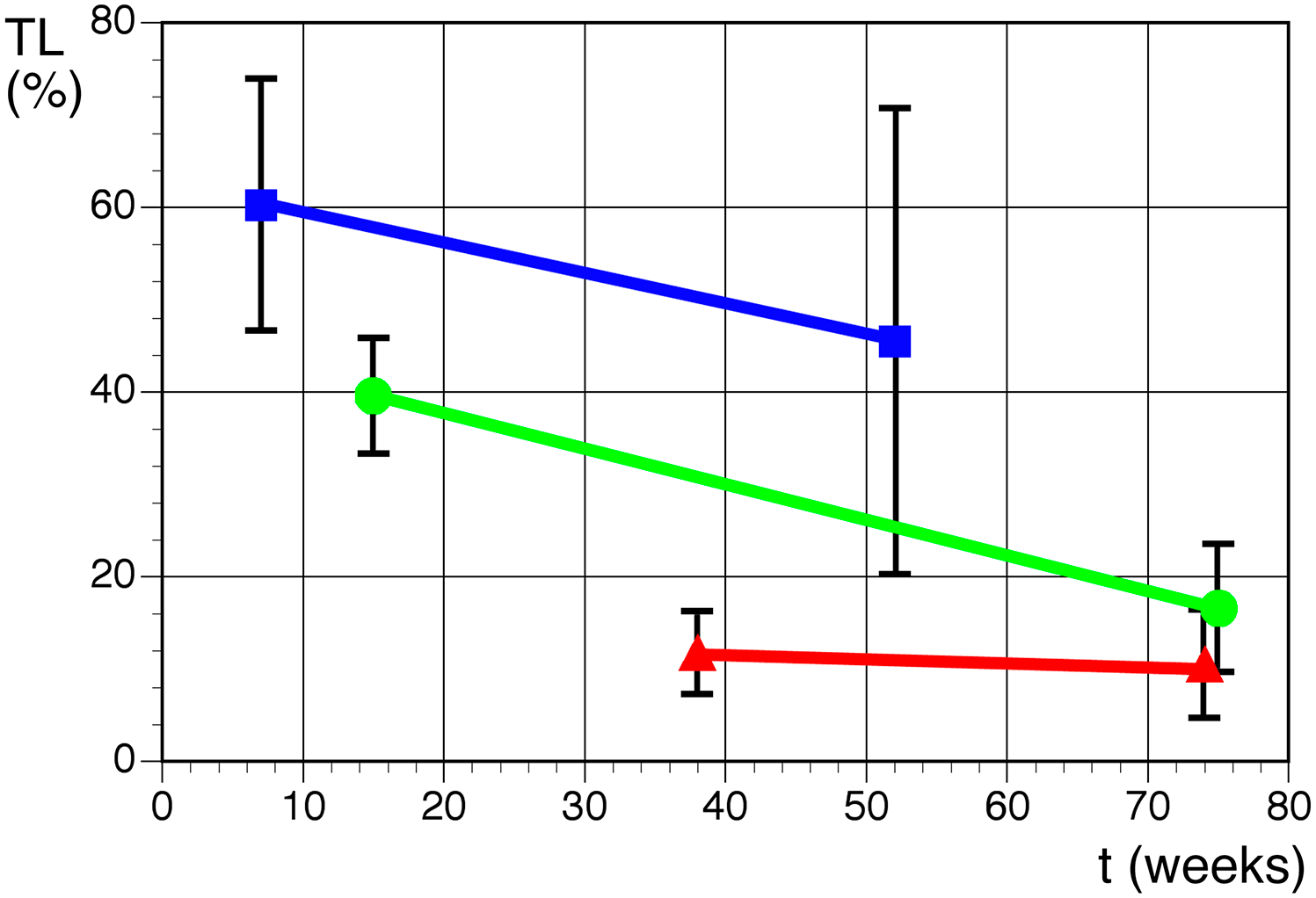
Average TL as a percentage of internal control (arbitrary units of T/C ± SD) measured by Q-FISH as a function of culture time in weeks in three LCLs: line N1 (blue), line A1-1 (green), and line A2 (red). Telomere length shortens with culture time. Abbreviations: TL, telomere length; LCL, lymphoblastoid cell line; Q-FISH, quantitative fluorescence in situ hybridization.
Discussion
This long-term study on 10 human LCLs detected karyotypic changes with fascinating fluctuating levels of aberrant clones, increases in telomeric fusions, and the acquisition of particular trisomies over 3 years of culture. Our observations are compared herein with the literature on cytogenetic changes known to occur during the long-term culture of LCLs.
Genomic Instability in Early LCLs Was Not Confirmed
In a study with two EBV-transformed cell lines, Lacoste et al. 22 found that after a short culture time of only 4 weeks, high levels of non-clonal aberrations (e.g., translocations, deletions, and fragments) emerged. This initial genomic instability decreased with culture time. However, the observation period was limited at only 25 weeks of culture. In contrast, our results demonstrate the occurrence of a low level of chromosomal breakage within the first 25 weeks and emergence of chromosomal rearrangements with prolonged culture.
We are unable to explain this discrepancy between the studies. However, different in vitro environmental conditions could influence the genomic stability of cultured cells in a variety of aspects. For example, the incidence of aneuploidies in certain human and mouse cell lines has been reported to vary with oxygen concentration. 23 The latter could be a possible explanation for these discrepancies.
Trisomy 12 Was the Most Common Acquired Aberration
Non-clonal trisomy 12 in short-term cultures of phytohemagglutinin-stimulated peripheral blood lymphocytes (T-cells) is a very rare and random event. 11 Therefore, the high rates of trisomy 12 observed here in the LCL long-term culture of B-cells are a surprising development. Interestingly, trisomy 12 has been reported as the most frequent numerical abnormality in LCLs. 11 This study documented trisomy 12 in approximately 20% of analyzed cell lines. However, following investigation of early passages (<5), only two of the seven LCLs with trisomy 12 demonstrated clonality for the aberration. 11 In a follow-up study, with a single LCL cultured over a longer period, the trisomy 12 clone expanded to compose 100%. 9 Our results are in full concordance with these observations. All 10 LCLs in this study acquired trisomy 12 in a considerable proportion of cells. In the majority of cell lines, the percentage of cells with trisomy 12 reached 100%. In a single cell line, N3, the trisomy 12 clone had quickly expanded to compose two thirds of analyzed cells after 1 year of culture. Only 4 months later, this cell line contained 100% 46,XY cells. However, it must be considered that non-chromosomal genomic changes could have favored the selection of this numerical inconspicuous clone. In other investigations of EBV-induced LCLs, trisomy 12 was among the numerous rearrangements observed, but not as frequent as in our experiments.1,7,24 It is very likely that at least under in vitro conditions, clones with trisomy 12 have a higher growth potential compared with those with normal karyotypes.
Interestingly, trisomy 12 is also the predominant aneuploidy observed in cultures of human pluripotent stem cells. Due to increased replication, clones with trisomy 12 grow faster than those with a normal karyotype. This effect is not a consequence of overexpression of genes on chromosome 12; instead, genome-wide changes in gene expression have been observed. 25 Of note, LCL clones with trisomy 12 have demonstrated tumorigenic potential when injected in nude mice. 9
Influence of EBV on Chromosomal Instability
The contribution of EBV to the observed chromosomal instability after prolonged culture of LCLs is a matter of debate. The mechanism by which EBV may induce genomic instability and thus oncogenesis is not yet understood. Several mechanisms have been proposed, that is, induction of oxidative stress, telomere dysfunction, and DNA damage (Kamranvar et al. 26 and references therein).
Transformation of B-lymphocytes without EBV is possible, but is rarely performed. Venuat et al. 27 studied two such lines for more than 3 years, but did not describe in depth set-up procedures. In this study, karyotypic stability was observed for approximately 2 years before numerical aberrations appeared in both lines; one line showed trisomy 12 in addition to trisomy 3.
For primary human mesothelial cells infected by SV40, it has been shown that the primary event for development of neoplastic phenotypes is the occurrence of non-clonal preneoplastic aneuploidy. Cells with karyotypes destabilized by aneuploidy evolve spontaneously into neoplastic clones at low rates. Thus, the oncogenic action of SV40 virus is indirect, as an initiator of the evolution of cancer-causing karyotypes. 28
Clonal Rearrangements Occur Before Immortalization
The statement of Sugimoto et al. 4 that cellular immortalization in EBV-induced LCLs is accompanied by aneuploidy without exception is certainly true. However, our results show that chromosomal changes during the immortalization process occur only in addition to the previously acquired rearrangements of the cell line. In contrast, many previous studies suggest telomerase activation and the (first) appearance of aneuploidy occur simultaneously. 4 Possibly, intervals between chromosome preparations were too long to detect rearrangements separately from telomerase activation and immortalization in these studies.4,6,7
Within our study of 10 LCLs, only a single cell line reached the postimmortal stage, displaying a near-tetraploid karyotype at the end of the observation period. All lines accumulated aberrant clones, although three lines stopped proliferating during the period of observation and were thus clearly preimmortal.
Our data from EBV-induced LCLs are in agreement with the observations from SV40-transformed fibroblast cultures. In both cases, it was shown that genetic alterations can be detected at early stages after expression of viral oncogenes, before crisis and the establishment of immortal lines. 29 In testing their alternative theory of virus-induced neoplastic transformation, Bloomfield and Duesberg 28 analyzed karyotypes of SV40-infected human mesothelial cells 1 and 2 months after infection. They found that SV40 transforms normal human cells by inducing preneoplastic aneuploidy (as observed in 13 of 20 cells analyzed). By karyotypic evolution, neoplastic clones with individual clonal but flexible karyotypes arose, stabilized by selections for variants with cancer-specific autonomy. 28
Our observations on EBV-infected lymphocytes showed that a similar process takes place with karyotypic changes long before an eventual immortalization step.
Aberrations Based on Changes in the Structure of 16qh
Undercondensation of the satellite 2–rich heterochromatic segments in chromosomes 1q and 16q was observed in LCLs derived from patients with chromosome breakage syndromes 13 and in LCLs from healthy donors due to strong alphoid DNA demethylation during the course of prolonged culture. 14 The methylation status of heterochromatic regions varied within the cell population and showed no correlation with clonal evolution. Satellite 3–rich regions, present in the heterochromatin of chromosomes 9 and Y, were never found in an undercondensed state despite strong demethylation. 14 Our results largely confirm this observation concerning aberrations caused by 16qh undercondensation.
Formation of Dicentrics and Telomere Length
Telomeric sequences at the chromosomal ends are lost in each cell division, under in vitro and in vivo conditions. When telomere length has reached a critically short-length threshold, and likely also when broken dicentric chromosomes are present, the DNA damage checkpoint pathway is activated, which leads to a non-proliferative state, telomere-initiated senescence. 30 Under certain circumstances (defective p53 and pRB pathways), cells can bypass this barrier for another 20 to 30 cell divisions. 31 Then, end-to-end fusions between chromosomes with short telomeres may occur until a second proliferate block, crisis, is promoted by genome instability. 31 Therefore, in most cases, the uncapping of chromosomal ends finally leads to apoptosis. However rarely, further rounds of cell division in these genetically altered cells are possible by either upregulation of telomerase or ALT. These two mechanisms for telomere elongation are not normally active in cultured fibroblasts. For unlimited growth potential, the majority of human cancer cells utilize telomerase-dependent telomere elongation. In contrast, in immortalized cells and a small fraction of cancer cells, ALT based on break-induced telomere synthesis 10 is used exclusively. It was further shown that the viral latent membrane protein 1, expressed in EBV-transformed lymphocytes, simultaneously modulates multiple signal transduction pathways in B-cells to enhance telomerase activity. 32
Also for EBV-transformed human B-lymphocytes, it was suspected that the formation of dicentrics, that is, telomeric fusions, may correlate with critically short telomeres. In the absence of telomerase activity, cells enter a proliferative crisis and die. 3 Chromosome stability could be achieved only by activating telomerase. Then, in the postimmortal stage, the telomeres are short, but stable and telomeric fusions should be rarely observed.
We found a clear increase in the number of telomeric fusions after a culture period of 1 year (52 weeks). In 6 of 10 LCLs investigated, one to four telomeric fusions per 30 metaphase spreads were found (median: 2). A high number of telomeric fusions indicated proliferative crisis. The average value calculated from three cultures which stopped proliferation was 0.25 telomeric fusions per metaphase spread in the last experiment before crisis (6, 7, and 10 per 30 metaphases). Furthermore, the single immortal LCL showed very few telomeric fusions after crisis. From our experiments, we, therefore, also speculate that critically short telomeres lead to dicentric chromosomes (telomeric fusions) before crisis. Such a situation will only rarely be overcome by activation of telomerase or ALT, leading finally to the immortal state.
Consequences of Aneuploidy
Surprisingly, it was shown that artificially generated trisomic murine embryonic fibroblasts showed in vitro cell proliferation delay and an altered cell metabolism irrespective of which chromosome was present in the trisomic state. 33 Effects of aneuploidy were also detected in human cells. By examination of immortalized human colorectal and retinal pigment epithelium cell lines, Passerini et al. 34 found that the presence of extra chromosomes leads to increased DNA damage and sensitivity to replication stress. The observed genetic instability was similar regardless of which trisomy was present in the cells. In contrast, in human pluripotent stem cells, it was shown that trisomy 12 results in a global overexpression of genes, leading to a strong selection advantage. 25 Surprisingly, trisomy 12 in the mosaic state apparently also leads to changes in gene dosage effects in vivo with the resulting fetal overgrowth in addition to other clinical symptoms. 35
Therefore, in our experiments, negative effects might prevail for the short-lived aberrant LCL clones with trisomies other than 12, whereas clones with trisomy 12 might outcompete all other clones due to their increased proliferation rates. In addition to karyotypic changes, proliferation rates of aberrant clones might be influenced by acquired gene mutations.
Today, it is quite clear that the assumption that LCLs have an almost indefinite life span 36 no longer holds true. This incorrect and long-living dogma is reminiscent of the struggle of Leonard Hayflick, 37 who spent many difficult decades to convince the scientific community that cells, especially fibroblasts taken into cell culture, are not per se immortal. Certainly, compared with normal lymphocyte and fibroblast cultures, the time period in which a good mitotic index can be yielded from LCLs—approximately 1 year—is clearly longer. However, it should be considered that after about 6 months, the percentage of cells with aberrant karyotypes continuously grows. Karyotypic changes will occur long before crisis and cell death or immortalization. A maximum of 10% of the cell lines will proceed to the postimmortal stage characterized by a clearly aberrant karyotype. In the course of long-term culture, the fate of cell clones with chromosomal aberrations may be extinction, survival, or acquisition of additional rearrangements. At a certain time, a cell line may be composed of clones with different aberrations in varying proportions. This is comparable with karyotype evolution in SV40-infected cells where virus-induced preneoplastic aneuploidy leads finally to cancer-specific karyotypes. 28 LCLs could therefore be best considered as “models” for studying clonal evolution in hematopoietic neoplasia than for preservation of cells with certain genomic features.
Footnotes
Acknowledgements
For excellent supervision of the cell lines, cordial thanks go to the technical assistants G. Böhme, S. Denecke, J. Koch, and A. Quaiser, Department of Human Genetics, Magdeburg. We are also very grateful to Heather Williams, Viapath at King’s College Hospital, Haematological Malignancy Diagnostic Centre, Cytogenetics Laboratory, London, UK, for correction of the English language. This work is dedicated to Prof. P. Wieacker, the former head of the Department of Human Genetics at Magdeburg (1994–2007) and Münster (2007–2019) (both Germany), on the occasion of his retirement.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MV carried out the chromosome preparation and karyotype analysis, performed the data analysis and interpretation, and wrote the manuscript. IJ performed the Q-FISH analysis. TL and MZ helped with drafting the manuscript. All authors have read and approved the final manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.