
Abstract
Antigen masking in routinely processed tissue is a poorly understood process caused by multiple factors. We sought to dissect the effect on antigenicity of each step of processing by using frozen sections as proxies of the whole tissue. An equivalent extent of antigen masking occurs across variable fixation times at room temperature. Most antigens benefit from longer fixation times (>24 hr) for optimal detection after antigen retrieval (AR; for example, Ki-67, bcl-2, ER). The transfer to a graded alcohol series results in an enhanced staining effect, reproduced by treating the sections with detergents, possibly because of a better access of the polymeric immunohistochemical detection system to tissue structures. A second round of masking occurs upon entering the clearing agent, mostly at the paraffin embedding step. This may depend on the non-freezable water removal. AR fully reverses the masking due both to the fixation time and the paraffin embedding. AR itself destroys some epitopes which do not survive routine processing. Processed frozen sections are a tool to investigate fixation and processing requirements for antigens in routine specimens.
Introduction
Fixation and embedding for routine histopathological diagnosis is obtained by sequential steps. The first step is tissue preservation, described as “fixation” around 1890. 1 Subsequently, the water inside the tissue is replaced with molten paraffin through sequential immersion in polar compounds (alcohol, xylene), the last ones miscible with the wax. Once the wax-embedded tissue is solidified, sections are obtained, which are placed on a microscope glass slide to adhere. The last part of this latter process is often enhanced by oven-drying. Last is processing the tissue section with the reverse order of reagents to do a water-miscible tissue histochemical stain or IHC.
During the whole process, tissue antigens undergo physicochemical modifications which results in masking of the mostly linear epitopes carried by the tissue components (reviewed in the study by Dapson 2 ). Procedures that reverse the antigen masking have been established and are commonly used.3,4
The chemistry of epitope masking itself is poorly understood. Molecular modifications of the antigen-carrying proteins upon fixation and embedding have been proposed, mostly based on studies of isolated antigens, such as proteins, with tests in formaldehyde-containing solution. The modifications leading to antigen masking are intrinsic to the protein considered (intramolecular), or include the effect on other proteins located in close contact with the antigen-bearing one (intermolecular).
Contact of formaldehyde with the tissue components results in the initial formation of highly reactive hydroxymethyl groups, leading to methylene bridges between amino groups of proteins, 5 inter- and intramolecularly.6,7 Some of these bonds are partially resolved upon exposure to moderate heating; others have been found less reversible, particularly after transfer of the specimen to ethanol. 8 The effect of fixation has been hypothesized to result in the inversion of the electrostatic charges, reversal of the protein polarity, and insolubility.5,9 All the above are supposed to cause intramolecular loss of antigen availability for the paratope to bind.
Cross-linking of the proteins adjacent to the one carrying the antigen affects the availability of said antigen, 10 and it has been shown that the masking is dependent on the concentration of these bystanders. 11 In an artificial model, the intramolecular bonds are more relevant for masking than the intermolecular bonds, unless fixation occurs in an environment of highly concentrated, macromolecular proteins. 7
Recently, we documented epitope masking of reexposed epitopes in formalin fixed and paraffin embedded (FFPE) material upon loss of the non-freezable water, 12 an effect which can be prevented by disaccharides, acting as water substitutes.
Coagulating fixatives, such as Carnoy, 13 Methacarnoy, 1 and other fixatives without formaldehyde, 14 have been suggested as alternatives to cross-linking fixatives, because antigen masking is supposed to be absent or reduced by avoiding formaldehyde.
It is unclear which agent or which passage during processing causes the antigen masking; therefore, we set up experiments aimed at elucidating this phenomenon by using frozen sections affixed to slides and by processing them identically to whole tissue blocks.
Materials and Methods
Tissues
Fully anonymous human leftover tissue (normal colon, tonsil, endometrium, and myometrium; two ovarian endometrioid carcinomas; one high-grade serous carcinoma; and two endometrial endometrioid carcinomas) was exempt from the San Gerardo Institutional Review Board approval as per Hospital regulations (ASG-DA-050 Donazione di materiale biologico a scopo di ricerca e/o sperimentazione, May 2012). The specimens were frozen in a Leica CM1850 cryostat (Leica Microsystems GmbH, Wetzlar, Germany) or a MICROM HM500 O (Heidelberg Instruments Mikrotechnik GmbH, Heidelberg, Germany). Four-µm sections were cut and placed on polylysine-coated glass slides (Thermo Fisher Scientific, Menzel-Glaser Superfrost Plus; Bio-Optica, Milan, Italy). Routinely processed companion tissues were used for staining references and have been described extensively. 15
Tissue Processing
All experiments were performed on sections obtained from flash-frozen material, cryosectioned, affixed on positively charged glass slides, and processed analogously to whole tissue biopsies (Table 1). Comparison with routinely processed tissue, FFPE was not performed because uninformative on each step of the process.
Processing Steps of the Tissue Sections.
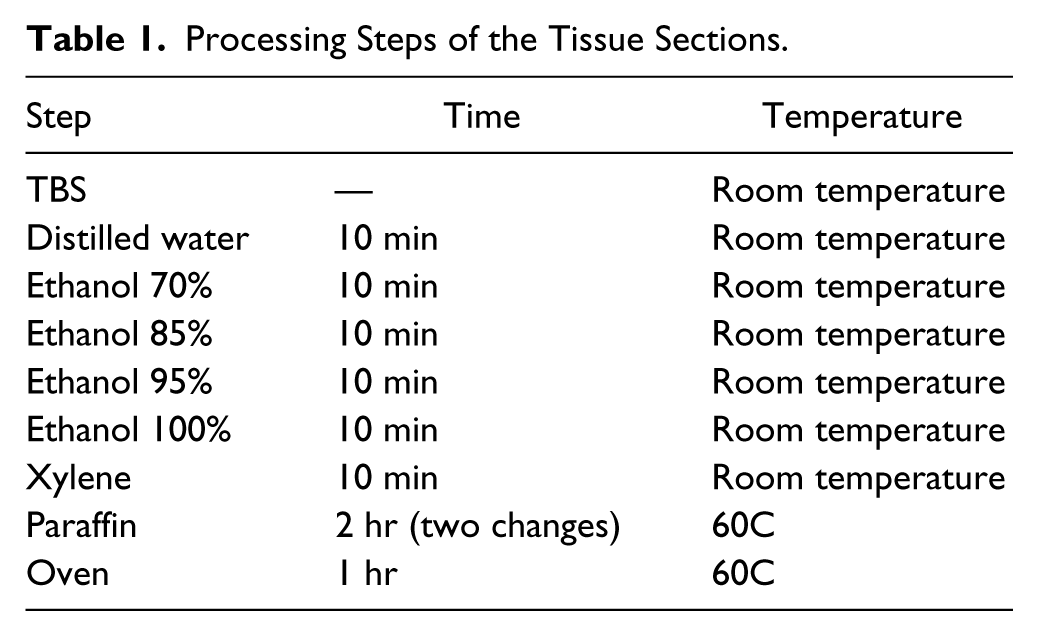
Cryosections to be fixed in 10% buffered formalin (FA; formaldehyde, 4%; monobasic sodium phosphate, 0.2%; dibasic sodium phosphate, 0.8%; methanol, 0.1%; distilled water; Bio-Optica) were immersed in the fixative immediately after cutting for 30 min, 24, 48, or 72 hr at room temperature (RT) or for 30 min, 1 hr, or 2 hr at 60C. Times above 12 hr were calculated within a ±15% to 20% range of the 24 hr multiple for practical reasons.
Each slide was processed analogously to a tissue specimen (Table 1 and Fig. 1) and brought back to water as per the dewaxing protocols used for immunostaining and hematoxylin and eosin staining. This protocol ensures complete dewaxing as observed on a daily basis with the routine diagnostic activity (not shown). The process was interrupted at discrete steps during the process (TBS, ethanol, xylene, paraffin, antigen retrieval [AR], dry; see Fig. 1) and returned to buffer with the appropriate reverse steps.
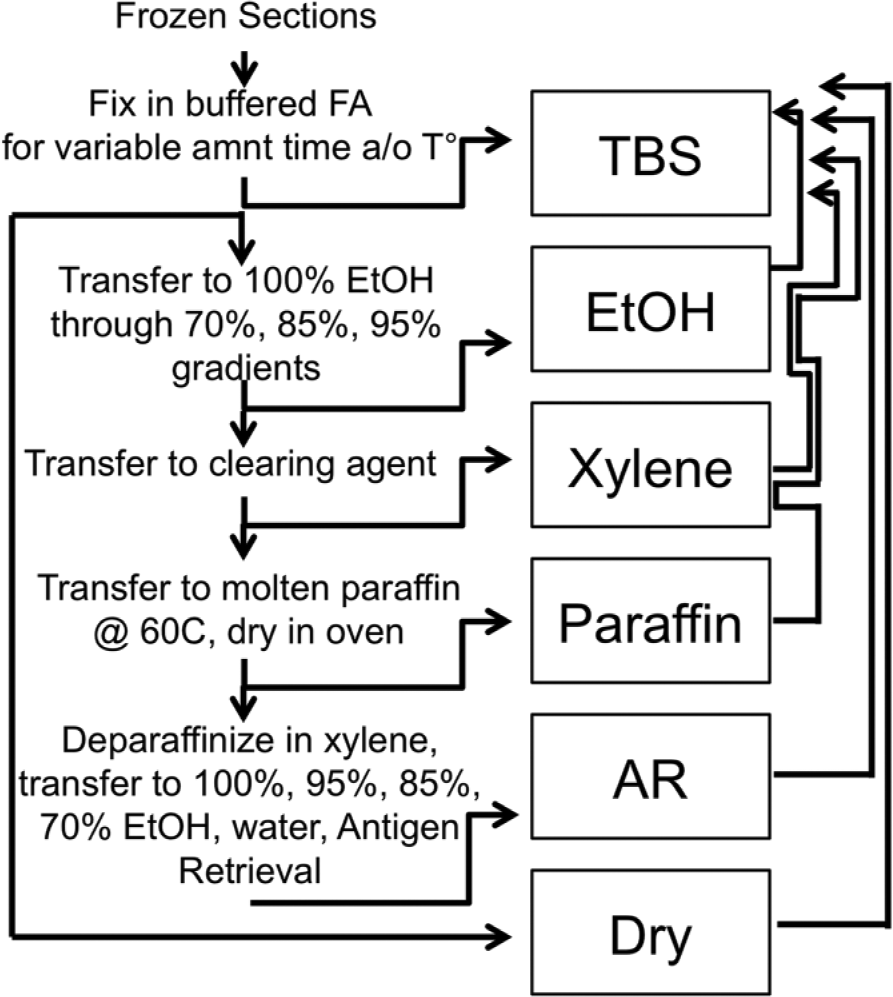
Scheme of the processing of frozen tissue sections. On the left are depicted the steps that a frozen section goes through, represented by vertical arrows pointing downward. A horizontal arrow pointing rightward illustrates each step where a group of sections interrupts the process, step which is named after the boxed name. On the right, an upward-pointing arrow depicts which process the section goes through before being brought back to buffer (TBS). Abbreviations: FA, formalin; EtOH, ethanol, AR, antigen retrieval.
Experiments with acetone fixation were abandoned because of the poor tissue stabilization (not shown) and the lack of use of acetone in routine whole tissue processing.
Experiments on the effect of denaturing agents on FA-fixed frozen tissue sections were performed by comparing ethanol 100% with a 30-min incubation in 0.2% Tween-20 (Sigma-Aldrich, Milan, Italy) or 0.2% v/v sodium dodecyl sulfate (SDS) in TBS, followed by several washings.
Antibodies
Primary and secondary antibodies are listed in Tables 2 and 3, respectively. They were used at the appropriate dilution. 16
Primary Antibodies Used.
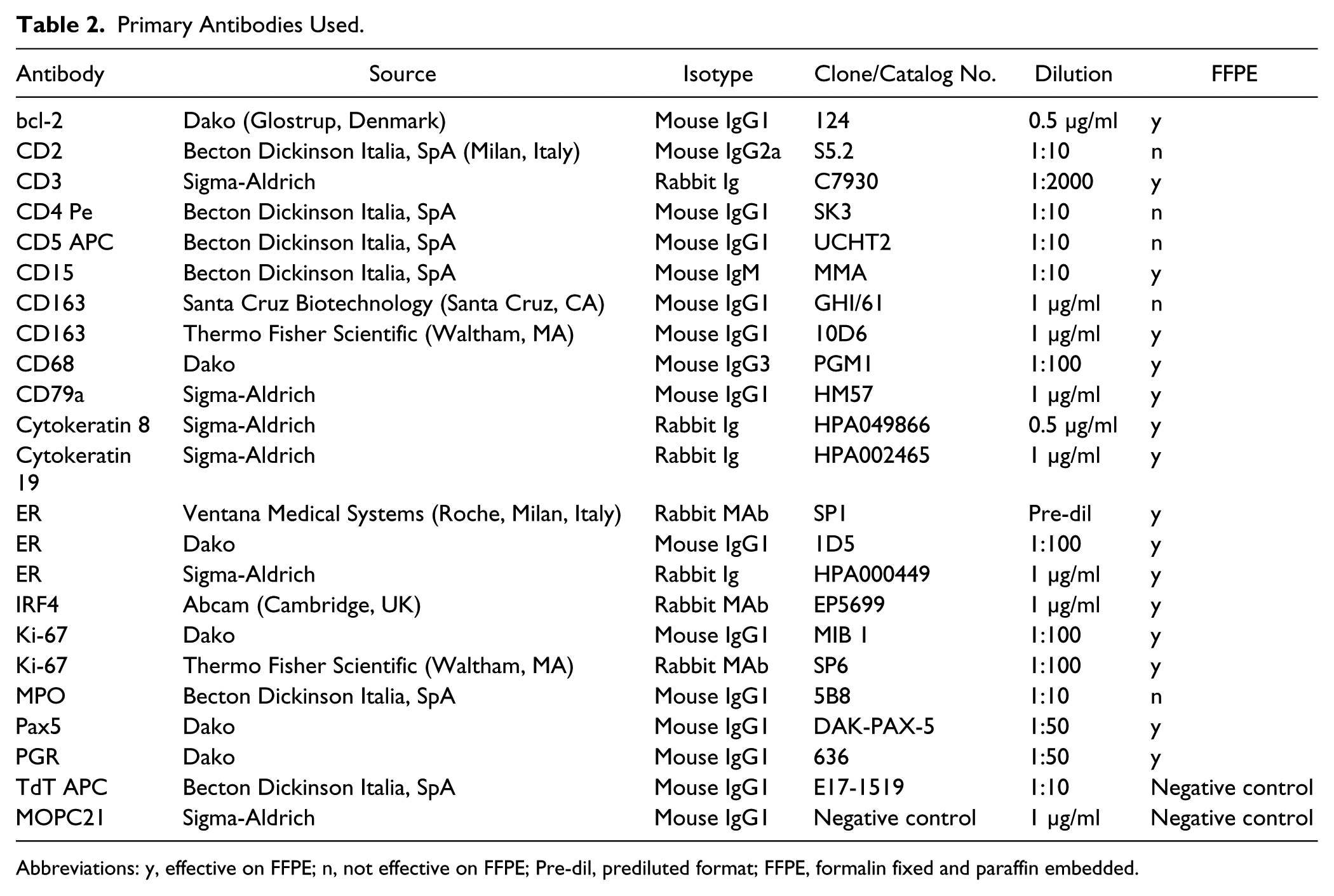
Abbreviations: y, effective on FFPE; n, not effective on FFPE; Pre-dil, prediluted format; FFPE, formalin fixed and paraffin embedded.
Secondary Antibodies Used.
Abbreviation: Pre-dil, pre-diluted format; HRP, horseradish peroxidase.
Immunostaining
Nonspecific background was blocked by immersing the sections in 5% defatted milk in TBS, as published, followed by inhibition of endogenous peroxidase. 15
AR was performed before the immunostaining as published, 15 when appropriate: Sections in distilled water were inserted into radio-transparent slide holders (model #S2029; Dako) and transferred to an 800-mL glass container filled with the retrieval solution (10-mM EDTA in Tris buffer pH 8; Sigma-Aldrich). The container was irradiated in a household microwave oven at full power for 8 min, followed by 20 min of intermittent electromagnetic radiation to maintain constant boiling.
Immunohistochemistry
Primary antibodies, optimally diluted, were applied overnight, washed in TBS, counterstained with a horseradish peroxidase–conjugated polymer (Dako), washed, developed in DAB, and mounted. For quantitative image analysis, sections were not counterstained.
Immunofluorescence
Experiments aimed at defining fixation time and temperature-dependent effect on masking and the effect of AR were performed by (1) fixing the sections, (2) staining in multiplex immunofluorescence, (3) coverslipping the sections with glycerol–polyvinyl alcohol solution containing antifade and 4′,6-diamidino-2-phenylindole (ProLong Gold; Thermo Fisher Scientific, Waltham, MA) to which 10% sucrose was added, (4) acquiring the images, (5) unmounting the sections, (6) performing AR, (7) restaining for the very same antigens, and (8) mounting with a coverslip as above and acquiring again the signal. Exposure times were kept the same for the same channel across all the experimental points to compare intensities.
The Aperio ScanScope FL Slide scanner (Leica Microsystems Srl, Milan, Italy) is equipped with an Olympus 20X/0.75 Plan SApo objective, an X-Cite exacte mercury lamp (Lumen Dynamics Group, Inc, Ontario, Canada), a linear time delayed integration sensor, and a quad-band filter set in the Pinkel configuration (DA/FI/TR/Cy5-4X-B; Semrock, Inc, Rochester, NY). The filter set consists of four excitation filters (FF01-387/11, FF01-485/20, FF01-560/25, and FF01-650/13) in a separate motorized filter wheel, and one filter cube in a motorized turret. The cube itself contains a quad-band emission filter (FF01-440/521/607/700) and a quad-band dichroic filter (FF01-410/504/582/669-Di01) (Semrock, Inc). One additional Semrock filter was separately purchased, an emission and dichroic orange filter (Cy3.5-A-Basic FF01-620/52-25 and FF585-Di01). All filters featured the zero-pixel-shift option. Details of the sensitivity and a graphic image of the filter set have been previously published. 17 Individual single stain images in light and fluorescent microscopy were acquired with the ImageScope software (Aperio).
Controls were inbuilt for the effect of AR on the fluorochromes, and for the capture by the second round of staining of the first antibody layer remaining after AR (see Supplemental Fig. S1).
Digital Image Analysis
IHC Images
Two to three 1600 × 1200 pixel images, 100× magnification, obtained with a digital camera (DP21; Olympus Italia Srl, Segrate, Italy) were inverted, and the pixel density histograms (8 bit, 0–255 channels) were obtained with Fiji (http://fiji.sc/) and saved as an Excel spreadsheet. The whole image was used for analysis except for CD79a (restricted to B-cell follicles) and Ki-67 in colon (germinal centers were excluded). Histograms were aligned on the negative background peak, and the resulting experimental curves were plotted. Cumulative percentage pixel numbers of the total in each channel were accrued over 255 channels (see Supplemental Fig. S2). Fewer channels were displayed for graphic clarity on a case-by-case basis. The channel position where 90% of the pixels are found was recorded and graphically represented for comparison of treatments. The analysis of channel position for 75% of the pixels showed superimposable results (not shown).
Immunofluorescence Images
Histograms data of fluorescence images as 8-bit gray levels (0–255) and of at least 2 × 2 mm were obtained with Fiji, exported in an Excel spreadsheet, and plotted as detailed above.
Results
Fixation Time- and Temperature-Dependent Antigen Masking
Fixation in FA in surgical pathology is usually performed by immersion at RT for 24 up to 72 hr. 18 However, much shorter fixation time and higher temperature are suggested as a practical alternative, to be performed in automated processors. 19 To assess the effect of fixation on immunoreactivity, frozen sections were fixed for 30 min, 24, 48, and 72 hr at RT (Fig. 2, left part) and for 30 min, 1 hr, and 2 hr at 60C (Fig. 2, right part). The resulting pre-AR immunoreactivity (Fig. 2 and Supplemental Fig. S3) was in general low. The effect of fixation at 60C was similar, although the masking was more pronounced for some antigens (IRF4, Ki-67, CD68) than for others, particularly for the longer hot fixation time. AR substantially increased the immunoreactivity (Fig. 2 and Supplemental Fig. S3). The increase was, however, heterogeneous across the panel of antigens and treatments. Longer RT fixation (48 hr) produced the best post-AR increase with most antigens, but a short, hot fixation also produced a noticeable increase. The extent of post-AR increase varied among the antigens and the treatments (Supplemental Fig. S4), with some antigens (e.g., IRF4 or Pax5) barely adding up to the control (see Supplemental Fig. S1).
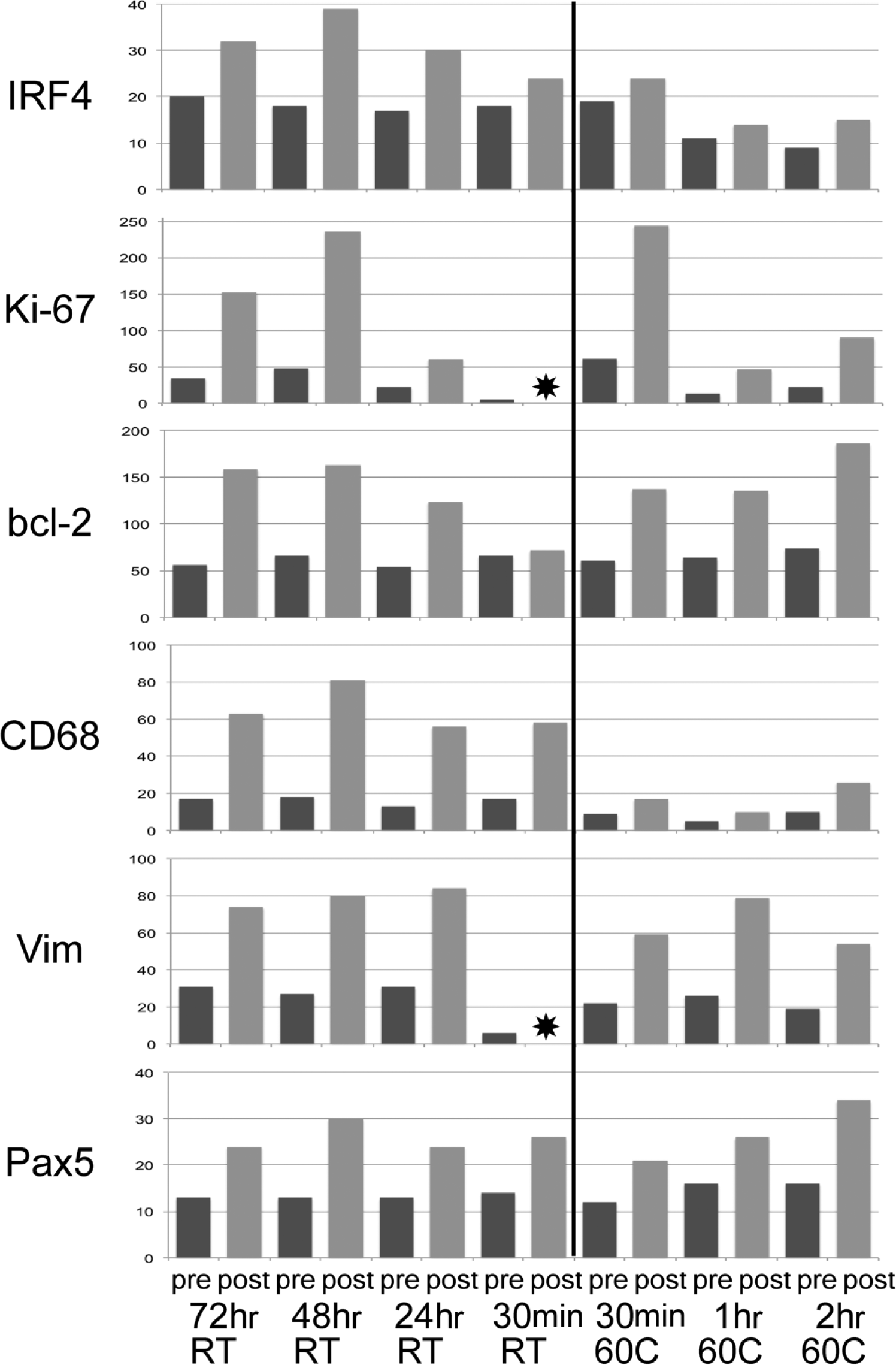
Effect of fixation time and temperature on antigens before and after antigen retrieval (AR). The channel position for 90% of the positive pixels before (darker bars) and after AR (lighter bars) is depicted for six antigens fixed at the time and temperature indicated at the bottom. An asterisk marks a value not available. A vertical line separates the RT treatments (left) from the fixation at 60C (right). Note a time-dependent detrimental effect of high temperature for some (IRF4, Ki-67) but not all antigens. Lower fixation time negatively affected IRF4, Ki-67, and bcl-2. Abbreviation: RT, room temperature.
We tested on normal uterus two commonly used hormone receptors (ER and PgR) with a limited range of time and temperature fixation conditions: 30 min, 24, 48, and 72 hr at RT and 2 hr at 60C. There was little difference in staining intensity for ER after AR, both in the myometrium and in the endometrial glands (Fig. 3). The masking effect of each treatment before AR was similar, and a trend toward worse results was noticed after AR for the longest fixation time (not shown). The increase after AR was similar for ER and PgR in each treatment, differently, for example, for bcl-2, which did not benefit from a short fixation (Fig. 4), as shown before. Tests on five ER+ cancers showed a more pronounced preference for longer fixation time (Supplemental Fig. S5).
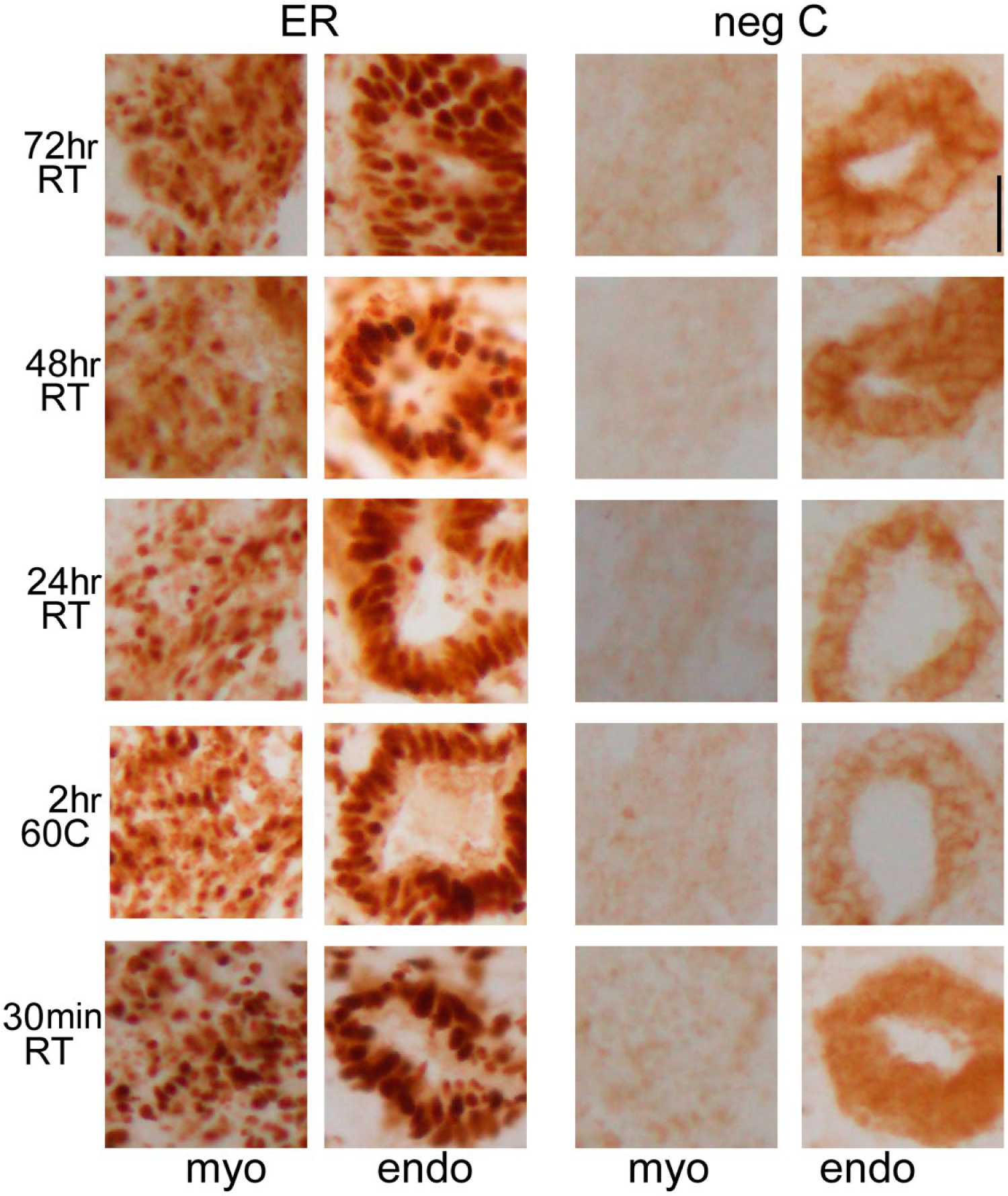
Effect of fixation time and temperature on ER and negative control staining in human uterus. Representative fields of human uterus frozen sections are shown fixed for the time and temperature shown at left and antigen retrieved. Separate staining for myometrium (myo) and endometrium (endo) is depicted. A mouse anti-ER monoclonal antibody has been used. Note the little variation in nuclear staining over the experimental points and the cytoplasmic staining of the negative control in the endometrial glands. Scale bar: 500 µm. Abbreviations: ER, estrogen receptor; RT, room temperature.
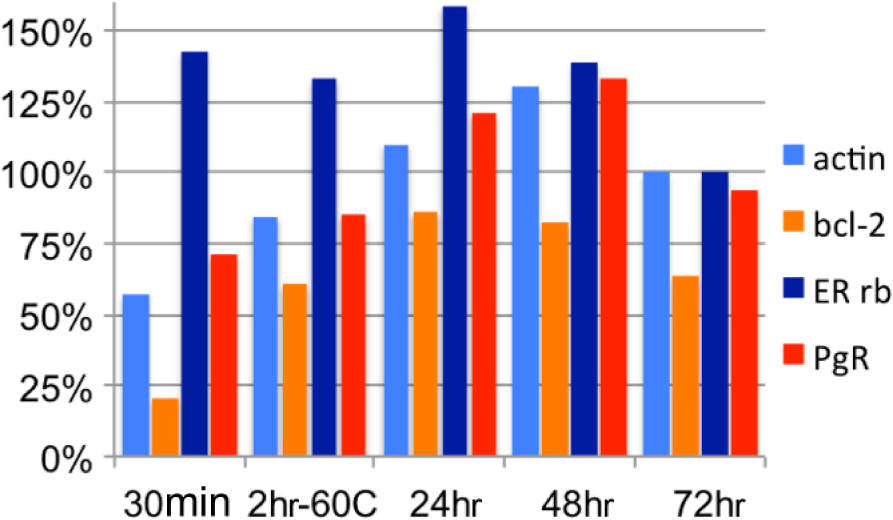
Percent changes in intensity of staining after AR for antigens expressed in the human myometrium. The ratio between the channel value for 90% of the positive pixels in immunofluorescence before and after AR is shown. A rabbit anti-ER has been used for this experiment. Note that bcl-2 and actin show the least increase for the shortest fixation time, while hormone receptors are less affected. Abbreviation: AR, antigen retrieval.
Masking Associated With Hydrophobic Solvents and Water Removal
Passages in water containing increasing concentrations of ethanol are necessary to allow hydrophobic clearing agents such as xylene to permeate the tissue. This step is sometimes referred to as “dehydration.” The whole process in routine tissue processing results in antigen masking.
We tested seven antigens on 30-min FA-fixed frozen sections for variations in immunoreactivity at each of the steps involved in tissue processing (Fig. 5 and Supplemental Fig. S6). Each antigen showed masking between the xylene and the paraffin step, to various extent. The temperature of the clearing agent was marginally affecting the masking (Supplemental Fig. S7), having a mild enhancing effect on shortly fixed antigens.
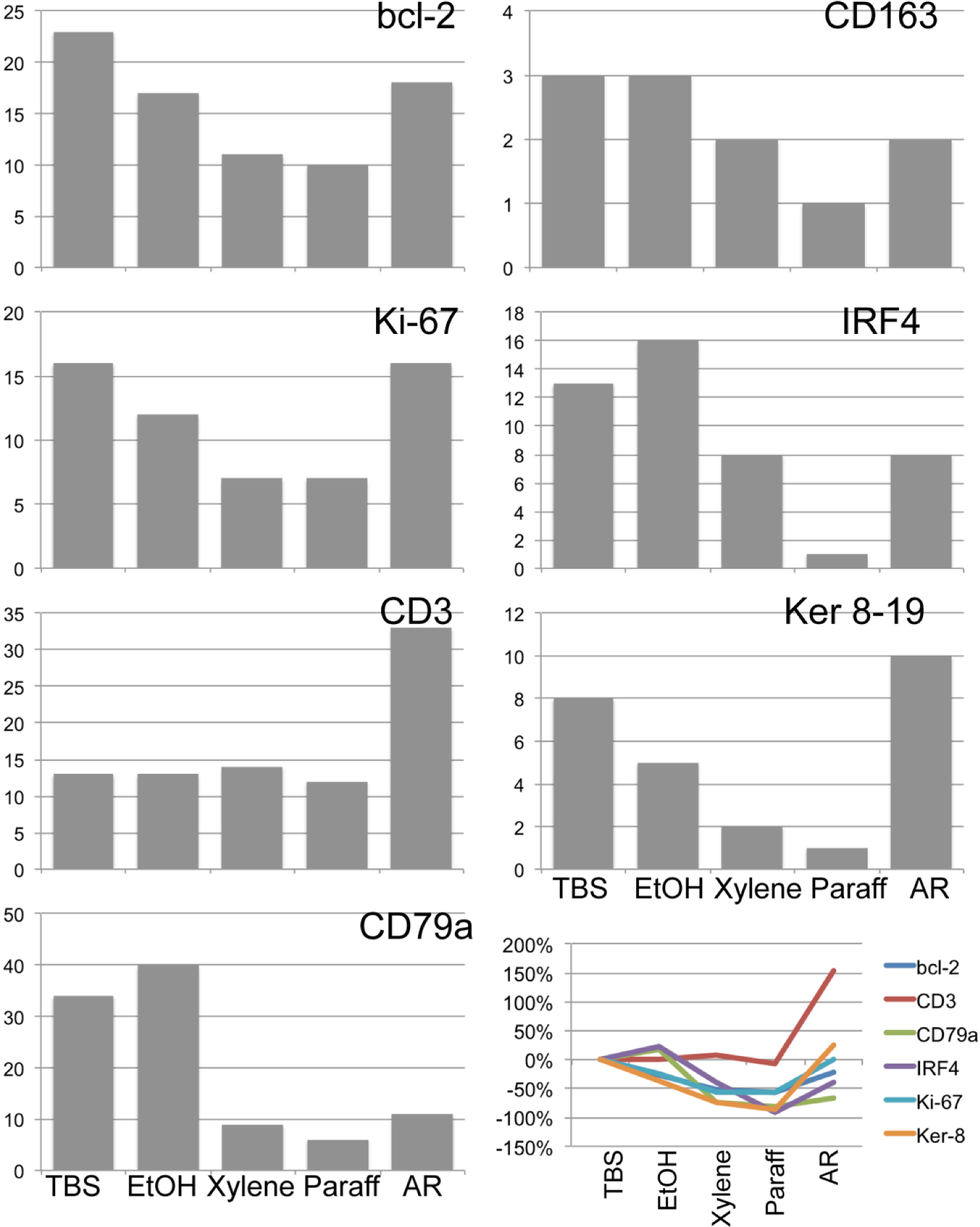
Effect of processing on antigenicity on frozen sections by immunofluorescence. Intensity graphs for seven antigens on a frozen tonsil tissue, fixed for 30 min in formalin at room temperature and processed as per Fig. 1, are shown. The channel position for 90% of the positive pixels is depicted for the step indicated at the bottom. The percent change between the channel values for TBS compared with the one for each processing step is shown in the lower right corner. Abbreviations: EtOH, ethanol; AR, antigen retrieval.
After the AR, not all the antigens regained the immunoreactivity they had at the beginning of the processing. Some antigens lost an additional fraction of immunoreactivity, on the top of the FA-dependent masking (e.g., CD79a, bcl-2, IRF4). Other antigens, instead, gained in immunostainability (CD3, Ki-67; Fig. 5).
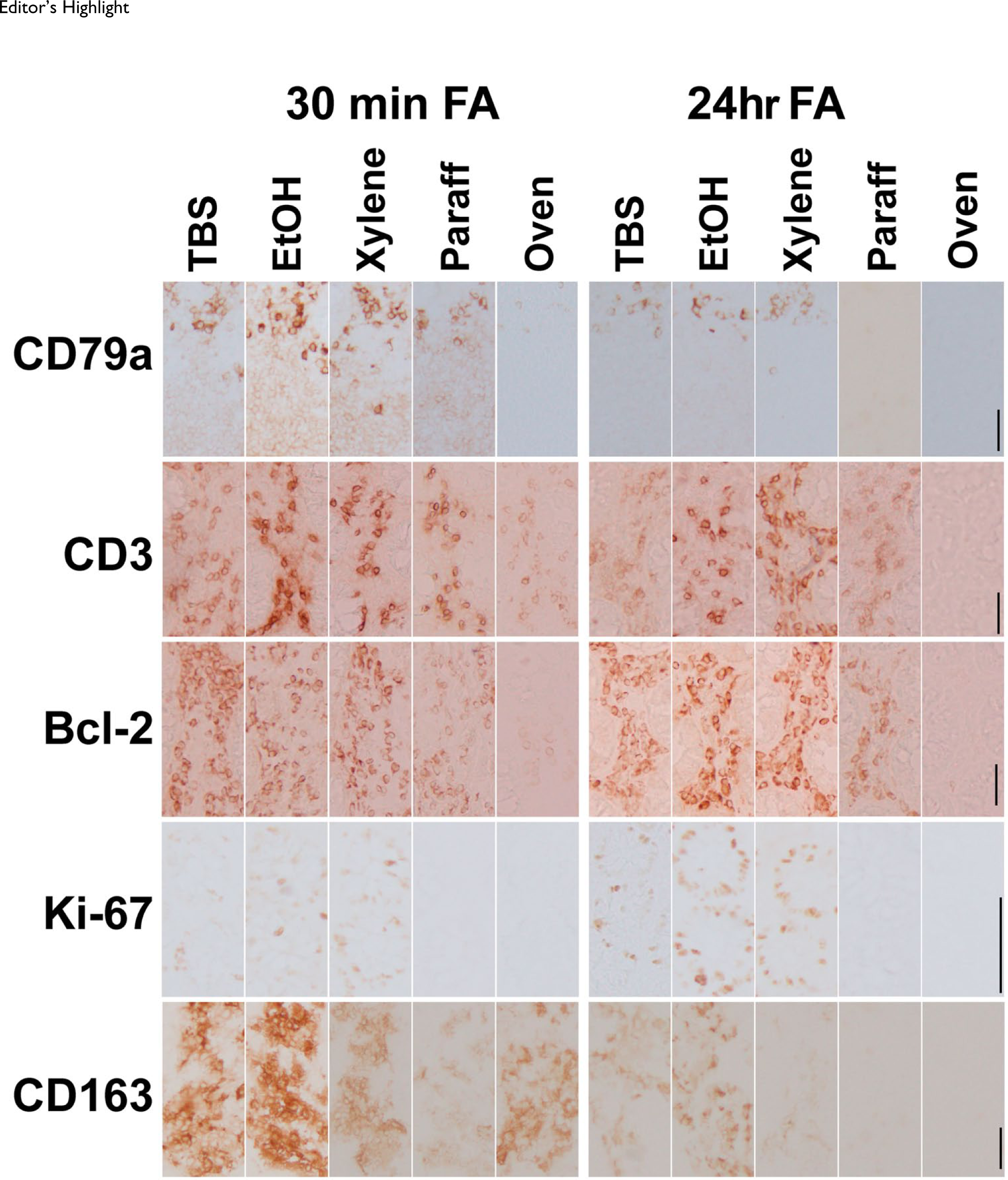
We repeated the test by using two fixation conditions (30 min and 24 hr) and IHC (Fig. 6, Fig. 7, and Supplemental Fig. S8). The results were superimposable, with a more pronounced detrimental effect for the longer fixation time for CD79a, CD163, and CD3, but not bcl-2 and Ki-67. However, differently from the previous experiment in which we used immunofluorescence, there was a noticeable increase of immunoreactivity after the ethanol step.
Editor’s Highlight
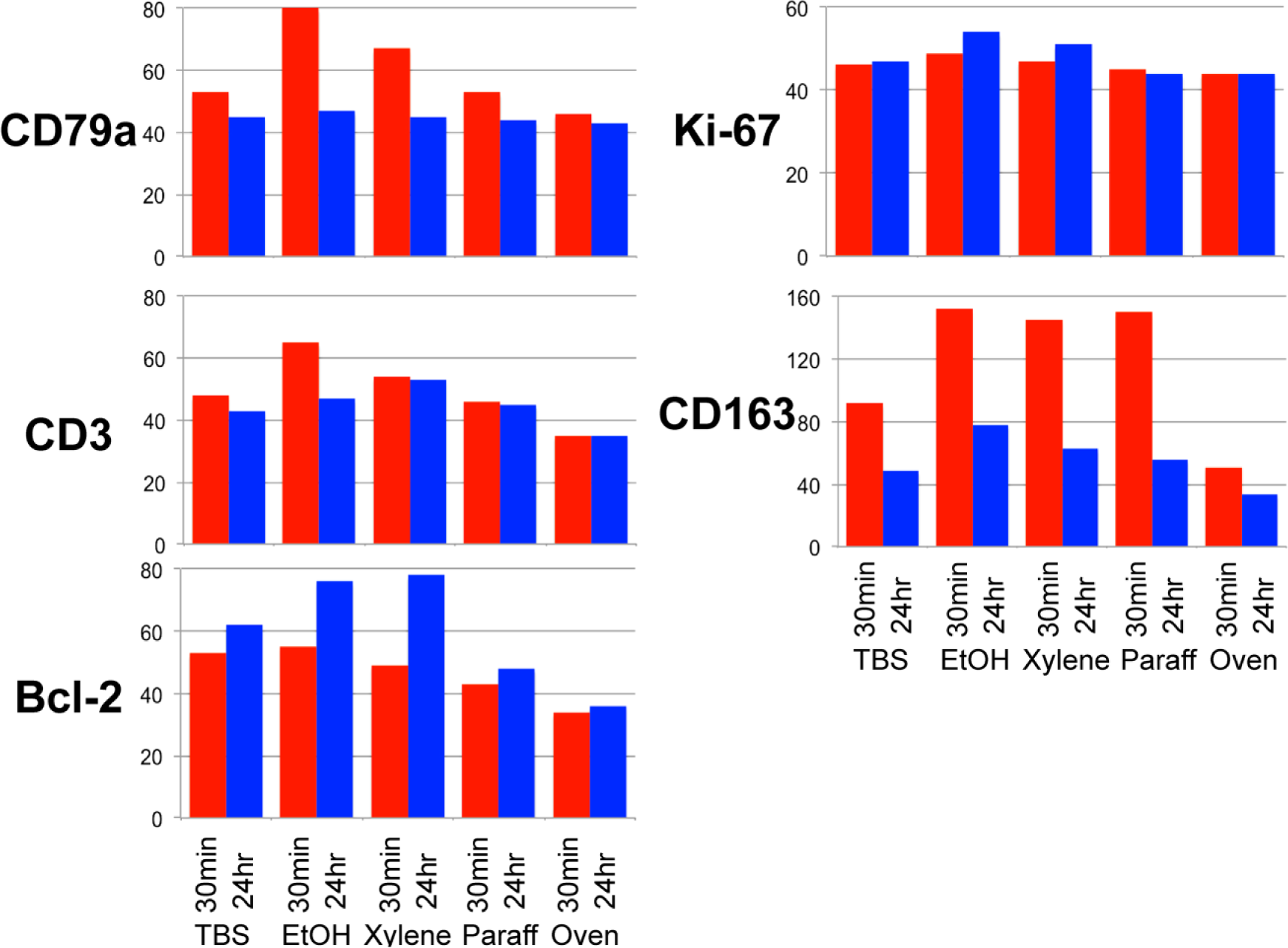
Effect of processing on antigenicity on frozen sections by IHC. Intensity graphs (channel for 90% of positive pixels) for five antigens fixed in formalin at room temperature for 30 min (lighter bars) or 24 hr (darker bars), processed along the steps shown (see Fig. 1), and immunostained for the antigens shown at the left. Note an intensity increase with the EtOH step and a decrease upon entering the paraffin step. Bcl-2 and Ki-67 show an increased staining after the alcohol and the xylene step with the longer fixation. No antigen retrieval was performed. Abbreviation: EtOH, ethanol.
Ethanol is often replaced by proprietary cheaper alcohol mixtures during processing. We thus investigated on whether the same enhancing effect was observed with some of these substitutes, and indeed, we obtained a variable amount of staining enhancement, compared with the starting step (Fig. 8). Alcohol act as a denaturant because of the ability to interfere with the water associated with the protein. Because of this, denaturing property is also referred to as a “coagulant.” We wanted to assess other denaturing agents such as detergents. Two detergents used in immunotechniques (Tween-20 and SDS) at the dilution commonly used were compared with ethanol, and we did observe an enhancing ability for them as well (Fig. 9).
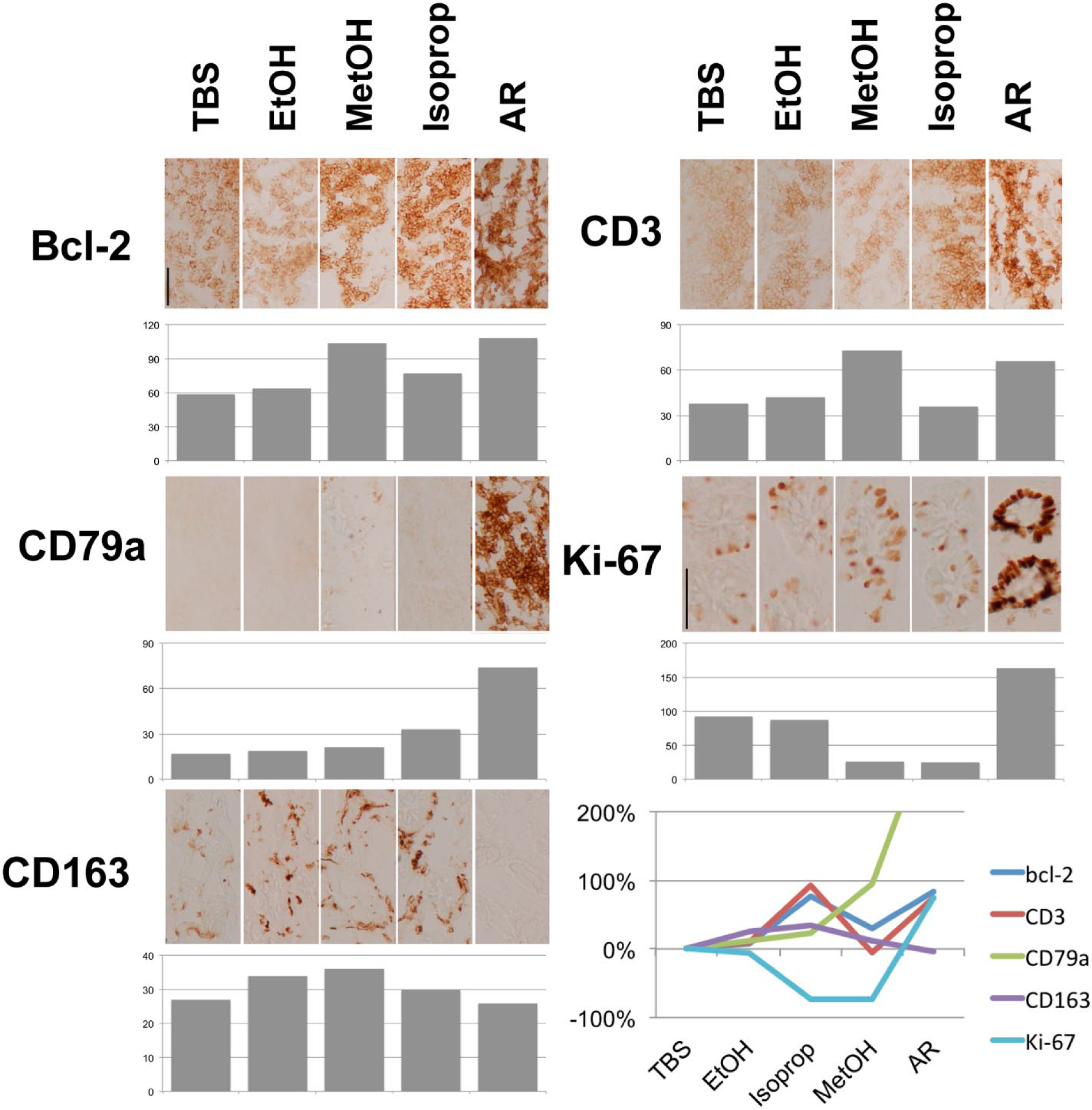
Effect of alcohol treatment on formalin (FA)-fixed tissue sections. The effect of three alcohol treatments (EtOH, isopropanol, and MetOH) is shown for five antigens, indicated at the left of each panel. The channel for 90% pixels for each antigen is shown below each IHC panel. In the lower right corner, the graph depicts the increase in immunoreactivity after the TBS step expressed as a percentage. Sections were fixed in FA for 24 hr at room temperature. AR treatment is shown for comparison. Note the variable degree of enhancement obtained by the alcohols. The CD163 epitope is destroyed by AR treatment. Scale bar = 100 µm. Abbreviations: EtOH, ethanol; MetOH, methanol; AR, antigen retrieval.
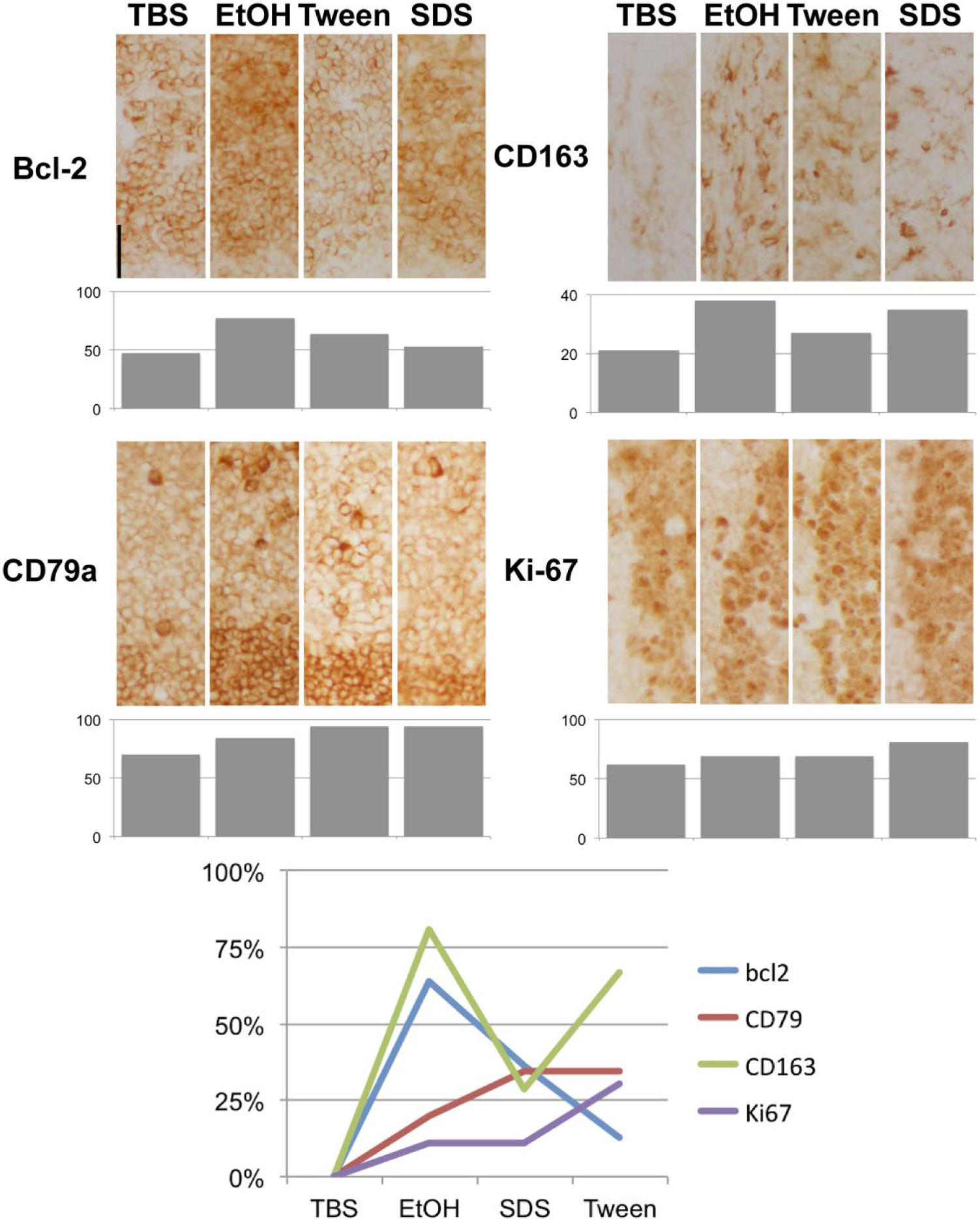
Effect of EtOH and detergents on immunoreactivity in formalin-fixed frozen sections. Frozen sections from tonsil tissue were fixed in buffered formalin (30 min, room temperature) and stained after treatment with TBS, graded EtOH, 0.2% Tween 20, or 0.2% SDS. Representative images are shown. No counterstain. Quantification bars are shown below each IHC panel. At the bottom, the percentage of the increase compared with TBS treatment. Various degrees of enhancement can be obtained by either ethanol or detergent treatment, compared with TBS. Scale bar = 500 µm. Abbreviations: EtOH, ethanol; SDS, sodium dodecyl sulfate.
In the previous experiments, we used a processing-sensitive epitope of CD163, detected by the antibody GHI/61, 16 and we noticed a negative staining after AR (Fig. 8 and Supplemental Fig. S6). We tested additional four antibodies (CD2/S5.2, CD4/SK3, CD5/UCHT2, and MPO/5B8) known not to react with FFPE material and used in live cell flow cytometry assays. When tested on FA-fixed frozen sections, they stained the expected target, but the stain almost completely disappeared after AR (Fig. 10), as observed with the CD163 GHI/61 antibody. Incidentally, we found that the epitope for another CD163 antibody (clone 10D6) was totally dependent on AR treatment on FA-fixed frozen sections (not shown).
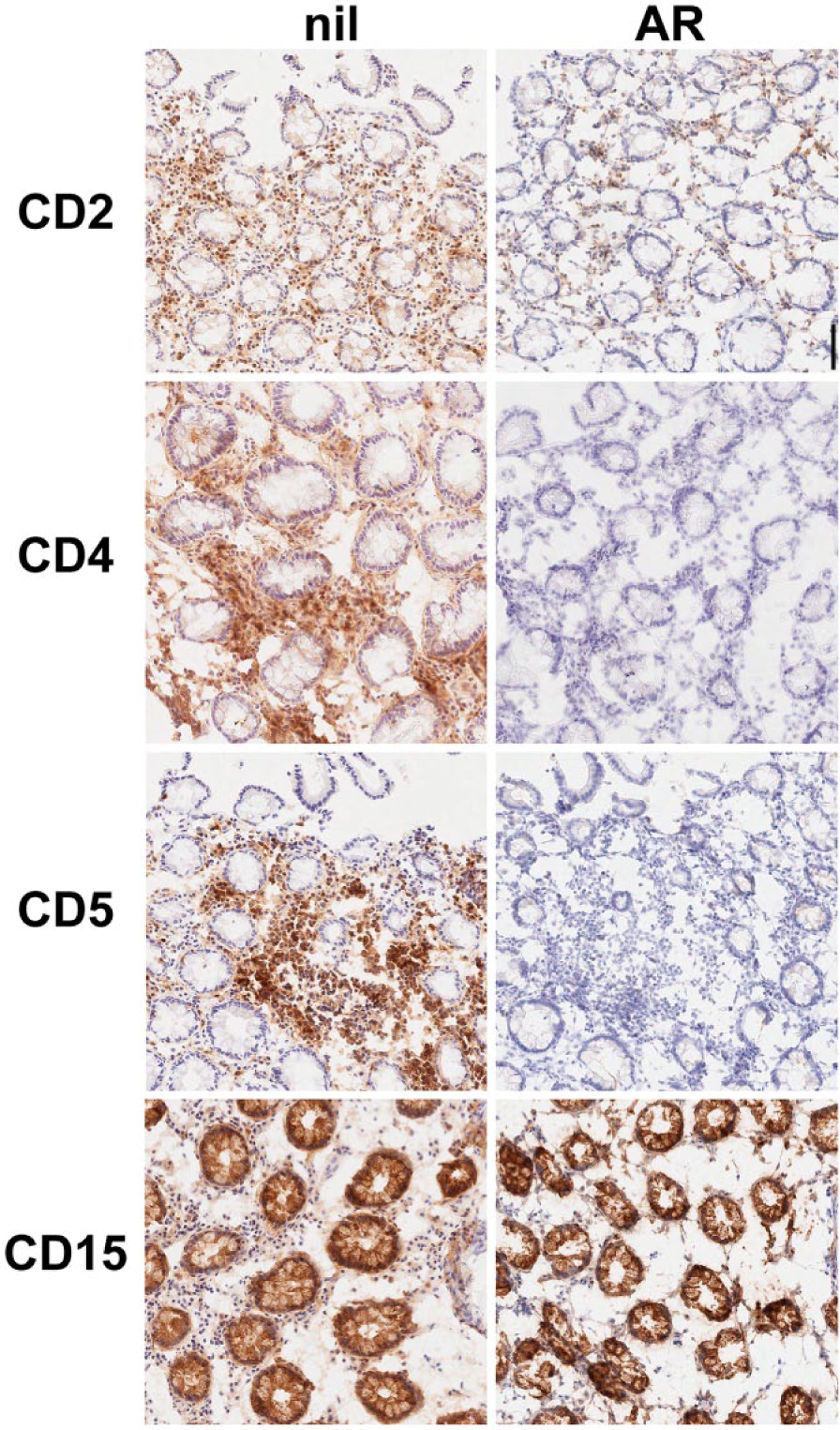
Effect of AR on putative conformational epitopes. Frozen sections from colon tissue were fixed in buffered formalin (30 min, room temperature) and either stained without (nil) AR or after AR. This latter treatment greatly reduces (CD2) or abolishes the immunostainability. A carbohydrate epitope (CD15/MMA) is enhanced by AR treatment. Nuclear hematoxylin counterstain. Scale bar = 500 µm. Abbreviation: AR, antigen retrieval.
Discussion
By dissecting the steps by which a tissue is processed, from fresh to being embedded in paraffin, we were able to observe changes in the immune availability of a handful of antigens at each step. We are aware of one single previous study addressing the same topic, using two cell lines on cytology preparations 20 : Our data are identical in tissue to the findings of Otali et al. 20 on cell lines.
Longer Fixation Time Allows Better Antigen Retrieval
Longer FA fixation times allow optimal post-AR detection of most of the antigens we tested: This is in the range of what has been recommended by the Pathology and Oncology Societies 18 (24–48 hr at RT) and the clinical guidance recommendation, accepted by the Food and Drug Administration (https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfstandards/detail.cfm?standard__identification_no=29272) from Clinical and Laboratory Standards Institute (CLSI; Quality Assurance for Design Control and Implementation of Immunohistochemistry Assays; Approved Guideline—Second Edition; CLSI document I/LA28A2 [ISBN 1-56238-745-6]; last accessed September 20, 2016). Much shorter times and higher temperatures were a mixed blessing, as about half of the antigens tested were negatively affected. We did not test a two-time protocol, 19 which is based on the rationale of allowing penetration of the cold fixative first, and then hot fixation. However, we used 5-µm sections which should make the penetration issue negligible.
By using a sensitive and quantitative immunofluorescent assay, we could detect and measure the masking effect of FA fixation and we found minimal differences among the range of fixation times at RT, but not when temperature was set at 60C; high temperature was often associated with an increased masking, not fully reversible after AR.
The recommended fixation times by the Pathology and Oncology Societies 18 run somehow against what has been considered the gold standard for IHC. In the pre-AR era, a handful of ubiquitous antigens detectable on routinely processed tissue have been considered as controls for overfixation, 21 contributing to the suggestion to keep FA fixation as short as convenient and avoid FA fixation time longer than 48 hr. 22 However, Shi et al. 23 suggested an overnight fixation as good as a short 30-min fixation in FA as a standard for frozen sections. Chung et al. 24 analyzed the amount and quality of RNA in tissues and observed optimal fragment yield between 4- and 48-hr fixation. Otali et al. 20 found similar evidence for a stabilization of the post-AR immunoreactivity with longer fixation time. We can confirm that the optimal fixation time for the antigens we tested is between 24 and 48 hr. We did observe, however, a great antigen-to-antigen variability with shorter schedules or higher temperatures, a factor to be considered if using a faster process.
The Post-AR Efficiency in Restoring Prefixation Antigen Levels Varies
We were able to measure the extent of the gain obtained by the AR step on a handful of antigens. We have demonstrated a broad range of increase in immune availability, in some cases barely above the unheated sample, and in a significative small group of antigens, the disappearance of the epitope.
After 1991, immunostaining procedures almost univocally call for an AR step. As a consequence, the notion that some antigens survive FFPE processing is somehow lost, and the intensity of staining after AR is seen as a mere measure of the efficiency of the reversal of the masking process. Here, we see a more nuanced picture, where heat is most likely the greatest player in the outcome of the process, not only post fixation but probably during fixation as well, influencing the amount of antigen available for detection in an antigen-specific fashion. 25 In other words, heating a FA-fixed specimen does not inevitably result in the full recovery of the original antigenicity but in a predetermined outcome which depends mostly on the epitope conformation itself.
We used an AR solution of one single composition and pH, not tailoring the retrieval conditions for each individual antibody, 26 a condition which was determined originally for FFPE material. This may have introduced some experimental noise in our experiments.
However, the results on frozen sections pointed to a consensus in antigen availability modifications upon changing the processing steps for all the antigens tested; thus, we expect that tailoring the AR condition for each antibody would not change the results.
The loss of an epitope upon AR is a novel finding. Remarkably, these epitopes are recognized by antibodies raised against targets on live cells, such as the ones tested in flow cytometry or in in vivo immunotherapy. These epitopes may be discontinuous epitopes,27,28 while most epitopes detected in FFPE material are linear. 29 A hypothesis to be tested is that discontinuous epitopes, such as the ones recognized by antibodies against intact cells, are destroyed by high temperatures after being cross-linked by formaldehyde.
Antigen Masking Occurs When the Specimen Is Transferred to Hydrophobic Compounds
Our frozen section model allows monitoring the antigen availability during processing. We observed antigen masking during the passage from the alcohol into the clearing agent to the paraffin. This masking was additional and cumulative to the masking due to formalin fixation. 2 This latter effect is due to the substitution of ethanol with hydrophobic substances (clearing agents, paraffin). These latter are required to progressively remove ethanol and residual water. The resulting effects are changes of the availability of antigens in the tissue, because of the spatial disposition of newly introduced hydrophobic residues or loss of non-freezable water. 12
There are few but intriguing clues on what occurs upon transferring cross-linked antigen from alcohol to paraffin. Fowler et al. 8 processed a tissue surrogate made of purified proteins and agar and observed a considerable increase of the presence of large oligomers upon transfer to the paraffin. They also were able to distinguish the enhanced cross-linking effect of FA fixation in the presence of high concentration of proteins from the postfixation molecular changes. 8 The masking effect upon transfer to paraffin seems to affect only FA-fixed material, as it is not observed with sections fixed in Carnoy. The most likely mechanism may be the creation of large multimeric complexes, where the intermolecular cross-links have a major masking effect, compared with the effect on immunoreactivity of intramolecular bonds in smaller complexes. 7
Another explanation of the mechanism leading to further masking during processing is the progress of intra- and intermolecular cross-linking during the process, favored by high temperatures. 8 We have to consider that our sections were soaked in TBS, an organic buffer able to bind to and block residual reactive FA-induced residues. Furthermore, processing the section in xylene at 60C modestly and heterogeneously affected the immune availability (see Supplemental Fig. S7) of the epitopes.
These changes and the fixation-dependent masking are reversed by the heat-based AR process.
Transfer of the Specimen to Alcohols Does Not Result in Antigen Masking
An intriguing observation is the enhancing immunostaining effect observed in IHC, but not in immunofluorescence, after the alcohol step, mimicked by the use of detergents.
Reports about an immunodetection enhancing effect of alcohols in combination with a cross-linking fixative are very scarce and applied to a different technique, flow cytometry. 30 Alcohols are usually used as the sole fixative, usually on cultured cells, 31 in alternative to a more common short combined formaldehyde-detergent fixation. Alcohol may act in this scenario as a denaturant, because of its ability to dissociate. 13
We tested, in comparison with ethanol, the effect of detergents such as Tween-20 and SDS, widely used purposely for cell membrane permeabilization on cultured cells and frozen sections. SDS and Tween-20 indeed unmask the FA-fixed antigen, as previously shown on FFPE sections. 16 Fixation with a cross-linking fixative is enough to provide ample antibody access to the cell structure, including the nucleus. 32 Detergents and alcohol may enhance the immunoreactivity not by permeabilizing the tissue as previously reported but by altering the protein structure and consequently the epitope spatial disposition by controlled denaturation. Lipid and protein extraction may be a mechanism by which alcohol or detergents reexpose an antigen.
The fact that the effect was noticed only with the use of a polymer as detection system, several orders of magnitude larger in size than a fluorochrome-conjugated antibody, points, however, at a more coarse mechanism, involving the macrostructure of the tissue, not the antigen itself. In other words, alcohols and detergents may allow more or larger polymer molecules to bind to the primary antibodies bound to the antigens.
The frozen section model we have used to dissect the effect of many variables during tissue processing may not be fully superimposable to a surgical specimen. We assumed that the thickness of the specimen (5 µm) would eliminate the penetrance factor during processing. However, a section affixed to a glass slide is very asymmetrical in terms of access and exchange of reagents, as demonstrated by the comparison of staining by free diffusion or forced microfluidic flow. 33
Another difference we noticed is that stress-stripping, 15 a 2-hr, 60C FA-fixed frozen section, with or without AR, leads to destruction and loss of the tissue (not shown), while an FFPE is not modified. Thus, the whole process and most likely FA-induced bonds plus dehydration in the whole tissue introduce modifications which are only partially reversed by AR, with the double bonus of AR and tissue stabilization.
Footnotes
Acknowledgements
We wish to thank Ms. Rossella Gendusa, Sara Malachina, Loredana Tusa, and Antonella Musarò for technical help, Dr. Franco Ferrario for continuous support, Dr. Dario Cerri (Central Pharmacy Services, San Gerardo) for providing fixatives samples, and Dr. Fabio Rossi (Blood Transfusion Laboratory) for providing essential reagents.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
GC, CRS, and GB designed the experiments. BEL and LD provided essential material. GB, LR, LD, FMB, and CRS performed immunohistochemical tests and histopathology preparations. GC, SR, MM, LD, FMB, MMB, and GB scored the IHC preparations. GC, MM, and SR wrote the manuscript. All authors have read and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: C.R.S. and M.M.B. are funded by a GlaxoSmithKline clinical research with the Università di Milano-Bicocca (HGS 1006-C1121 / BEL114054) and by the Fondazione per la Ricerca Scientifica Termale (FoRST), IV call grants (Project “Lymphopoiesis in Secondary Lymphoid Tissue”). F.M.B. is funded by the MEL-PLEX research training program (“Exploiting MELanoma disease comPLEXity to address European research training needs in translational cancer systems biology and cancer systems medicine,” Grant Agreement No. 642295, MSCA-ITN-2014-ETN, Project Horizon 2020, in the framework of the Marie Skłodowska-Curie Actions). This project has been supported by Departmental University of Milano-Bicocca and Hospital funds.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.