
Abstract
The adenomatous polyposis coli (APC) gene is a known tumor suppressor gene, and mice with mutations in Apc (ApcMin/+) spontaneously form multiple intestinal neoplasms. In this model of human colorectal cancer (CRC), it has been reported that CD4+ T-cell-derived interleukin 17 (IL-17) promotes intestinal tumor development, but it is not known if the Apc mutation actually directly alters T-cell function and subsequently tumor immunosurveillance. To investigate the ApcMin/+ mutation on T-cell function, flow cytometric, histochemical, and immunofluorescent studies on both wild-type (Apc+/+) and ApcMin/+ mice were performed. We identified decreased levels of interferon gamma (IFN-γ+)IL-17+ double-positive CD4+ cells in the mesenteric lymph nodes and Peyer’s patches of ApcMin/+ mice. In addition, altered levels of CD8+ cells, and changes in CD8+ production of IFN-γ and granzyme B were observed. These T-cell alterations did modify tumor immunosurveillance, as the adoptive transfer of splenocytes from ApcMin/+ animals into a chemically induced CRC model resulted in the inability to prevent epithelial dysplasia. These results suggest an altered T-cell balance in ApcMin/+ mice may disrupt intestinal homeostasis, consequently limiting intestinal tumor immunosurveillance.
Keywords
Introduction
Colorectal cancer (CRC) is the third most common type of cancer in the United States, affecting both males and females, as well as a variety of races and ethnic groups. 1 Early onset (age 35–40) of CRC can occur in individuals with an autosomal dominant mutation in the adenomatous polyposis coli (APC) gene. 2 The APC gene is a known tumor suppressor gene that mediates β-catenin destruction and cytoskeletal rearrangements in intestinal epithelial cells. Loss of APC results in increased cellular proliferation, decreased migration, and defective mitosis, all of which greatly increase the risk of cancer in the highly proliferative environment of the intestinal epithelium. 3 In humans, the inherited condition known as familial adenomatous polyposis (FAP) is due to a mutation in the APC gene and is characterized by the presence of adenomatous polyps occurring throughout the colon, which later develop into CRC.2,4
Inactivation of the Apc gene, the mouse homologue of human APC, in C57BL/6 mice has yielded a laboratory model of human FAP. 5 This mutation of the murine Apc gene leads to the formation of multiple intestinal neoplasia (Min) in the small intestine and colon of the heterozygous mutant mice; thus, these animals are known as ApcMin/+, whereas their wild-type littermates are identified as Apc+/+. ApcMin/+ mice develop approximately 30 visible tumors, 1 to 8 mm in size, in both the small intestine and colon, with little to no metastasis, and typical survival is approximately 120 days. 5 ApcMin/+ mice are also known to develop splenomegaly, a result of increased hematopoiesis, particularly of splenic hematopoietic cells and megakaryocytes, 6 as well as thymic atrophy, a result of reduced mesenchymal progenitor cells in the bone marrow, which leads to decreased thymic T cells and splenic natural killer and immature B cells. 7
The impact of the Apc mutation on intestinal epithelial cell proliferation, migration, and division has been well studied and is clearly the primary underlying cause of the development of FAP. However, several immune deficiencies have also been described in the ApcMin/+ mouse, suggesting that a failure of the immune system to control epithelial cell tumorogenesis may also contribute to the development and progression of tumor growth.6-8 Of note, it has been described that ApcMin/+ T-cell-derived interleukin 17 (IL-17) promotes tumor progression, and cultured ApcMin/+ CD4+ T cells produce less interferon gamma (IFN-γ), suggesting an important role of the immune response in controlling the Apc mutation. 8
Although the role of IL-17 and T helper 17 (Th17) cells has been investigated in the ApcMin/+ model of FAP, the role of IFN-γ-secreting CD4+ and CD8+ T cells remains unclear. The antitumor response of CD4+ IFN-γ-secreting Th1 cells and IFN-γ has been shown to be important in protection from other models of CRC, and from disease in human patients.9,10 This Th1 antitumor response has been shown to increase CD8+ cytotoxic T-cell influx into tumors, 9 and CRC patients with higher levels of Th1 cells in CRC tumors have prolonged survival. 10 In addition, Th1-derived IFN-γ increased CRC tumor cell apoptosis when delivered in conjunction with oxaliplatin 11 or ionizing radiation therapy. 12 Finally, murine studies have shown that a shift from a Th1 to a Th2 response promotes tumor progression in CRC.13,14 Interestingly, IFN-γ+IL-17+ CD4+ cells have been described and are thought to be able to further develop into full Th1 or Th17 cells depending on the local environment. 15 In contrast to Th1 cells, Th17 cells have been shown to increase colonic tumor progression.13,14 Recently, it has been demonstrated that in the ApcMin/+ model of CRC, there is an altered microenvironment that appears to modify the differentiation program of wild-type bone marrow immune cells, but immune cells derived from ApcMin/+ mice were not extensively evaluated. 16 These previous studies highlight the importance of an effective Th1 antitumor response to prevent or suppress tumor development in the context of CRC. 14 However, as the full IFN-γ/Th1/CD8+ response has not yet been investigated in the ApcMin/+ model of CRC, we designed a series of experiments using flow cytometric and immunofluorescent methods to further explore the role of the ApcMin/+ mutation on CD4+ and CD8+ T-cell function. This included an investigation of the levels of IFN-γ-producing CD4+ and CD8+ T cells both in vivo and in an ex vivo cell culture system. These studies revealed several important observations, most notably that ApcMin/+ mice have decreased levels of IFN-γ+IL-17+ double-positive CD4+ cells and decreased levels of total CD8+ cells and IFN-γ+granzyme B (GnzmB)+ double-positive CD8+ cells. This suggests that in addition to epithelial cell alterations and the protumor Th17 environment caused by the Apc mutation, the decreased IFN-γ-producing antitumor T cells may also contribute to the development of CRC in this model.
Materials and Methods
Animals
C57BL/6.Apc+/+ (Apc+/+) littermate controls and C57BL/6.ApcMin/+ (ApcMin/+) as originally described by Moser et al. 5 were purchased from Jackson Laboratory (Bar Harbor, ME). Male and female Apc+/+ and ApcMin/+ mice were used at approximately 100 days old. C57BL/6.Rag1−/− mice were purchased from Jackson Laboratory at 5 weeks of age, maintained in our mouse colony for 2 weeks, and then used for adoptive transfer/colon cancer induction experiments. All mice were bred and maintained under specific-pathogen-free (SPF) conditions in Thoren Isolator racks (Hazleton, PA) under positive pressure. The number of mice used for each experiment is indicated in the figure legends. The Institutional Care and Use Committee of the University of Alabama at Birmingham (UAB) approved all experiments. SPF conditions at UAB include absence of the following organisms, as determined by serological screening: mouse parvoviruses, including MPV-1, MPV-2, and minute virus of mice; mouse hepatitis virus, murine norovirus, Theiler’s murine encephalomyelitis virus; mouse rotavirus (epizootic diarrhea of infant mice); Sendai virus; pneumonia virus of mice; reovirus; Mycoplasma pulmonis; lymphocytic choriomeningitis virus; mouse adenovirus; ectromelia (mousepox) virus; K polyomavirus; and mouse polyomavirus. Testing and other methods were as described at http://www.uab.edu/research/administration/offices/ARP/ComparativePathology/SupportServices/Pages/HealthSurveillance.aspx.
Cell Isolation
Spleen, mesenteric lymph nodes (MLNs), and Peyer’s patches (PPs) were removed and a single-cell suspension prepared by mechanical disruption. Small intestinal and colonic tissue was collected and cells isolated using the Miltenyi Biotec Mouse Lamina Propria Dissociation Kit (Miltenyi Biotec, Auburn, CA) following the manufacturer’s protocol. Briefly, PPs and fat were removed; the intestinal/colonic tissue was then opened longitudinally and cut into 0.5-cm pieces. To isolate small intestinal intraepithelial lymphocytes (IELs), the intestinal tissue was then incubated 2 × 30 min at 37C with gentle shaking in HBSS containing 5% newborn calf serum (NCS; HyClone, Logan, UT), 5-mM EDTA (Sigma-Aldrich, St. Louis, MO), and 1-mM dithiothreitol (Sigma-Aldrich). Media containing IELs was collected, washed, and further purified and collected at the interface of a 40%/75% Percoll (GE Healthcare Life Sciences, Fairfield, CT) gradient. The remaining tissue was then incubated with the components of the Miltenyi Biotec Lamina Propria Dissociation Kit in HBSS containing Ca2+ and Mg2+ and 5% NCS for 30 min at 37C with gentle shaking. After incubation, tissue was dissociated using the m_intestine_01 program on the Miltenyi Biotec gentleMACS Dissociator, releasing lamina propria cells. Cells were washed, and further purified and collected at the interface of a 40%/75% Percoll gradient. Cells were washed in PBS supplemented with 2.5% NCS and prepared for downstream experiments.
Cell Separation
Splenic CD4+CD62L+ naïve T cells were isolated via MACS (Miltenyi Biotec) magnetic beads following the manufacturer’s protocol. Briefly, CD4+ cells were negatively selected, followed by positive selection of CD62L+ cells. Splenic CD8+ T cells were isolated via BD Biosciences Mouse CD8 T Lymphocyte Enrichment Set–DM (BD Biosciences, San Jose, CA) following the manufacturer’s protocol. Briefly, the CD8+ cells were negatively selected, and the CD8+ cells were used in further experiments.
T-Cell Polarization and Culture
Splenic CD4+CD62L+ cells were isolated and separated as described above. An amount of 1 × 105 freshly separated cells were then added to each well of a 96-well flat-bottom plate coated with anti-CD3 (1 µg/ml; Table 1) and anti-CD28 (0.5 µg/ml) in 200-µl Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal calf serum (HyClone), 100 IU/ml penicillin (Mediatech, Manassas, VA), 100 µg/ml streptomycin (Mediatech), GlutaMAX (Thermo Fisher Scientific, Carlsbad, CA), and 5-µM 2-mercaptoethanol (Sigma-Aldrich) for 72 hr at 37C in various polarization conditions—Th0: media only; Th1: 10-µg anti-IL-4 (clone 11B11) and 10 ng/ml recombinant mouse (rm) IL-12; Th17: 10 µg/ml anti-IFN-γ (clone XMG1.1), 10-µg anti-IL-4, 5 ng/ml recombinant human (rh) transforming growth factor beta (TGF-β), 10 ng/ml rmIL-1β and 25 ng/ml rmIL-6; and Th1/Th17: 10 µg/ml anti-IL-4, 10 ng/ml rmIL-12, 5 ng/ml rhTGF-β, 10 ng/ml rmIL-1β and 25 ng/ml rmIL-6. After 72 hr, supernatants were collected and cells were rested for 24 hr with IL-2 (10 ng/ml) before staining as described below.
Antibody Information.
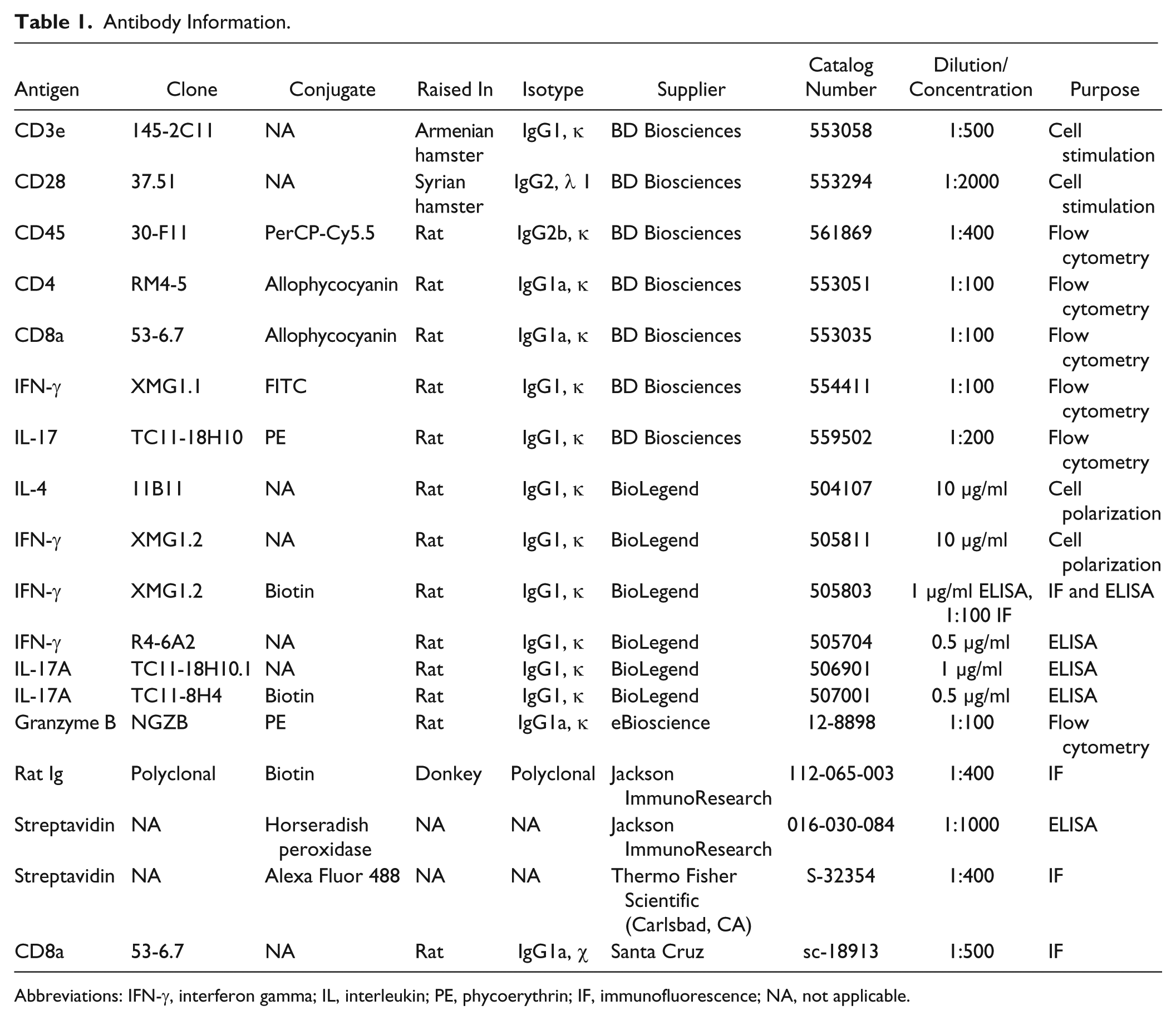
Abbreviations: IFN-γ, interferon gamma; IL, interleukin; PE, phycoerythrin; IF, immunofluorescence; NA, not applicable.
Splenic CD8+ cells were isolated as described above. An amount of 1 × 105 freshly separated CD8+ cells were added to a 96-well flat-bottom plate coated with anti-CD3 (1 µg/ml) and anti-CD28 (0.5 µg/ml) for 72 hr at 37C with 10 ng/ml IL-2. After 72 hr, supernatants were collected and cells were rested with fresh media and IL-2 before staining as described below. All cytokines and antibodies were purchased from BioLegend (San Diego, CA), except for anti-CD3 and anti-CD28, purchased from BD Biosciences.
Flow Cytometry Staining
For cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (750 ng/ml) (Sigma-Aldrich) in the presence of GolgiStop (BD Biosciences) for 5 hr before staining. Cells isolated from spleen, MLN, PPs, small intestinal lamina propria (SILP), colonic lamina propria (CoLP), and small intestinal IEL compartments were stained with CD45 PerCP-Cy5.5 (clone 30-F11; BD Biosciences; Table 1), anti-CD4 allophycocyanin (clone RM4-5; BD Biosciences), or CD8α allophycocyanin (clone 53-6.7; BD Biosciences). Following cell surface staining, cells were permeabilized with the eBioscience (San Diego, CA) Foxp3 Staining Buffer Kit and stained with anti-IFN-γ FITC (clone XMG1.1; BD Biosciences), anti-IL-17 PE (clone TC11-18H10; BD Biosciences), or GnzmB PE (clone NGZB; eBioscience). All samples were then run on a BD FACSCalibur flow cytometer and analyzed with FlowJo Software (Tree Star, Inc., Ashland, OR).
Immunofluorescent Staining
Cytospins
Cytospins were performed at 700 rpm for 4 min, and cells were fixed in 100% ethanol at −20C for 1 hr. Nonspecific protein binding was blocked using 1% BSA, 0.3% Triton X-100 (Thermo Fisher Scientific, Carlsbad, CA), and 0.2% powdered milk in PBS for 15 min at room temperature. Endogenous biotin was blocked using a biotin blocking kit: avidin for 15 min at room temperature followed by biotin for 15 min at room temperature (Vector Laboratories, Burlingame, CA). All staining was done in 1% BSA and 0.2% powdered milk in PBS. Cells were then stained using biotinylated anti-IFN-γ (clone XMG1.1, 1:100 dilution; BioLegend) overnight at 4C and visualized with Streptavidin, Alexa Fluor 488 (Thermo Fisher Scientific, Carlsbad, CA) for 60 min at room temperature in the dark. Sections were then counterstained with Hoechst H33258 (Thermo Fisher Scientific, Carlsbad, CA) for 10 min at room temperature in the dark. Images were captured using a Zeiss Axio Scope and Zeiss AxioCam with AxioVision 4.7 software (Carl Zeiss, Inc., Peabody, MA).
Frozen
Splenic frozen sections were prepared by fixation with 2% paraformaldehyde for 2 hr followed by 3 days in 20% sucrose at 4C and embedded in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA). Endogenous peroxidases were blocked with 0.3% H2O2 for 10 min at room temperature, and nonspecific protein binding was blocked using 1% BSA, 0.3% Triton X-100, and 0.2% powdered milk in PBS for 15 min at room temperature. Endogenous biotin was blocked using a biotin blocking kit: avidin for 15 min at room temperature followed by biotin for 15 min at room temperature (Vector Laboratories). All staining was done in 1% BSA and 0.2% powdered milk in PBS. Sections were stained overnight at 4C using rat anti-mouse CD8α (clone 53-6.7, 1:100 dilution; Santa Cruz, Dallas, TX) and visualized with biotinylated donkey anti-rat Ig (1:100 dilution; BD Biosciences) for 60 min at room temperature followed by Alexa Fluor 488–conjugated streptavidin (1:400 dilution; Thermo Fisher Scientific, Carlsbad, CA) for 60 min at room temperature. Sections were then counterstained with Hoechst H33258 for 10 min at room temperature. Images were captured using a Zeiss Axio Scope and Zeiss AxioCam with AxioVision 4.7 software (Carl Zeiss, Inc.)
Elisa
Cell culture levels of IFN-γ and IL-17A were measured using ELISAs. Briefly, Immulon (Thermo Scientific, Rockford, IL) 96-well plates were coated with capture antibody (0.5 µg/ml IFN-γ: clone R4-6A2; 1 µg/ml IL-17A: clone TC11-18H10.1) at 4C overnight. Plates were then washed with 0.05% Tween 20 (Thermo Fisher Scientific, Carlsbad, CA) in PBS and blocked with 10% NCS in PBS. After washing, samples or known standards were added and the plates incubated for 2 hr at room temperature. Standards consisted of seven known dilutions and a blank, all run in triplicate (IFN-γ standard started at 5 µg/ml and diluted 1:2, IL-17A standard started at 2.5 µg/ml and diluted 1:2). Samples were washed off, and a biotinylated detection antibody (1 µg/ml IFN-γ: clone 1 µg/ml XMG1.2; 0.5 µg/ml IL-17A: clone TC11-8H4) was added and incubated at room temperature for 1 hr. Plates were washed, and horseradish peroxidase–conjugated streptavidin was added for 30 min at room temperature. Plates were washed, and 3,3′,5,5′-tetramethylbenzidine (Thermo Scientific) was added and developed for 15 min in the dark. The reaction was stopped by the addition of 0.05-M H2SO4. After development, the plates were read on a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm. All capture and detection antibodies were purchased from BioLegend. Results were analyzed using SoftMax (Molecular Devices) software.
Proliferation Assay
CD4+CD62L+ and CD8+ cells were isolated as described above. An amount of 1 × 105 freshly separated cells were added to each well of a 96-well flat-bottom plate coated with anti-CD3 (1 µg/ml) and anti-CD28 (0.5 µg/ml) in 200-µl RPMI supplemented with 10% fetal calf serum (HyClone), 100 IU/ml penicillin (Mediatech), 100 µg/ml streptomycin (Mediatech), GlutaMAX (Thermo Fisher Scientific, Carlsbad, CA), and 5-µM 2-mercaptoethanol (Sigma-Aldrich). Cells were cultured for 72 hr at 37C (CD8 culture contained 10 ng/ml IL-2). After 72 hr, the RayBiotech Quick Cell Proliferation Assay was performed. Briefly, culture volume was adjusted to 100 µl/well, and 10-µl water-soluble tetrazolium salt reagent was added. Cells were incubated for 210 min followed by 1 min of shaking. Absorbance was measured at 450 nm, using 650 nm as the reference wavelength on a VersaMax microplate reader (Molecular Devices). Results were analyzed using SoftMax (Molecular Devices) software.
Adoptive Transfer and AOM/DSS Induction of Colon Cancer
Splenocytes from male Apc+/+ and ApcMin/+ donors (approximately 8 weeks old) were isolated into single-cell suspensions as described above. Red blood cells were lysed using a 9:1 mixture of 140-µM NH4Cl and 17-mM Tris. Cells were washed in PBS and resuspended at 2 × 107 cells/ml in sterile PBS. Two hundred fifty µl (5 × 106 cells) was injected intraperitoneally (i.p.) into male C57BL/6.Rag1−/− recipient mice (n=5 for each donor population) 2 days before the start of azoxymethane/dextran sulfate sodium (AOM/DSS) treatment (day −2). On day 0, C57BL/6.Rag1−/− recipient mice were injected i.p. with 10 mg/ml of 0.5 mg/ml AOM (Sigma-Aldrich). Two percent DSS (Alfa Aesar, Ward Hill, MA) was administered in the drinking water on days 5 to 9, 26 to 30, and 47 to 51, with regular drinking water in between DSS treatments. Recipient mice were sacrificed on day 73 and tissue prepared for histological analysis (below).
Tumor Counts and Tissue Preparation
After sacrifice, longitudinal strips of the ileum and colon (proximal and distal) were pinned to dental wax, with the luminal side facing up. The ileum and colon were then rinsed with 1× PBS and placed in 10% formalin fixative (Thermo Fisher Scientific, Waltham, MA) for 24 hr. After 24 hr, the formalin was removed and a 0.05% methylene blue stain (Sigma-Aldrich) was added to the intestinal segments. Samples were rocked in methylene blue for 1 min to ensure equal distribution of the stain. The tissue was then rinsed three times in 70% ethanol, while on the rocker, for 3 min per rinse. The tissue was then viewed under a Zeiss Stemi 2000-C dissecting microscope to determine the number of tumors within each tissue segment. Tumors were identified by a greater retention of methylene blue compared with surrounding tissue. After a microscopic tumor count, the intestinal segments were fixed and embedded in paraffin. Tissue was cut into 5-µm sections and stained with standard hematoxylin and eosin for histological examination. Experimental conditions were concealed until after the slides were examined. For each colonic segment, crypt epithelial hyperplasia, goblet cell loss, superficial and crypt epithelial degeneration and loss, crypt exudate, inflammatory cell accumulation in lamina propria and submucosa, submucosal edema, mucosal ulceration, transmural inflammation, fibrosis, and dysplasia were evaluated. Severity of each change was scored 0, 1, 2, or 3 for absent (normal), mild, moderate, and severe, respectively. The distribution of each change present also was scored 1, 2, 3, or 4 for ≤25%, 25% to 50%, 50% to 75%, or 75% to 100%, respectively, of the segment affected. Lesion scores for each segment were calculated as the sum of severity scores multiplied by distribution scores, with changes indicating severe inflammation or injury, including crypt epithelial degeneration, ulceration, transmural inflammation, and dysplasia, weighted by a factor of 1. An overall colonic lesion score was calculated as the average of the scores for the two colonic segments.
Statistical Analysis
Statistical significance was evaluated by Student’s t test. (Graph Pad QuickCalcs, La Jolla, CA). Error bars represent standard deviation.
Results
Decreased Percentages of IFN-γ+IL-17+ CD4+ Cells in ApcMin/+ Intestinal Tissue
To begin the investigation of intestinal Th1 cells in ApcMin/+ mice, cells were harvested from spleen, MLN, PPs, SILP, CoLP, and IEL. IFN-γ-secreting Th1 and IL-17A-secreting Th17 cells were identified after treatment with PMA, ionomycin, and GolgiStop via flow cytometry (Fig. 1A). ApcMin/+ mice were observed to have splenomegaly (Supplemental Table 1); however, no significant changes were observed in the percentage of total CD4+ T cells or in Th1 or Th17 percentages between Apc+/+ and ApcMin/+ littermates. There was a significant decrease (p≤0.01) in the percentage of IFN-γ+IL-17A+ double-positive CD4+ T cells in both the PPs and IEL compartments (Fig. 1A and B).
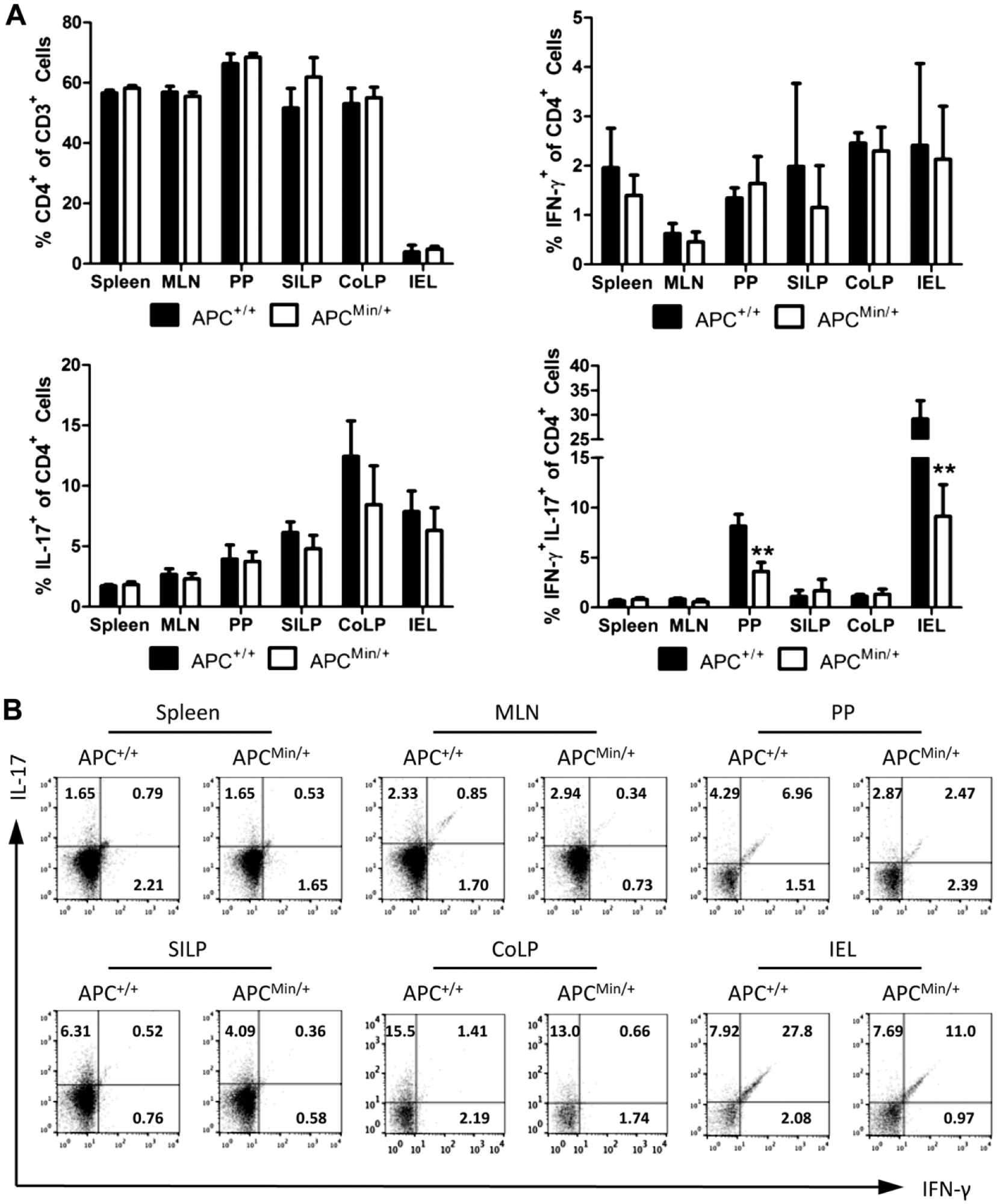
Decreased levels of IFN-γ+IL-17+ double-positive CD4+ T cells in the intestines of ApcMin/+ mice. (A) Percentages of total CD3+CD4+ T cells, IFN-γ+ single-positive, IL-17+ single-positive, and IFN-γ+IL-17+ double-positive CD4+ T cells from spleen, MLN, PPs, SILP, CoLP, and small intestinal IEL of Apc+/+ and ApcMin/+ mice measured by flow cytometry. (B) Representative flow cytometry plots of the IFN-γ versus IL-17 staining. Cells were gated on lymphocytes (based on forward and side scatter), CD45, and CD4. n=4 mice per group. Representative of two independent experiments. **p≤0.01. Abbreviations: MLN, mesenteric lymph node; PPs, Peyer’s patches; SILP, small intestinal lamina propria; CoLP, colonic lamina propria; IEL, intraepithelial lymphocyte; IFN-γ, interferon gamma; Apc, adenomatous polyposis coli; IL-17, interleukin 17.
Disrupted Polarization of Th1 Cells in ApcMin/+ Cultures
We next sought to investigate the ability of the IFN-γ+IL-17+ cells to be polarized to either an antitumor Th1 cell or a protumor Th17 cell. Naïve splenic CD4+CD62L+ cells were isolated and cultured in the presence of various T-cell-polarizing cytokines and antibodies to generate Th1, Th2, Th17, or Treg cells. In addition, a combination of Th1/Th17 cytokines was used to determine the polarization of CD4 T cells in an environment of competing conditions. There were no changes in the percentages of IFN-γ+ or IFN-γ+/IL-17+ double-positive cells after culture, regardless of the T-cell-polarizing conditions (Fig. 2A). As has been previously reported, increased levels of IL-17A cells were observed in the Th0, Th1, and Th2 ApcMin/+ cultures (Fig. 2A, p≤0.05). 8 However, upon the analysis of the ratio of Th1 cells to Th17 cells, it became clear that there was an inhibition of Th1 generation in both Th0 and Th1 cultures (Fig. 2A, p≤0.01). Although no significant in vitro changes in IFN-γ or IL-17A cytokine secretion were measured via ELISA (Fig. 2B), the decrease in Th1 polarization was confirmed by immunofluorescence analysis of IFN-γ on a cytospin of cultured CD4+ cells (Fig. 2C).
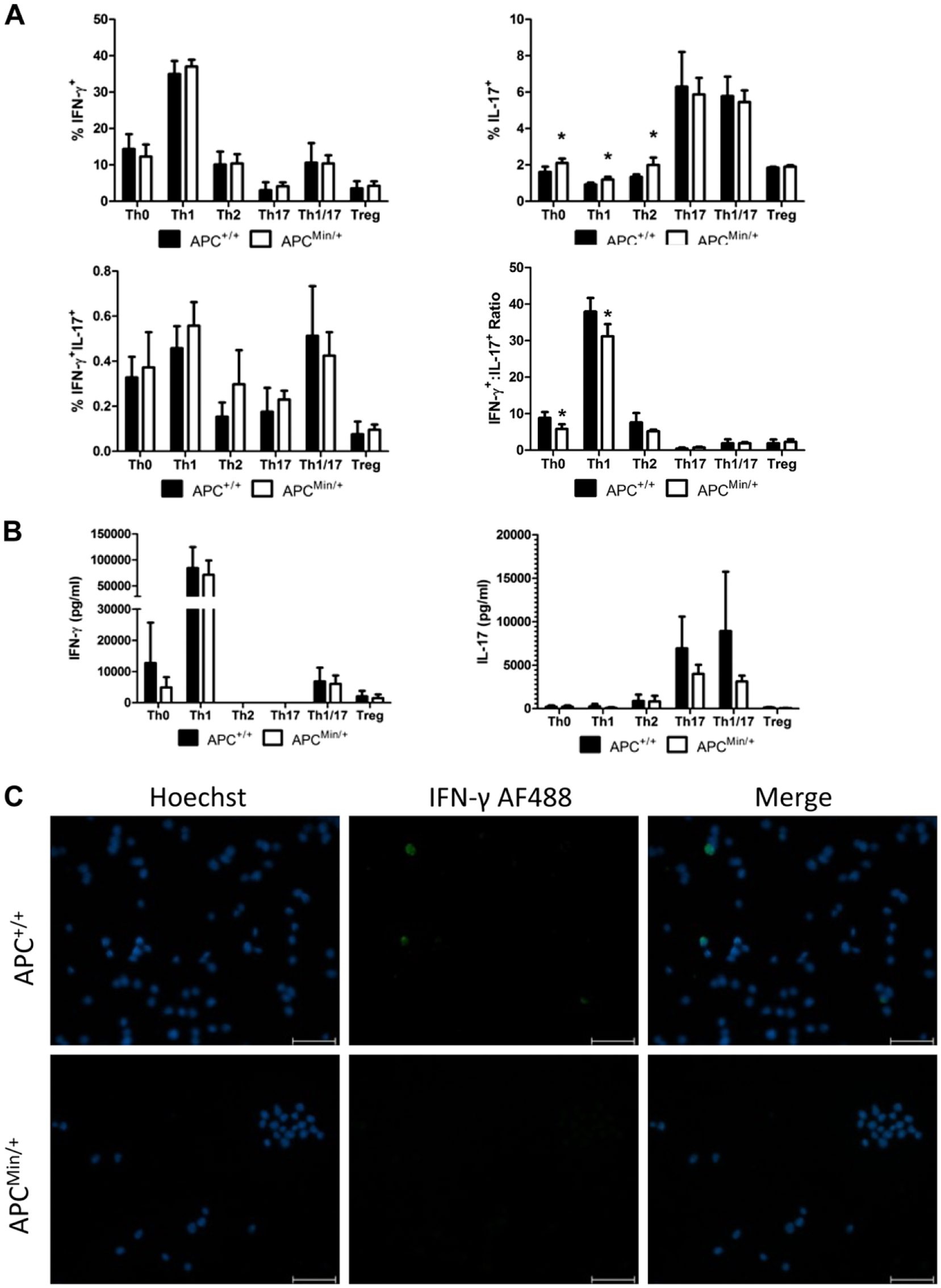
In vitro cell polarization reveals increased Th17 polarization and decreased IFN-γ/IL-17 ratio. Splenic CD4+CD62L+ cells were isolated and activated for 72 hr with anti-CD3 and anti-CD28 stimulation in the presence of helper T-cell-polarizing cytokines and rested for an additional 24 hr in the presence of IL-2. (A) Flow cytometric analysis of IFN-γ and IL-17 expression in cultured cells after stimulation with phorbol 12-myristate 13-acetate and ionomycin in the presence of GolgiStop. (B) ELISA analysis of IFN-γ and IL-17 levels from the supernatants of cultured cells harvested after the first 72 hr of culture. (C) Cytospin of cultured cells and subsequent immunofluorescent staining of IFN-γ. Scale bar = 50 µm. n=4 mice per group. Representative of two independent experiments. *p≤0.05. Abbreviations: Th17, T helper 17; IFN-γ, interferon gamma; IL, interleukin; Apc, adenomatous polyposis coli.
Alteration in CD8+ Cells in ApcMin/+ Mice
To continue the analysis of T cells in the ApcMin/+ mouse, CD8+ cells were also analyzed via flow cytometry. After PMA, ionomycin, and GolgiStop treatment, cells isolated from spleen, MLN, PPs, SILP, CoLP, and IEL were stained for CD8 and the traditional CD8 effector molecules IFN-γ and GnzmB, both of which are involved in antitumor responses. Decreased percentages (p≤0.05) of CD8+ cells were detected in both the spleen and SILP of ApcMin/+ mice, indicating a diminished antitumor CD8+ population in vital organs (Figs. 3A and B). In addition, those CD8+ cells in the SILP of ApcMin/+ mice seem to produce GnzmB, without the production of IFN-γ (Fig. 3A). A much lower percentage of IFN-γ+GnzmB+ cells were detected in the PPs of ApcMin/+ animals. Because CD8+ cells expressing both IFN-γ and GnzmB are thought to generate a more robust response, this lack of IFN-γ+GnzmB+ CD8+ cells could significantly impact immune tumor surveillance in this model. These data are supported by ex vivo culture of CD8+ cells and the corresponding decrease in IFN-γ+GnzmB+ cells observed by flow cytometry after 72-hr culture with PMA/ionomycin stimulation, with no difference in IFN-γ secretion as measured by ELISA (Fig. 4A and B).
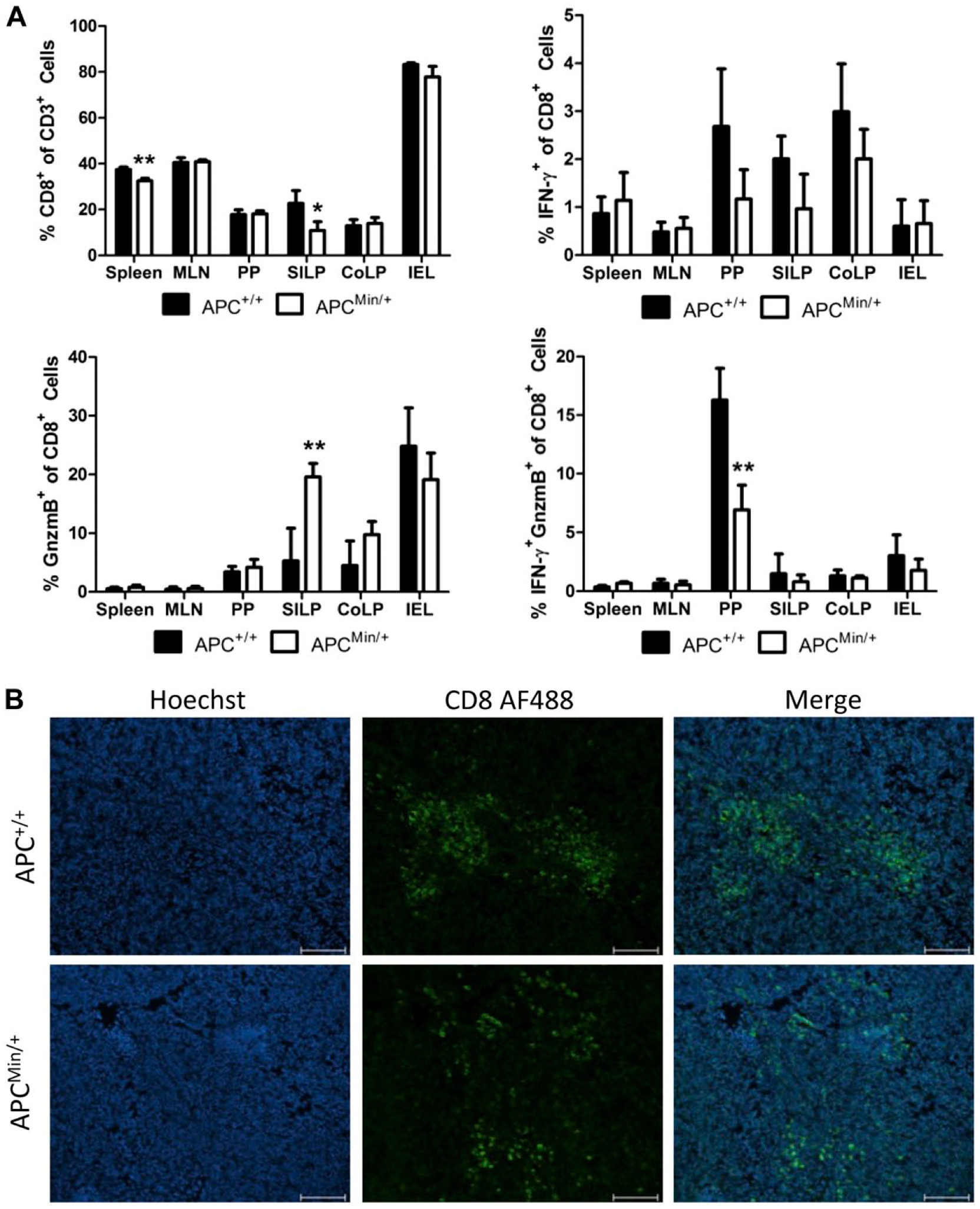
Decreased levels of splenic and small intestinal CD8+ cells and decreased CD8+IFN-γ+GnzmB+ cells in PPs in ApcMin/+ mice. (A) Percentages of total CD3+CD8+ T cells, IFN-γ+ single-positive, GnzmB+ single-positive, and IFN-γ+GnzmB+ double-positive CD8+ T cells from spleen, MLN, PPs, SILP, CoLP, and IEL of Apc+/+ and ApcMin/+ mice measured by flow cytometry. (B) CD8 immunofluorescent staining of paraformaldehyde/sucrose–fixed splenic tissue from Apc+/+ and ApcMin/+ mice. Scale bar = 100 µm. n=4 mice per group. Representative of two independent experiments. *p≤0.05, **p≤0.01. Abbreviations: IFN-γ, interferon gamma; GnzmB, granzyme B; Apc, adenomatous polyposis coli; MLN, mesenteric lymph node; PPs, Peyer’s patches; SILP, small intestinal lamina propria; CoLP, colonic lamina propria; IEL, intraepithelial lymphocyte.
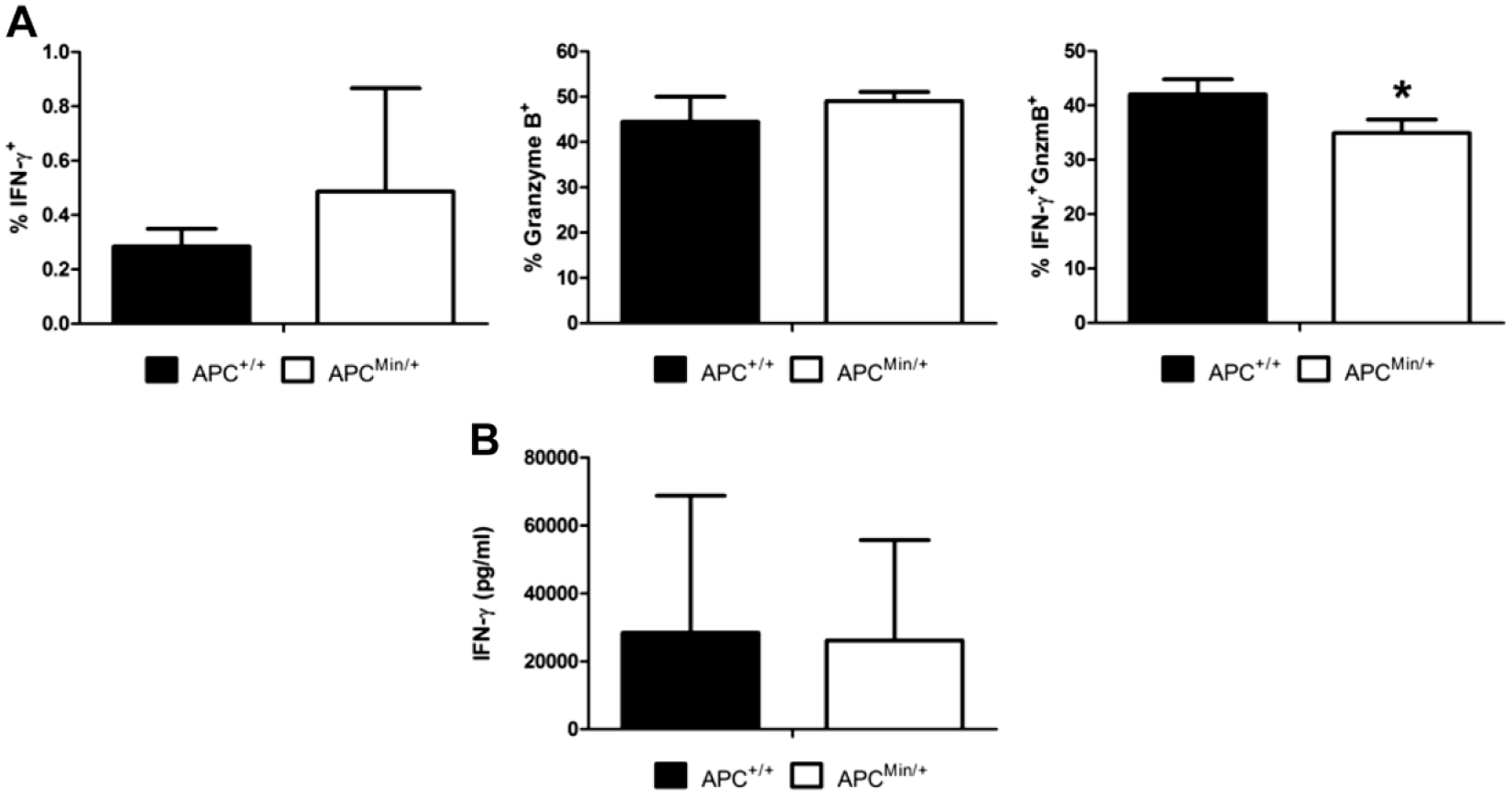
Low levels of in vitro production of IFN-γ+GnzmB+ in ApcMin/+ splenic CD8+ cells. Splenic CD8+ cells were isolated and activated for 72 hr with anti-CD3 and anti-CD28 stimulation in the presence of IL-2 and rested for an additional 24 hr with presence of IL-2. (A) Flow cytometric analysis of IFN-γ and GnzmB production from cultured cells after stimulation with phorbol 12-myristate 13-acetate and ionomycin in the presence of GolgiStop. (B) ELISA analysis of IFN-γ levels from the supernatants of cultured cells harvested after the first 72 hr of culture. Abbreviations: IL-2, interleukin 2; IFN-γ, interferon gamma; GnzmB, granzyme B; Apc, adenomatous polyposis coli.
Change in Proliferation of ApcMin/+ T Cells
The change in polarization of both CD4+ and CD8+ T cells in ApcMin/+ animals and cultures potentially generates an altered tumor surveillance response. However, although polarization is altered, proliferation in response to stimuli could restore the antitumor potential of both and CD8+ cells. Therefore, we next sought to investigate the proliferation potential of Apc+/+ and ApcMin/+ T cells in ex vivo culture. Cells were stimulated with plate-bound anti-CD3 (1 µg/ml) and anti-CD28 (0.05 µg/ml) for 72 hr to activate cells. After activation, the RayBiotech Quick Cell Proliferation kit was used to measure the proliferation of the cell cultures. To mimic our CD4 T-cell polarization conditions, CD4+ T-cell proliferation was analyzed in the absence of exogenous IL-2, and ApcMin/+ CD4+CD62L+ cells were found to proliferate significantly (p≤0.05) more than the wild-type cells (Fig. 5A). Interestingly, CD8+ cells, which were cultured in the presence of IL-2 to mimic the conditions used above for cytokine secretion, showed significant decrease (p≤0.05) in their ability to proliferate relative to Apc+/+ littermates (Fig. 5B).
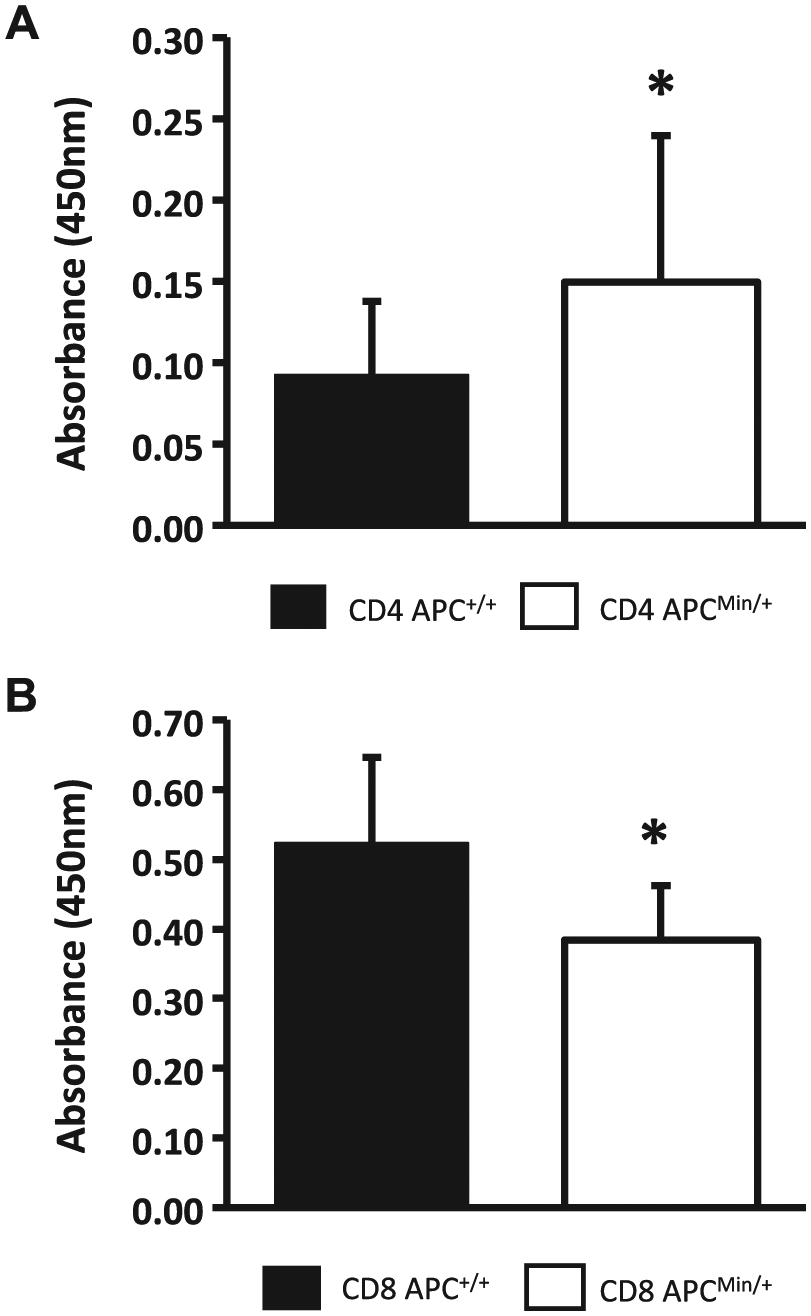
In vitro culture of CD4+CD62L+ cells and CD8+ cells leads to altered proliferation when stimulated with anti-CD3 and anti-CD28. Splenic T cells (CD4+CD62L+ or CD8+) were isolated via magnetic separation and cultured for 72 hr in the presence of 1 µg/ml plate-bound anti-CD3 and 0.5 µg/ml plate-bound anti-CD28. CD8 cells were additionally cultured in the presence of 10 ng/ml interleukin 2. (A) ApcMin/+ CD4+CD62L+ cells show increased proliferation when compared with Apc+/+ littermates. (B) CD8+ cells isolated from ApcMin/+ mice display decreased proliferation as compared with Apc+/+ littermates. Abbreviation: Apc, adenomatous polyposis coli.
Differential Response of ApcMin/+ Splenocytes to Colon Cancer in vivo
We next investigated whether the T-cell changes detected in vitro would translate to an altered response to colon cancer challenges in vivo. To test this, we devised a novel model of CRC utilizing an adoptive transfer protocol previously described by our laboratory,17,18 coupled with a well-described method for inducing colon cancer using AOM and DSS.19,20 Splenocytes were isolated from ApcMin/+ or wild-type Apc+/+ littermates, and 5 × 106 cells were i.p. transferred into immunodeficient C57BL/6.Rag1−/− recipient mice. Two days after transfer, the mice were injected with AOM, and then DSS was administered in drinking water at regular intervals (Fig. 6A). Animals were sacrificed 73 days after the AOM injection. After sacrifice, colonic tissue was spread open, fixed in formalin, and stained with methylene blue, and tissue was examined grossly for tumor formation. No obvious tumors were observed upon gross macroscopic observation in the recipients of either Apc+/+ or ApcMin/+ splenocytes, although there were minor surface epithelial irregularities in mice that had received ApcMin/+ splenocytes (Fig. 6B). However, upon microscopic examination, tissue damage was found to be much more profound in the recipients of ApcMin/+ splenocytes (Fig. 6C), and small microscopic neoplastic regions were identified in colon sections. Mice that had received Apc+/+ splenocytes only displayed minimal submucosal inflammation and edema, whereas four of five mice that had received ApcMin/+ splenocytes displayed microscopic colonic damage, including areas of inflammation, dysplasia, ulceration, and abnormal epithelium (Fig. 6C and D).
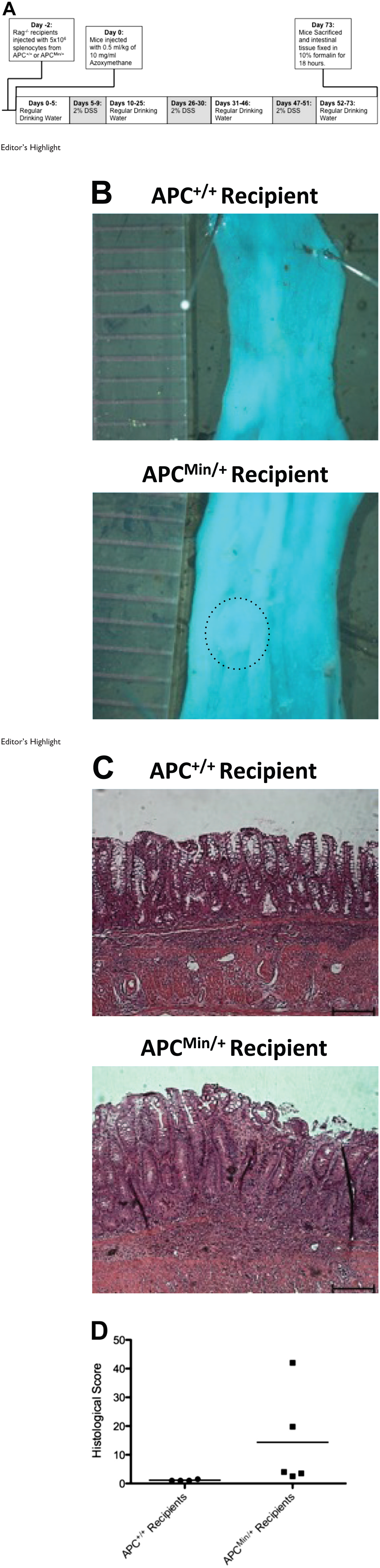
Adoptive transfer of ApcMin/+ splenocytes into Rag1−/− mice does not protect against the development of azoxymethane/dextran sulfate sodium (AOM/DSS)–induced colorectal cancer. (A) Schematic representing the adoptive transfer of Apc+/+ or ApcMin/+ splenocytes to Rag1−/− recipient mice and the induction of colorectal cancer using AOM and DSS. (B) Representative whole-mount images of distal colon sections from Rag1−/− recipient mice after the treatment protocol. Ruler indicates 1-mm divisions. (C) Representative hematoxylin and eosin staining of colon sections from Rag1−/− recipient mice after the treatment protocol. Scale bar = 100 µm. (D) Summary of the histological scoring of the Rag1−/− recipient mice after the treatment protocol. Abbreviation: Apc, adenomatous polyposis coli.
Discussion
The ApcMin/+ model of CRC has provided an excellent mechanism to study the role of epithelial alterations in the development of colon cancer, particularly human FAP. Deficiencies in Apc lead to several epithelial cell alterations, including increased cellular proliferation, decreased epithelial cell migration within the crypt, and altered mitosis. However, it has been unclear whether the ApcMin/+ mutation also impacts normal tumor immunosurveillance, rendering them less effective in controlling the increased rate of tumor formation in cells with Apc mutations. Our studies demonstrate that ApcMin/+ T cells have an altered immunological response, which hinders the ability of ApcMin/+ mice to mount the appropriate response to epithelial dysplasia and tumor progression. The mechanisms involved in this altered immunosurveillance in ApcMin/+ mice appear to include both altered T-cell polarization and proliferation. Although previous studies have implicated the role of Th17 cells in cancer development in these mice, our data also identify Th1 cells and CD8 cells as important players in this system.
Initial experiments revealed decreased percentages of IFN-γ+IL-17+ CD4+ cells in the PPs and IEL of the ApcMin/+ mice compared with wild-type littermates. These cells are known to further develop into either Th1 or Th17 cells depending on the cytokine milieu in the local environment. Previous studies have indicated that Th17 cells may promote colonic tumor development; however, numerous studies support the fact that Th1 cells promote an antitumor environment. A decrease in IFN-γ+IL-17+ cells in the ApcMin/+ intestinal tissue may therefore limit the ability of the animals to mount a Th1-based antitumor response, suggesting that animals with defective Apc genes are less equipped at mounting an effective immunological response to the development of cancerous cells. In addition, a decreased Th1/Th17 ratio in Th0- and Th1-polarized CD4+CD62L+ cultures was observed, further supporting the conclusion that ApcMin/+ animals have an immunological impairment preventing an appropriate tumor immunosurveillance response.
Although CD4+ T cells are necessary in coordinating the tumor immunosurveillance response, CD8+ cytotoxic T lymphocytes are vital in the actual destruction of tumorogenic cells. Not only were decreased levels of lamina propria CD8+ cells observed in ApcMin/+ small intestinal tissue, but those cells also displayed decreased levels of IFN-γ and GnzmB coexpression. Both IFN-γ and GnzmB are necessary for CD8+ cells to effectively eliminate potential cancerous cells. This decrease in CD8+ T cells also results in an increase in the CD4/CD8 ratio, which has been correlated with dysplastic intestinal mucosa in patients. 21 Thus, with a lower percentage of CD8+ cells, and less functionally effective CD8+ cells, tumorogenesis within the ApcMin/+ intestine would be much less inhibited, leading to increased formation of tumors and CRC.
Interestingly, experiments demonstrated an increase in ApcMin/+ CD4+CD62L+ proliferation in the absence of IL-2. This suggests that ApcMin/+ CD4+ T cells are better able to respond to stimuli, and when combined with our data demonstrating that under multiple differentiation conditions (Th0, Th1, or Th2) a higher percentage of cells produce IL-17, it indicates that more procancer Th17 cells would be available. Likewise, the ability of ApcMin/+ CD8+ cells to proliferate in the presence of IL-2 is inhibited, leading to fewer available antitumor CD8+ cells. Both of these results suggest mechanisms further limiting the ApcMin/+ animal’s response to tumor generation; however, additional studies are needed to study these mechanisms further.
These results have been supported by in vivo studies. Through the adoptive transfer of Apc+/+ or ApcMin/+ splenocytes into C57BL/6.Rag1−/− mice, and the subsequent chemical induction of colon cancer, our data suggest that mice receiving ApcMin/+ splenocytes are less protected from intestinal damage. Although gross tumor formation was not observed, increased inflammation and epithelial dysplasia in these mice suggest that they are more susceptible to formation of precancerous lesions, supporting the conclusions from the ex vivo studies.
Taken together, these data show that there are numerous immunological defects occurring as a result of Apc deficiencies in ApcMin/+ mice. Apc deficiencies are already known to increase the rate of colonic epithelial tumor formation. These additional defects suggest decreased tumor surveillance in animals with Apc mutations. Potential causes for these immunological defects may include changes in bone marrow hematopoietic stem cells, or a role in WNT signaling in T-cell differentiation. Because WNT signaling is known to be disrupted by Apc mutations, this may be the most likely cause. Increased epithelial tumorogenesis coupled with these newly described limitations in the antitumor immune response may help explain the accelerated formation of CRC within ApcMin/+ mice. These conclusions are supported by the recent findings of Barone et al., 16 which indicate that bone marrow–derived stem cells are involved in colon carcinogenesis in the ApcMin/+ model.
In patients suffering from FAP, one treatment option is to remove the colon to prevent the development of CRC. However, these patients are also at risk for other cancers, including desmoid, small intestinal, pancreatic, and papillary thyroid. 22 Our results indicating that animals with Apc mutations have altered antitumor immune responses provide an impetus for further research on the use of immune-modulating treatment strategies for the prevention of CRC and other tumors in FAP patients. 23 These avenues of exploration could include, but would not be limited to, trials of the new monoclonal antibodies against immune checkpoint molecules. 24
Footnotes
Acknowledgements
The authors thank Mason Harris for animal husbandry.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SMT and RGL designed the study, and developed the methodology. SMT, SAH, JGD, and CAM collected the data. SMT, SAH, JGD, CAM, and RGL performed the analysis and wrote the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by National Institutes of Health (NIH) grants R01 DK059911 and P01 DK071176, the University of Alabama at Birmingham Digestive Diseases Research Development Center grant P30 DK064400, a RayBiotech Innovative Research Grant Award, the Crohn’s and Colitis Foundation (26971), and the Richard A. Elkus, M.D., Eminent Scholars Program in GI Oncology administered by the UAB Comprehensive Cancer Center (P30AR050948). S.M.T. was supported by the Howard Hughes Medical Institute Med into Grad Fellowship and the University of Alabama at Birmingham Carmichael Fund. S.A.H. was supported by the UAB PREP Scholars Program (R25 GM086256). Aspects of this project were conducted in biomedical research space that was constructed with funds supported in part by NIH grant C06RR020136.