
Abstract
Small interfering RNA (siRNA) therapeutics have advanced from bench to clinical trials in recent years, along with new tools developed to enable detection of siRNA delivered at the organ, cell, and subcellular levels. Preclinical models of siRNA delivery have benefitted from methodologies such as stem-loop quantitative polymerase chain reaction, histological in situ immunofluorescent staining, endosomal escape assay, and RNA-induced silencing complex loading assay. These technologies have accelerated the detection and optimization of siRNA platforms to overcome the challenges associated with delivering therapeutic oligonucleotides to the cytosol of specific target cells. This review focuses on the methodologies and their application in the biodistribution of siRNA delivered by lipid nanoparticles.
Keywords
Small interfering RNA (siRNA) therapeutics have been developed and tested in preclinical models (Blagbrough and Zara 2009; Manjunath and Dykxhoorn 2010; Seth et al. 2012) and in clinical trials (Burnett et al. 2011; Bhavsar et al. 2012; Burnett and Rossi 2012) in recent years. The 19–21nt (nucleotide) double-stranded siRNA mediates cleavage of its target messenger RNA (mRNA) by incorporating into the RNA-induced silencing complex (RISC), a ubiquitous machinery in mammalian cells. siRNA-activated RISC exploits the endogenous RNA interference (RNAi) pathway to prevent translation of disease-causing proteins for therapeutic benefit (Sepp-Lorenzino and Ruddy 2008; Snead and Rossi 2012). Naked siRNA without formulation or encapsulation, where topical (intravitreal) and local (intranasal) administration is the major route of delivery, has also been summarized (Morin et al. 2009). Naked siRNA is unstable, with a plasma half-life of less than 10 min (Soutschek et al. 2004; Gao et al. 2009), due to rapid degradation by nucleases in the blood after systemic injection (Grimm 2009; Takahashi et al. 2009). Therapeutic siRNAs can be chemically stabilized to achieve improved pharmacokinetics in vivo (Morrissey, Blanchard, et al. 2005; Morrissey, Lockridge, et al. 2005; Kawakami and Hashida 2007; Abrams et al. 2010; Stanton and Colletti 2010).
Systemic delivery of siRNA has been an area of extensive investigation in recent years (Kawakami 2008; White 2008; Li L and Shen 2009; Peer and Shimaoka 2009; Tseng et al. 2009). Different RNA delivery vehicles have been studied, including lipid nanoparticles (LNPs) (Judge et al. 2009; Abrams et al. 2010; Tao et al. 2010; Basha et al. 2011), polymers (Rozema et al. 2007; Kim et al. 2009), cell-degradable multilayered polyelectrolyte films (Dimitrova et al. 2008), nanocages (Yavuz et al. 2009), aptamer-based approaches (McNamara et al. 2006; Dassie et al. 2009; Thiel and Giangrande 2009), peptide-mediated delivery (Jafari and Chen 2009), glucan-encapsulated siRNA particles (Aouadi et al. 2009), and other non-viral (Chen Y and Huang 2008) and viral vectors (Crowther et al. 2008; Guibinga et al. 2008; Manjunath et al. 2009). Among these platforms, LNPs are the most extensively studied class of RNA delivery vehicle. Numerous reviews have been published for lipid-based nanoparticles for siRNA delivery (Li W and Szoka 2007; Zuhorn et al. 2007; Fenske and Cullis 2008; Li SD and Huang 2008; Moreira et al. 2008; Tseng et al. 2009; Wu SY and McMillan 2009; Ewert et al. 2010; Schroeder et al. 2010; Musacchio and Torchilin 2011; Gindy et al. 2012), as well as for siRNA tumor targeting delivery (Tseng and Huang 2009).
In this review, we focus on lipid nanoparticle–delivered siRNA, not naked modified or other carrier-formulated siRNA. We illustrate the barriers to the use of LNP-siRNA delivery to target cells. Several technologies that have accelerated the challenge of overcoming physiological and cellular barriers toward the realization of siRNA therapeutics will be highlighted. We also briefly summarize the published biodistribution reports of siRNA therapeutics in preclinical rodent models and non-human primates (NHPs).
LNP Composition and Function
LNPs usually are composed of three to four lipid components (Fig. 1): (1) a cationic lipid that contains a cationic head group, a lipophilic tail group, and a connecting linker; (2) a PEGylated lipid; (3) cholesterol; and (4) a helper lipid. One such lipid nanoparticle, LNP201, has been extensively investigated (Abrams et al. 2010; Pei et al. 2010; Tao et al. 2010; Bartz et al. 2011; Dharmapuri et al. 2011; Shi et al. 2011; Tadin-Strapps et al. 2011; Wei et al. 2011). Fig. 1 illustrates the components of LNP201 encapsulating an siRNA against the ubiquitously expressed Sjögren syndrome antigen B (Ssb) mRNA. The lipid components of LNP201 are a cationic lipid (CLinDMA, 30–50 mol%), a PEGylated lipid (DMG-PEG2K, 2–6 mol%), and cholesterol (20–50 mol%).
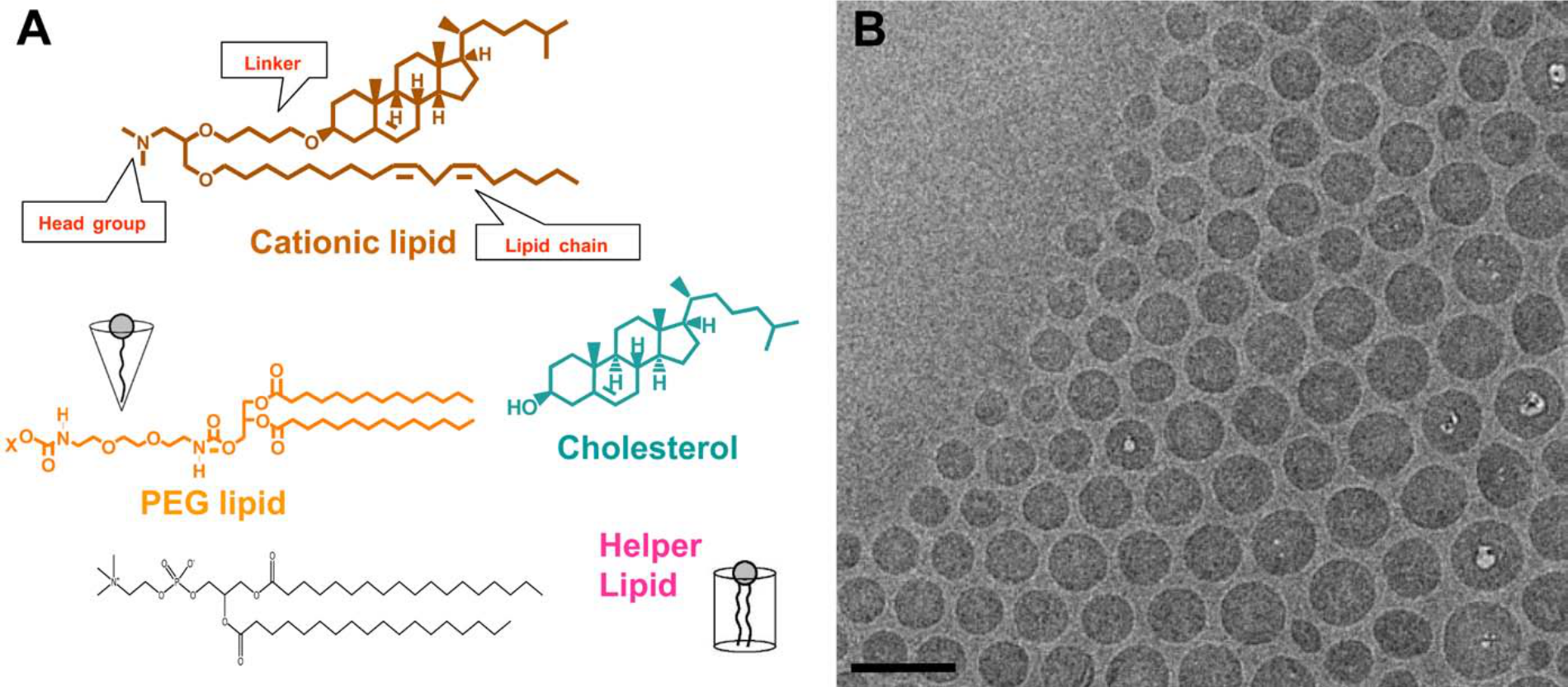
(A) The components of a lipid nanoparticle (LNP): a cationic lipid (CLinDMA, 30–50 mol%), a PEGylated lipid (DMG-PEG2K, 2–6 mol%), cholesterol (20–50 mol%), and possibly a helper lipid. (B) Cryo–electron microscopy (Cryo-EM) image of LNP to show the shape, size, and uniformity of lipid-siRNA nanoparticles. Bar = 100 nm. (With permission from Matthew Haas and Ye Zhang, unpublished data.)
Cationic lipids have been used to formulate doxorubicin (DOXIL), which was approved by US Food and Drug Administration (FDA) in the mid-1990s for the treatment of Kaposi sarcoma and possibly refractory breast and ovarian cancers (Laginha et al. 2005; La-Beck et al. 2011). Cationic lipids may be advantageous for cell internalization compared with neutral and anionic lipids because they can facilitate particle-cell association where a positive surface charge is required for negatively charged cell membranes. Cationic lipids also enable endosomal escape and cytosolic delivery after internalization. The formation of charge-neutral ion pairs with ionic endosomal membranes (Huang Z, Guo, et al. 2006; Huang Z, Li, et al. 2006) and/or the conversion of lamellar phases to non-lamellar phases between lipids in LNP and lipids in the cell membrane could enhance endosomal escape (Hafez et al. 2001; Hafez and Cullis 2001).
Polyethylene glycol (PEG) lipids can improve particle-circulating time in the bloodstream after intravenous injection because polymer PEG provides stealth to prevent opsonization and clearance of nanoparticles by the mononuclear phagocytic systems (Klibanov et al. 1991), also called the reticuloendothelial system (Sapra et al. 2005). A disadvantage of PEGylation could be its neutralization of the positive surface charge, which is required for siRNA uptake by cells in the context of negatively charged cell membranes and cationic lipids. Carefully monitoring the percentage of PEG lipids to achieve prolonged circulation time and intact siRNA transfection efficacy is one of the critical steps in optimizing and maximizing LNP-siRNA pharmaceutical properties (Li W et al. 2005). Tao et al. (2010) compared the PEG lipid ratio in LNP201-like lipid nanoparticles. When comparing two sets of LNPs with a 2% and 5.4% molar ratio of PEG lipid (PEG-DMG) incorporated into the same head cationic lipid (ClinDMA) and same linkers (butyl, hexyl, or octyl), 2% PEG LNPs were more efficacious than the corresponding LNPs with identical components but containing 5.4% PEG.
Cholesterol is another critical component of the formulation, as it is required for stabilization of the lipid bilayer in the assembled nanoparticle and also may be directly involved in siRNA diffusion into the cell membrane (Lu et al. 2009). Neutral helper lipids or fusogenic lipids (Huang et al. 2012) could be added to the LNP to further advance cellular uptake and endosomal escape properties. A detailed summary of LNP medicinal chemistry containing different cationic lipids, PEG lipids with different molecular weights, and helper lipids has been reviewed (Stanton and Colletti 2010).
siRNA Delivery Barriers and Detection Technologies
In order for LNPs to effectively deliver siRNA to the pharmacologically active compartment of target cells, numerous physiological barriers must be overcome (Fig. 2). First, the LNP must protect the siRNA from serum endonucleases while in circulation. Second, the LNP must accumulate in the target tissue. Third, the LNP must enable internalization of the siRNA into target cells. Finally, the siRNA must partition within the cell to the cytosol (endosome/lysosome escape), where it can associate with RISC and find its cognate siRNA target. It is critical to have methodologies to evaluate each of these steps to advance the platform technology. We will address the methods used currently in three sections below.
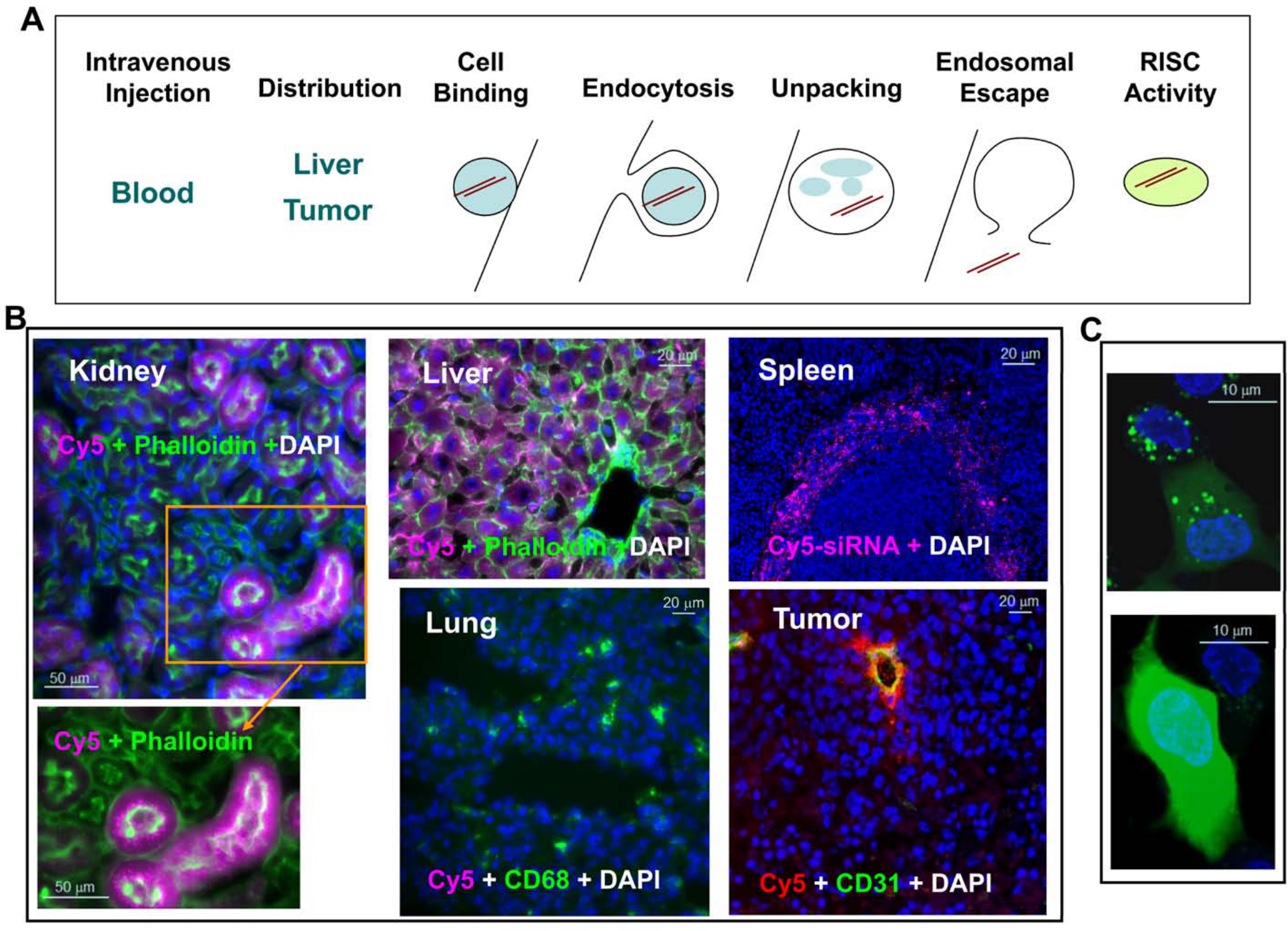
Barriers to lipid nanoparticle (LNP)–mediated small interfering RNA (siRNA) delivery and assay development. (A) Delivery is the key challenge to be addressed before an siRNA therapeutic can be fully realized. Successful delivery must enable bypass of multiple biological barriers, including blocking blood nuclease digestion of siRNA by chemical modification of the siRNA duplex, improving target-specific tissue biodistribution and cellular uptake, increasing cell binding via electrostatic interaction of the LNP and cell membrane or ligand and cell surface receptor, enhancing receptor-mediated endocytosis, increasing the efficiency of unpacking siRNA from the lipid nanoparticle delivery vehicle, enhancing the endosomal escape of siRNA to the cytosol, and improving siRNA loading into the RNA-induced silencing complex (RISC) in the cytosol for target mRNA cleavage (courtesy of Matt Stanton and Steve Colletti). (B) Tissue distribution of LNP-siRNA-Cy5 in liver, spleen, lung, kidney, and KB3 (a nasopharyngeal carcinoma cell line) tumor xenograft. LNP-siRNA labeled with Cy5 was intravenously injected into a mouse at 3 mg/kg. Different tissues were collected 2 hr postdosing and cryosections were analyzed by epifluorescence microscopy. All images were taken at ×20 magnification. siRNA-Cy5 (purple) mainly distributed to the liver, the red pulp of the spleen, and the proximal tubules of the kidney but not much to the lung. Phalloidin (green) outlines cell membrane and CD68 (green) stains macrophages. In the tumor section, siRNA-Cy5 (red) was mainly located at CD31-stained vessels and adjacent KB3 tumor cells but did not penetrate very far. (C) Dextran endosomal escape assay measuring the endosomal release of dextran (green) from punctate endosomes into diffused signals in the cytosol in HepG2 cells in vitro (with permission from Eileen Walsh and Bonnie Howell, unpublished data). Bars = 50 µm (B: kidney); 20 µm (B: liver and lung); 20 µm (B: spleen and tumor); 10 µm (C).
In addition, some of the animal studies reported here are not reported elsewhere. In this study, all animal experiments were conducted in accordance with the standards established by the US Animal Welfare Act and approved by Merck & Co., Inc.’s Institutional Animal Care and Use Committee.
Approaches to Evaluate siRNA Overall Tissue Distribution
We will address the first two barriers—namely, siRNA blood and tissue distribution (Fig. 2)—in this section. Following systemic injection of siRNA particles in vivo, the total levels of the siRNA payload in blood and its distribution in different organs can be quantitatively addressed by stem-loop reverse transcription quantitative polymerase chain reaction (stem-loop RT-qPCR) (Stratford et al. 2008; Abrams et al. 2010; Seitzer et al. 2011). This method is applied to quantitatively measure siRNA in any biological sample in a high-throughput manner. One advantageous attribute of this technology is that, unlike many qPCR-based methods, the sample input can be whole blood or tissue rather than isolated nucleic acid (Seitzer et al. 2011). This increases precision, reduces cost, and increases the number of potential clinical applications. It is, however, critical to recognize that determination of siRNA concentration at the organ level does not report on the ability of the LNP to deliver to a target cell type within a heterogeneous tissue, nor does it report on the ability of the LNP to deliver to the cytsol where siRNA is activated by RISC.
The comparison of numerous technical variations of stem-loop RT-qPCR (Stratford et al. 2008) has led to a highly precise, sensitive, and widely accepted assay format. The methodology is cost-effective and can detect a linear range of 5 × 10−4 to 500 pg/µl siRNA. One liability of the qPCR approach is the difficulty in differentiating full-length siRNA from potential metabolites. This assay generally will still detect a one- or two-nucleotide truncation at the 3′ end without loss of signal but will not detect 5′-end truncations because of the fidelity requirement during the reverse transcription step. Interestingly, however, at least one example of step-loop qPCR reagents has proved to be specific in distinguishing single-nucleotide changes in mature microRNAs (Chen C et al. 2005). In short, the application of stem-loop RT-qPCR throughout the siRNA field has enabled the facile determination of LNP biodistribution properties. There are now numerous preclinical examples (Cheng et al. 2009; Abrams et al. 2010; Cheng et al. 2011; Shi et al. 2011; Tao et al. 2011) and at least one clinical example (Davis et al. 2010) of its application.
Application of PCR and many other analytical methods in preclinical models requires necropsy and tissue extraction. Sometimes it is advantageous to study pharmacokinetics (PK)/biodistribution in a live animal, in real time. Several live imaging technologies have been applied to study the biodistribution of siRNA delivery. The imaging modalities include but are not limited to non-invasive whole-body IVIS spectrum imaging by Xenogen (Alameda, CA) (Tao et al. 2011), non-invasive whole-body positron emission tomography (PET) (Hatanaka et al. 2010), and combination of PET with computed tomography (CT) (Mudd et al. 2010), single photon emission computed tomography (SPECT) imaging (Merkel, Beyerle, et al. 2009; Merkel, Librizzi, Pfestroff, Schurrat, Behe, et al. 2009; Merkel, Librizzi, Pfestroff, Schurrat, Buyens, et al. 2009), bioluminescent imaging (Bartlett et al. 2007; Bogdanov 2008; Tao et al. 2010), near-infrared fluorescence imaging (Asai et al. 2011), magnetic resonance imaging (Mikhaylova et al. 2009), intravital imaging on live animals (Ofek et al. 2010; Ra et al. 2010), and others (Bogdanov 2008; Hong et al. 2010; Kang and Wang 2010). Although these technologies enable visualization of siRNA biodistribution processes in living animals, their application is often limited by inadequate spatial resolution, sensitivity, dynamic range, and stability of the contrast reagents.
One powerful imaging technology is quantitative whole-body autoradiography (QWBA) using a radiolabeled lipid delivery vehicle (Fig. 3) or siRNA. QWBA is a comprehensive and quantitative method for assessing the tissue distribution of radiolabeled materials in preclinical species (Coe 2000; Kertesz et al. 2008). QWBA was used to examine the biodistribution of LNP201-siRNA at various time points by radiolabeling the lipid delivery vehicle, CLinDMA, with 14C and administering 14C-LNP201-siRNA to rats intravenously at various time points. Radiolabeled LNP201-siRNA–related material was observed in the blood, liver, spleen, kidney, lung, bone marrow, and other various tissues, with highest levels achieved in the liver and spleen following intravenous administration (Fig. 3; Vavrek, M and Koeplinger, K, unpublished data). Although QWBA can provide quantitative data in 50+ tissues as well as information on substructure distribution (Coe 2000), it is dependent on the availability of the radiolabeled test article. In addition, QWBA is inherently non-selective and does not distinguish between possible lipid metabolites (if the radioactive label is on lipid) and siRNA degradants (if the label is on siRNA duplex).
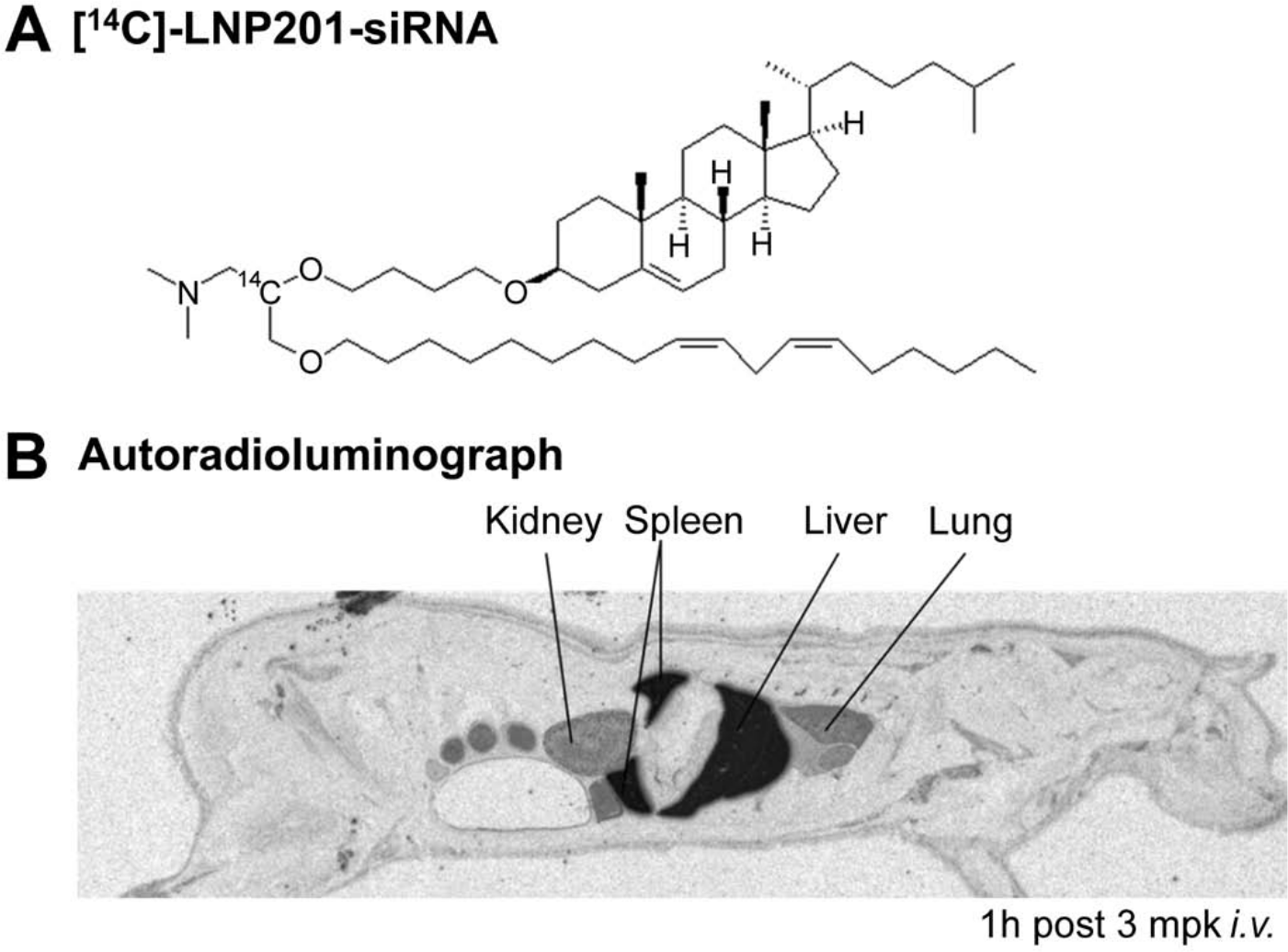
Quantitative whole-body autoradiography (QWBA) of 14C-labeled lipid nanoparticle (LNP)–small interfering RNA (siRNA) in rats. (A) The chemical structure of 14C-labeled lipid nanoparticle delivery system LNP201-siRNA. (B) The biodistribution of 14C-LNP201-siRNA was examined at various time points in rats following intravenous administration of 3 mg/kg siRNA and 100 µCi/kg; the autoradioluminograph image at 1 hr postdose is illustrated. Dark areas represent 14C-related material. For QWBA, at the terminal time point, rats were frozen rapidly in a dry ice/hexane bath after whole blood was sampled. Rats were embedded in 2% carboxymethycellulose, and 40-µm-thick sagittal cryo-sections were taken at various levels. Sections were exposed to phosphor imaging plates for 4 days and imaged with a Fuji FLA-5100 phosphor imager (Fujifilm; Tokyo, Japan). 14C-LNP-siRNA was delivered to the liver, spleen, lung, and kidney 1 hr after intravenous dosing, suggesting a quick and broad distribution of LNP201-siRNA after systemic administration. (With permission from Marissa Vavrek and Kenneth Koeplinger, unpublished data.)
Intravital imaging is another tool that has been applied to trace siRNA distribution in skin and xenograft tumors (Hickerson et al. 2012; Chong et al. 2013). This technique has been demonstrated to follow up fluorescent-labeled siRNA on the surface of live animals, such as skin and subcutaneous tumors, but the procedure of applying the detection probe is invasive for liver, kidney, and other internal organs. Also, the animals need to be stabilized by anesthesia and the images are acquired at lower objectives (×10 or ×20). Higher magnification (×40 and above) requires the animal to be in an extremely stable condition to avoid blurring of the images due to movement of the animal or organs.
To extend the evaluation of siRNA biodistribution at multiple time points (up to 48 hr) and at higher magnification (up to ×60), we implemented a histological platform to evaluate biodistribution of siRNA across tissues in vivo and across cell types by in situ immunofluorescence (IF) staining (Shi et al. 2011). At organ levels (Fig. 2B), we observed siRNA-Cy5 in liver, kidney, spleen, and tumor after tail vein injection of LNP201-siRNA-Cy5 in mice, with low to no Cy5 signal in lung, brain, muscle, and other organs. This technique is applied on fixed tissue sections and has been approved for semi-quantitation of cellular siRNA distribution (Shi et al. 2011). It is worth mentioning that fixation, tissue processing, and the staining procedure may lead to loss of siRNA. Different fixatives, such as ethanol, methanol, acetone, 10% formalin, or 4% paraformaldehyde, are recommended to be tested for individual siRNA carriers. Taken together, low-resolution imaging and biochemical qPCR approaches can be used to evaluate overall tissue distribution to address the first two barriers illustrated in Fig. 2A.
General conclusions of LNP-siRNA biodistribution emerged in numerous studies using the methods described above. The results from a variety of preclinical studies indicate that LNP is broadly distributed in all tissues, with preferential delivery and accumulation in the liver and spleen (Zimmermann et al. 2006; Davis et al. 2010; Abrams et al. 2010; Tao et al. 2010; Tao et al. 2011). The liver and spleen organ tropism of LNP has been reviewed (Tseng et al. 2009) and is mainly attributed to opsonization of LNP in the blood circulation by IgM, IgG, complements, fibronectins, and apolipoprotein E (ApoE). Hepatocytes have low-density lipoprotein receptors that mediate the internalization of ApoE-opsonized LNP, whereas macrophages in liver sinusoids or red pulp of the spleen engulf complement-opsonized nanoparticles because of the scavenger receptors on macrophages. About 60% of the total 14C-LNP201-siRNA dose is located in the liver and spleen 1 hr after tail injection in rats, as measured by QWBA using 14C-ClinDMA–labeled LNP-siRNA (Fig. 3). The majority of Cy5-labeled siRNA was trapped in sinusoids at 0.5 hr after intravenous injection and translocated into hepatocytes at 2 hr, as observed by collagen IV–outlined sinusoids in mouse liver sections (Shi et al. 2011). The extrahepatic distribution of LNP-siRNA has also been reported in tumor xenograft models (Liu et al. 1992; Medarova et al. 2007; Li SD and Huang 2009; Schadlich et al. 2011), monocytes (Leuschner et al. 2011), and peritoneal macrophages and splenic dendritic cells (Basha et al. 2011).
Analysis of siRNA at Cell and Subcellular Levels
Cellular binding and uptake would require methods capable of providing cellular resolution. In cultured cells in vitro, cell membranes can be detected by a membrane protein-specific binding ligand or antibody (Kruth et al. 2001; Morishima et al. 2010) or a membrane detection algorithm, such as ImarisCell’s Membrane Detection algorithm (http://www.bitplane.com/go//knowledge-base/how-to–/membrane-detection-of-cells). In this algorithm, nuclei locations determined by specific nuclear staining in the preceding step are used as seed points for an algorithm performing a cell membrane calculation.
Using fluorescent-labeled siRNA in combination with membrane detection, siRNA membrane association can be addressed. Internalized siRNA was captured by observation of punctate siRNA fluorescent (FL) signals (Fig. 2C, upper panel). The punctate FL signals can be further addressed by applying LysoTracker (Invitrogen; Carlsbad, CA) in the live cells, which labels late endosomes and lysosomes.
In animal models in vivo, we have developed a platform to evaluate siRNA location in different cells by co-staining cell type–specific markers in different compartments of the organ (e.g., siRNA trapped in sinusoids or translocated into liver parenchymal cells) and in the membrane, cytosol, or nuclei of a cell by counterstaining with DAPI for nuclei and with phalloidin for liver cell membranes (Shi et al. 2011). Phalloidin can outline the hepatocyte membrane in liver tissues, which has been reported by Mirus (Rozema et al. 2007). Co-localization analysis with other liver cell markers—for example, CD68 for Kupffer cells (KCs; the resident macrophage in the liver) and desmin for hepatic stellate cells—demonstrates the prompt engulfment of LNP-siRNA by KCs but not by stellate cells, suggesting that saturation or depletion of KCs could be one of the strategies for efficient LNP-siRNA delivery to hepatocytes. We observed the delivery of siRNA into KCs but no knockdown of target mRNA in isolated KCs. Similarly, siRNA was delivered mainly to splenic red pulp where macrophages are located, and no or a low level of target mRNA KD has been observed in spleen tissues (Abrams et al. 2010; Shi et al. 2011). The mechanism of less RNAi silencing with abundant uptake of siRNA in macrophages is still unclear, but it might be related to the quick transition from endosome to lysosome, leading to clearance of the siRNA in macrophages before siRNAs are able to escape from the endosome and be released to the cytosol for RISC loading and the silencing effect. This hypothesis could be further investigated using leupeptin, an inhibitor of lysosomal proteases; treating the cells with chloroquine, a weak base that inhibits proteolysis by raising the pH in endosomes and lysosomes (Smit et al. 1987); or inhibiting phagocytosis and endocytosis by cytochalasin B (Malawista 1971; Wagner et al. 1971; Al-Hallak et al. 2011) or methyl palmitate (Cai et al. 2005).
One advantage of IF staining and co-localization methods is the ability to determine the location of LNP-delivered siRNA at suborgan and cellular levels. The drawbacks are low throughput, inability to detect metabolites, and difficulty in achieving precise quantification of fluorescence intensity on IF-stained tissue sections. There are examples of applying such histological methods to investigate siRNA deposition in the liver (Shi et al. 2011), tumor (Yoshizawa et al. 2008; Mikhaylova et al. 2009), lung (Tayyari et al. 2011), or even subcellular compartments (Akita et al. 2010; Basha et al. 2011; Pittella et al. 2011).
Analyses of siRNA Endosomal Escape and RISC-Loaded siRNA
Because RISC and target mRNA all exist in the cytosol, successful translocation of siRNA from endosome to cytosol, termed endosomal escape, is the key step to achieving RNAi efficacy. Four possible mechanisms of endosomal escape have been reported: (1) pore formation in the endosome membrane by internal stress or internal membrane tension strong enough to enlarge or create pores in the lipid membrane (Huang HW et al. 2004); (2) the pH-buffering effect, also called proton sponge effect, which is caused by an extensive inflow of ions and water into the endosomal environment and subsequently leading to rupture of the endosomal membrane and release of entrapped siRNAs (Pack et al. 2000); (3) fusion in the endosomal membrane by a fusogenic peptide (Aroeti and Henis 1991; Wharton et al. 1998; Skehel et al. 2001; Russell et al. 2003); and (4) photochemical disruption of the endosomal membranes by a number of photosensitizers that localize primarily in the membrane of the endosomes and lysosomes and induce the formation of reactive singlet oxygen to destroy the membrane upon exposure to light (Selbo et al. 2000). Conjugation of endolytic peptides, such as cell-penetrating peptides (Lundberg et al. 2007), or chemical reagents, such as polyethylenimine (PEI) (Masotti et al. 2007), could facilitate siRNA endosomal escape and ensure cytosolic delivery of siRNA. A list of endosomal escape agents was summarized by Varkouhi and colleagues (2011). The cellular uptake and intracellular trafficking of oligonucleotides, including siRNA, also has been reviewed (Juliano et al. 2011). The methodologies summarized here could provide useful tools to detect the endosomal escape and prove beneficial for in vitro screening of endosomolytic agents.
In vitro evaluation of LNP endosomolytic properties has required specially designed biochemical assays. Colleagues at Merck implemented a horseradish peroxidase (HRP) endosomal escape assay (Bartz et al. 2011) and dextran imaging assay (E. Walsh and B. Howell, unpublished data) to address the critical step of siRNA release from its carriers. HRP and dextran endosomal escape assays are semi-quantitative techniques developed for indirect evaluation of siRNA entering the cytosol after endocytosis.
HRP is a mannose-containing glycolated enzyme with high affinity for mannose receptors. But its internalization is not completely pathway specific, and only a portion is taken up via receptor-mediated endocytosis (Ellinger and Fuchs 2010). Bartz et al. (2011) treated cells with both HRP and LNP-siRNA, resulting in endocytosis of HRP and LNP-siRNA into vesicles in cells. They then treated the cells with pore-forming toxin SLO (streptolysin O) at 4C for 10 min, and cell membrane unbound extra SLO was thoroughly washed out. The low temperature allowed specific binding of SLO to the cell membrane but no internalization, which avoided pore formation on internal membranes such as endosomal membranes. Next, they increased the incubation temperature to 37C when a transformation of SLO happened to form pores only on cell membranes where SLO attached. HRP or siRNA can leak into cell medium via the newly formed SLO pores from the cytosol if there is endosomal escape after LNP-siRNA/HRP endocytosis. The HRP activity can be measured by adding substrate of the HRP enzyme to the conditional medium, or the siRNA can be measured by stem-loop qPCR from the conditional medium after ultracentrifugation and removal of cells.
Another method for measuring endosome escape is an imaging-based assay using dextran as the surrogate readout. This requires the imaging systems to provide subcellular resolution. Dextran is a complex, branched polysaccharide composed of varying lengths of glucose chains. It is known to be a fluid phase marker and has partial uptake via clathrin-mediated endocytosis and/or macropinocytosis (Shurety et al. 1998). The translocation characteristics of dextran are inversely related to their molecular weight, with molecules more than 70 kDa able to cross the membrane via fluid phase endocytosis and molecules smaller than 40 kDa able to passively diffuse toward the cytoplasm (Matsukawa et al. 1997). When cells were treated with LNP-siRNA and FITC-labeled dextran (60 kDa), siRNA and dextran were internalized and bright punctate green fluorescent signals were observed under a confocal microscope in cells, indicating FITC-containing endosome formation (Fig. 2C, upper panel). In the acidic environment of the endosome (pH 5.5–6), siRNA may detach from the lipid, and if the cationic lipid or helper lipid facilitates siRNA translocation from the endosome to the cytosol, dextran-FITC can also go through 6-nm radius transmembrane pores generated by an unknown mechanism by LNPs, possibly through osmetic lysis of pinosomes (Gruber et al. 2004) or the proton sponge effect (Patil et al. 2009). Therefore, diffused green signals can be detected and measured in the entire cells (Fig. 2C, lower panel), which suggests endosomal escape of dextran/siRNA.
Others (Akita et al. 2010; Pittella et al. 2011) have applied confocal microscopy where they labeled siRNA with Cy5 (red) and stained siRNA-treated cells with LysoTracker (green), and they calculated the ratio of yellow pixels (co-localization of siRNA with LysoTracker), which was equal to yellow pixels divided by the sum of yellow and red pixels. If more yellow perinuclear signals were observed and the yellow co-localization ratio was higher, it indicated more endosomal/lysosomal captured siRNAs. If the red siRNA signals extensively spread in the cells in a non-perinuclear manner, it indicated more cytosolic siRNA, suggesting endosomal escape. Basha et al. (2011) also applied similar confocal microscopy techniques to quantify endosomal versus cytoplasmic siRNA levels using fluorescently labeled siRNA. They stained early endosomes with anti-EEA1 (early endosomal antigen 1) and lysosomes with anti-LAMP1 (lysosomal-associated membrane protein 1). They compared siRNA formulated in LNP containing four different ionizable cationic lipids and found that lipid DLinKC2-DMA (1,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane) exhibited a predominantly diffused cytoplasmic pattern of siRNA, indicating good endosomal escape, which correlated with greater efficacy. It is worth mentioning that LysoTracker is good for live cells, whereas antibody detection of endosomes is applied to fixed cells and fixed tissues.
Calculation of cytosolic siRNA fluorescent pixels or semi-quantitative HRP or dextran assays has not always correlated with the efficacy of siRNA in cells. This is perhaps because there is a bottleneck between cytosolic delivery and RISC association that has yet to be fully elucidated. The association of the oligonucleotide with RISC in the cytosol is a prerequisite for mRNA knockdown and the desired downstream pharmacology. Pei and colleagues (2010) established a biochemical method to detect the functional guide strand siRNA levels by Argonaute 2 (Ago2) immunoprecipitation followed by stem-loop RT-qPCR. Ago2, a member of the Argonaute superfamily, is the protein in RISC that binds to the guide strand of siRNA and its recruited target mRNA and catalyzes the cleavage of mRNA at the site complementary to the 10–11 nucleotide position of the guide strand (Carmell et al. 2002; Meister et al. 2004). Pei et al. carefully selected species-specific Ago2 antibodies to verify that the RISC-associated siRNA was not from a postlysis Ago2-siRNA association. Careful validation of this RISC-binding methodology ensured that post–cell lysis artifacts did not contribute to the Ago2-associated siRNA signal.
Implementation of the RISC-binding assay solidified the concept that mRNA knockdown requires the compartmentalization of the siRNA into a subcellular fraction (the cytosol). This is the reason that total siRNA in tissues does not always generally agree with efficacy (Abrams et al. 2010; Seitzer et al. 2011). Wei et al. (2011) demonstrated that target mRNA silencing in LNP-siRNA–treated livers correlated with the amount of siRNA bound to Ago2 but not with the siRNA measured in total liver lysates. Pei et al. (2010) also addressed that the amount of Ago2-associated siRNA guide strand coincided with the efficacy of target knockdown in a dose- and time-dependent manner. They also reported more than 30-fold less passenger strands than guide strands associated with Ago2 on day 1 after LNP-siRNA treatment in mouse. This result is consistent with the report that passenger strands usually unwind upon RISC loading of the guide strand and quickly degrade, which is a natural mechanism present in organelles, from worms to mammals, to reduce off-target effects (Filipowicz 2005).
HRP, dextran, and florescence imaging assays could provide tools for in vitro screening of LNP-siRNA drug optimization. Stem-loop RT-qPCR measures quantitatively the total siRNA levels in cells in vitro or in different tissues in vivo to facilitate the estimation of PK of siRNA therapeutics. QWBA and histological imaging techniques help visualize the siRNA in vivo. The RISC-loading assay assesses the amount of functional siRNA at both cell and tissue levels. Tissue and cell type distribution of siRNA largely define their therapeutic effect and toxicity. The intracellular fate of the siRNA after cellular internalization affects the efficacy of therapeutic siRNA. Therefore, careful assessment of functional siRNA in the right organ, tissue, cell type, and subcellular compartment is critical for the evaluation of siRNA on-target effects. The technologies summarized above are state-of-art technologies for proof-of-concept measurement of the efficacious siRNA levels after administration of therapeutic siRNAs in vitro and in vivo. Currently, stem-loop RT-qPCR is largely used in the field to address total siRNA levels in cells and tissues. We would suggest that researchers explore the RISC-loaded siRNA assay, which may better correlate siRNA PK with its efficacy.
siRNA Delivery Tested in NHPs
Although many successful preclinical studies have been reviewed using rodents as the models for testing lipid-based siRNA delivery (Wu and Nandamuri 2004; Tseng and Huang 2009; Ewert et al. 2010; Schroeder et al. 2010), there are very few reports of preclinical research conducted in NHPs. The successful applications of LNP-siRNA delivery in NHPs will pave the road to human clinical trials. A summary of siRNA nanoparticles tested in preclinical studies in NHPs is illustrated in Table 1. To our knowledge, this is the first time that siRNA therapeutics tested in NHPs have been summarized. In the reported NHP studies, siRNA levels were determined by stem-loop RT-qPCR, Northern blot, and imaging analysis (see Table 1). None of them applied the RISC-loading siRNA assay to detect functional siRNA levels. We recommend detecting total siRNA versus the RISC-loaded siRNA in rodent and NHP models. This comparison may shed light on understanding the species-specific efficient delivery of siRNA by different carriers due to species-specific barriers, if any.
Summary of siRNA Therapeutics Tested in Non-Human Primates
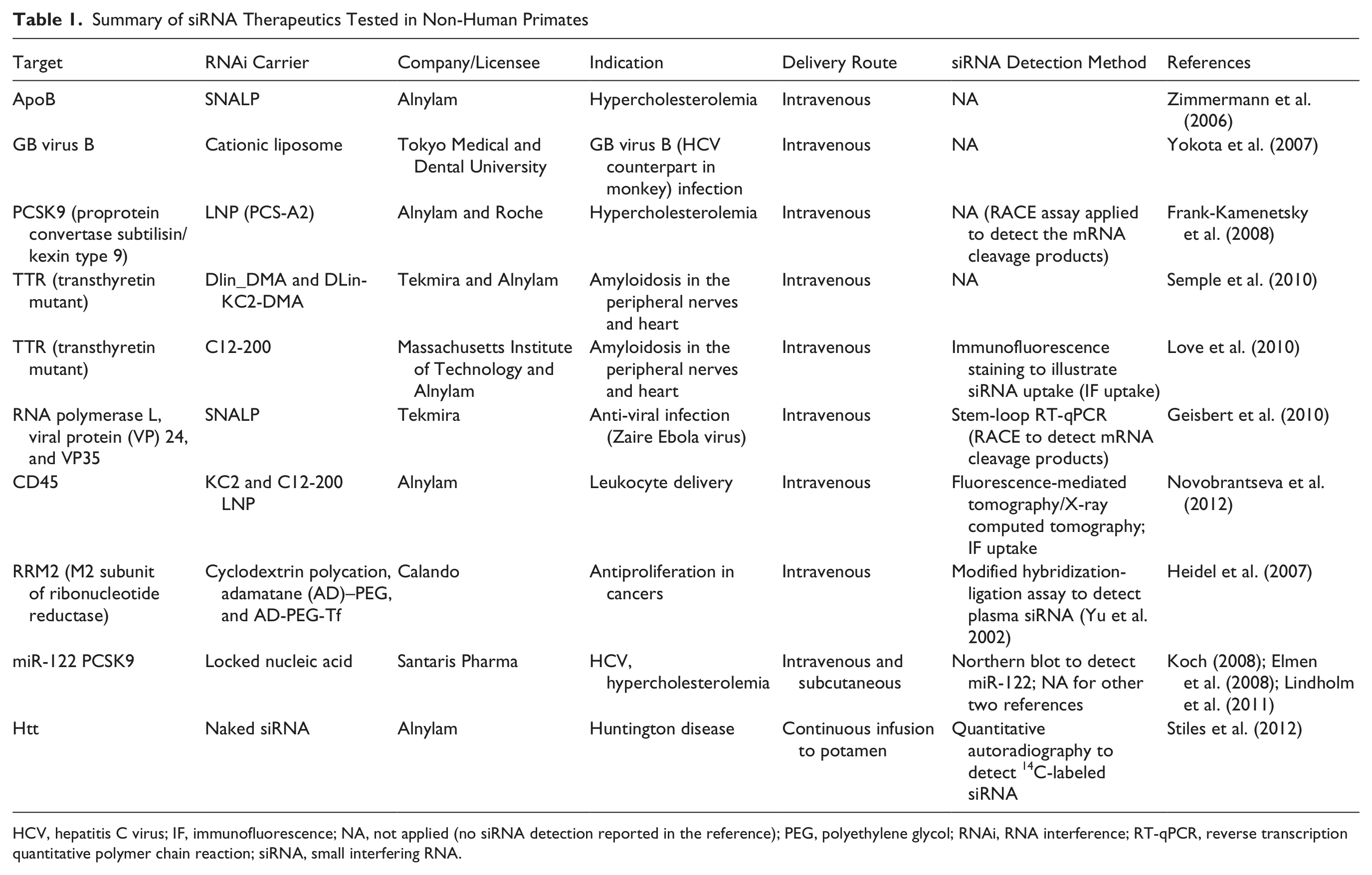
HCV, hepatitis C virus; IF, immunofluorescence; NA, not applied (no siRNA detection reported in the reference); PEG, polyethylene glycol; RNAi, RNA interference; RT-qPCR, reverse transcription quantitative polymer chain reaction; siRNA, small interfering RNA.
Conclusion and Perspective
siRNA-based therapeutics promise to provide benefits to patients with unmet medical needs, especially via knockdown of otherwise undruggable genes in human diseases. With more advanced lipid characterization and formulation, the emergence of appropriate tools to evaluate safety and efficacy of RNA therapeutics, and more RNA therapeutics in clinical trials, we are enthusiastic about overcoming siRNA delivery issues—for example, application of optimized lipid-based RNA delivery vehicles, engagement of endosomolytic and pH-sensitive linkers, chemical modification of siRNA nucleotides, conjugation of targeting moieties, and so on. We can fine-tune the design of lipid nanoparticles to lead to enhanced siRNA delivery, uptake, and efficacy at lower doses; reduced toxicity; and an increased therapeutic window. In this review, we summarize current frontiers on the development of lipid nanoparticle–based siRNA therapeutics, especially focusing on the discussion of technologies applied for the discovery of siRNA biodistribution evaluated by quantitative whole-body assessment and histologic imaging tools, stem-loop RT-qPCR, endosomal escape, and RISC loading. These technologies enable the correlation of pharmacokinetics and pharmacodynamics of siRNA therapeutic drugs and the detection of the targeted delivery of siRNA to the ideal specific cell types. The therapeutic siRNAs have been applied in rodents and NHP preclinical models by us and others, and they are being tested in human clinical trials. The strategies and technologies we discussed in this review will be beneficial to the development of LNP-siRNA in clinics now and in the future.
Footnotes
Acknowledgements
We thank the following colleagues at Merck who contributed to the data published in this review: Matthew Haas and Ye Zhang for cryo-EM studies, Matt Stanton and Steve Colletti for ![]() , schematic drawing of barriers to lipid nanoparticle delivery, Marissa Vavrek and Kenneth Koeplinger for QWBA studies, and Eileen Walsh and Bonnie Howell for dextran endosomal escape studies. We also thank Marissa Vavrek for her careful editing.
, schematic drawing of barriers to lipid nanoparticle delivery, Marissa Vavrek and Kenneth Koeplinger for QWBA studies, and Eileen Walsh and Bonnie Howell for dextran endosomal escape studies. We also thank Marissa Vavrek for her careful editing.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of Merck and receive financial support for the research and publication of this article from the company. Any potential conflicts of interests were resolved during the editorial process.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are employee of Merck and receive financial support for the research and publication of this article from the company.