
Abstract
Cancer-associated changes in cellular behavior, such as modified cell-cell contact, increased migratory potential, and generation of cellular force, all require alteration of the cytoskeleton. Two homologous mammalian serine/threonine kinases, Rho-associated protein kinases (ROCK I and II), are key regulators of the actin cytoskeleton acting downstream of the small GTPase Rho. ROCK is associated with cancer progression, and ROCK protein expression is elevated in several types of cancer. ROCKs exist in a closed, inactive conformation under quiescent conditions, which is changed to an open, active conformation by the direct binding of guanosine triphosphate (GTP)–loaded Rho. In recent years, a number of ROCK isoform-specific binding partners have been found to modulate the kinase activity through direct interactions with the catalytic domain or via altered cellular localization of the kinases. Thus, these findings demonstrate additional modes to regulate ROCK activity. This review describes the molecular mechanisms of ROCK activity regulation in cancer, with emphasis on ROCK isoform-specific regulation and interaction partners, and discusses the potential of ROCKs as therapeutic targets in cancer.
Keywords
Cancer is a leading cause of death worldwide (Ferlay et al. 2010; Bray et al. 2012). Initiation and progression of cancer are multistep events involving cellular transformation, tumor growth, neovascularization, invasion, and metastasis (Hanahan and Weinberg 2011). Alterations to the actin cytoskeleton cause changes in cell adhesion, migration, and invasion, resulting in epithelial-mesenchymal or mesenchymal-epithelial transition, metastasis, neoangiogenesis, and infiltration of immune cells. Moreover, reorganization of the actin cytoskeleton can affect gene expression, the cell cycle, vesicular trafficking, and remodeling of the extracellular matrix (ECM) (Ridley 2006; Singh et al. 2010; Skarp and Vartiainen 2010); this emphasizes the many important roles of the actin cytoskeleton in the behavior of both cancer and cancer-associated cells, such as fibroblasts and endothelial cells (Pietras and Ostman 2010; Hanahan and Coussens 2012).
The Rho-associated protein kinases (ROCKs or Rho kinases) are central regulators of the actin cytoskeleton downstream of the small GTPase Rho. Rho is a molecular switch cycling between guanosine diphosphate (GDP)– and guanosine triphosphate (GTP)–bound states under signaling through growth factors or cell adhesion receptors (Jaffe and Hall 2005). The ROCKs belong to the AGC family of classical serine/threonine protein kinases, a group that also includes other regulators of cell shape and motility, such as citron Rho-interacting kinase (CRIK), dystrophia myotonica protein kinase (DMPK), and the myotonic dystrophy kinase-related Cdc42-binding kinases (MRCKs) (Pearce et al. 2010). The main function of ROCK signaling is regulation of the cytoskeleton through the phosphorylation of downstream substrates, leading to increased actin filament stabilization and generation of actin-myosin contractility (Amano et al. 2010). ROCK signaling is required for many cytoskeleton-dependent processes, including, but not limited to, cell adhesion, motility, and phagocytosis. As regulators of such cellular processes, ROCKs are important players in smooth muscle contractility (and, therefore, vasculature tone) and in neuronal development and nerve regeneration (Schmandke et al. 2007; Wang et al. 2009). ROCK signaling plays critical roles in a range of human diseases and is being considered as a potential target for treatment for a number of diseases, including diabetic nephropathy (Komers 2011), as well as diseases in the central nervous system and the cardiovascular system (including spinal cord injury, vasospasm, hypertension, atherosclerosis, and myocardial hypertrophy) (Kubo et al. 2008; Satoh et al. 2011; Zhou et al. 2011). Recently, it has been recognized that ROCKs are also major modulators of tumor invasion (Liu S et al. 2009; Narumiya et al. 2009).
ROCK Structure and Conventional Activation
The two ROCKs, ROCK I (also known as p160ROCK and ROKβ) and ROCK II (Rho-kinase and ROKα), are 160-kDa proteins encoded by distinct genes. The mRNA of both kinases is ubiquitously expressed (Leung et al. 1996; Nakagawa et al. 1996), but ROCK I protein is mainly found in organs such as liver, kidney, and lung, whereas ROCK II protein is mainly expressed in muscle and brain tissue.
The amino acid sequences of the two ROCKs are highly homologous (~65%), and they exhibit the same overall domain structure (Fig. 1A). The kinase domain is located at the N-terminus, followed by a coiled-coil region containing a Rho-binding domain (RB). At the C-terminus is a pleckstrin homology domain (PH) split by a cysteine-rich region (CR) (Wen et al. 2008). The highest degree of similarity between ROCK I and ROCK II is found within the kinase domain (92% identity) (Nakagawa et al. 1996). The kinase domains of the ROCKs are distinct from most other protein kinases in that they require both an N- and a C-terminal extension segment for catalytic activity (Leung et al. 1996). The N-terminal extension segment brings two ROCK kinase domains together to form head-to-head homodimers (Fig. 1B) (Jacobs et al. 2006; Yamaguchi et al. 2006), a structural formation further supported by crystallization analyses of other domains of the ROCKs, which all form parallel homodimers (Shimizu T et al. 2003; Dvorsky et al. 2004; Wen et al. 2008; Tu et al. 2011). ROCK dimer formation through homophilic interactions via the coiled-coil domains and the kinase domains was also suggested by pull-down experiments of recombinant domains (Chen et al. 2002). The crystal structure of the coiled-coil domain of ROCK I suggests conformational flexibility, based on flexible amino acids that are either conserved or replaced by other flexible amino acids in ROCK II (Shimizu T et al. 2003; Tu et al. 2011). This correlates well with reports on autoinhibitory properties of ROCKs. In the autoinhibitory conformation, the C-terminal RB and PH domains bind to the kinase domain, rendering the kinase inactive (Fig. 1B) (Amano et al. 1999).
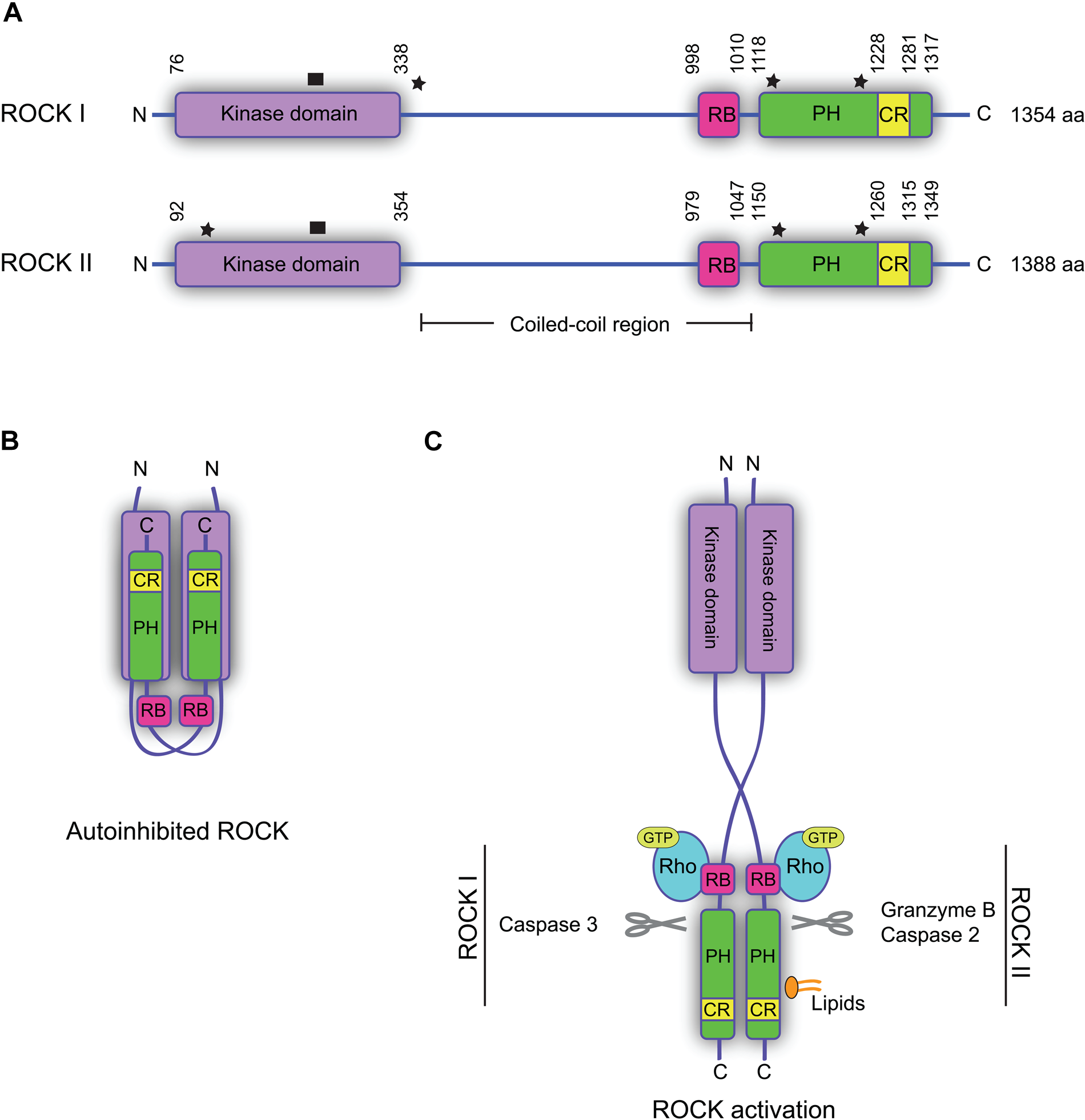
Structure and regulation of the Rho-associated protein kinases (ROCKs). (A) Domain structures of human ROCK I and II. The kinase domain is located in the N-terminus and is flanked by extension segments. RB, Rho-binding domain; PH, pleckstrin homology domain; CR, cysteine-rich region. Star: sites of somatic mutations associated with human cancers. ROCK I: Y405*, S1126*, and P1193S; ROCK II: W138*, Y1174*, and S1194P (*premature translation termination point). Black box: site of activation loop. (B) Model of autoinhibited ROCK, where the C-terminal part of one ROCK interacts with the kinase of the other and inhibits the activity. (C) Model of ROCK activation. Binding of Rho-GTP to the RB domain of both ROCKs, or binding of lipids to the PH domain of ROCK II, leads to a conformational change and activation of the kinases. Cleavage of the N-terminus of the PH domain makes ROCK constitutively active. Indicated proteases can cleave the ROCKs and release constitutively active forms.
The autoinhibitory conformation must be altered in order for the ROCKs to become active; however, as no means of detecting active ROCK directly exist, phosphorylation of downstream substrates is used as a measure of ROCK activity. The best-characterized mode of ROCK activation is the binding of Rho-GTP to the RB domain of the kinase (Fig. 1C). This induces a conformational change, making the kinase domain free and thereby active (Amano et al. 1999). The crystal structures of the ROCK catalytic domains indicate that, unlike many other protein kinases, phosphorylation of the activation loop of ROCK is not required for full catalytic activity (Fig. 1A) (Jacobs et al. 2006; Yamaguchi et al. 2006). In apoptotic cells, the ROCKs can also be activated by protease cleavage (by granzyme B and caspase-2 or -3) in the N-terminus of the PH domain (Coleman et al. 2001; Sebbagh et al. 2001, 2005; Sapet et al. 2006). This cleavage makes the kinases constitutively active, causing membrane bleb formation (Fig. 1C). In addition, binding of lipids, such as arachidonic acid or phosphatidylinositol (PI) phosphates, to the PH domain of ROCK II also results in kinase activation in the absence of Rho-GTP (Fig. 1C) (Feng et al. 1999; Yoneda et al. 2005). Although ROCK I is able to bind lipids, it remains to be clarified if this binding leads to kinase activation (Wen et al. 2008). Recently, autophosphorylation of ROCK II at Ser1366 was suggested to reflect the activation status of the kinase (Chuang et al. 2012). However, whether autophosphorylation at this site truly reflects the activity status still remains to be investigated. The effect of this phosphorylation, if any, on protein targeting or protein-protein interactions also remains uncharacterized.
Functional Roles of ROCKs
At the time when the ROCKs were first identified almost 20 years ago, they were suggested to be regulators of the actin cytoskeleton downstream of Rho (Leung et al. 1995; Ishizaki et al. 1996; Matsui et al. 1996). Since then, a range of substrates (Amano et al. 2010) and interaction partners for ROCKs have been identified. Regulatory binding partners of the two ROCKs are listed in Table 1. Many of the substrates/interaction partners are linked to regulation of the actin cytoskeleton, including ezrin/radixin/moesin (ERM), the LIM-kinases (LIMK), myosin light chain (MLC), and MLC phosphatase (MLCP).
Molecules Regulating ROCK by Direct Binding
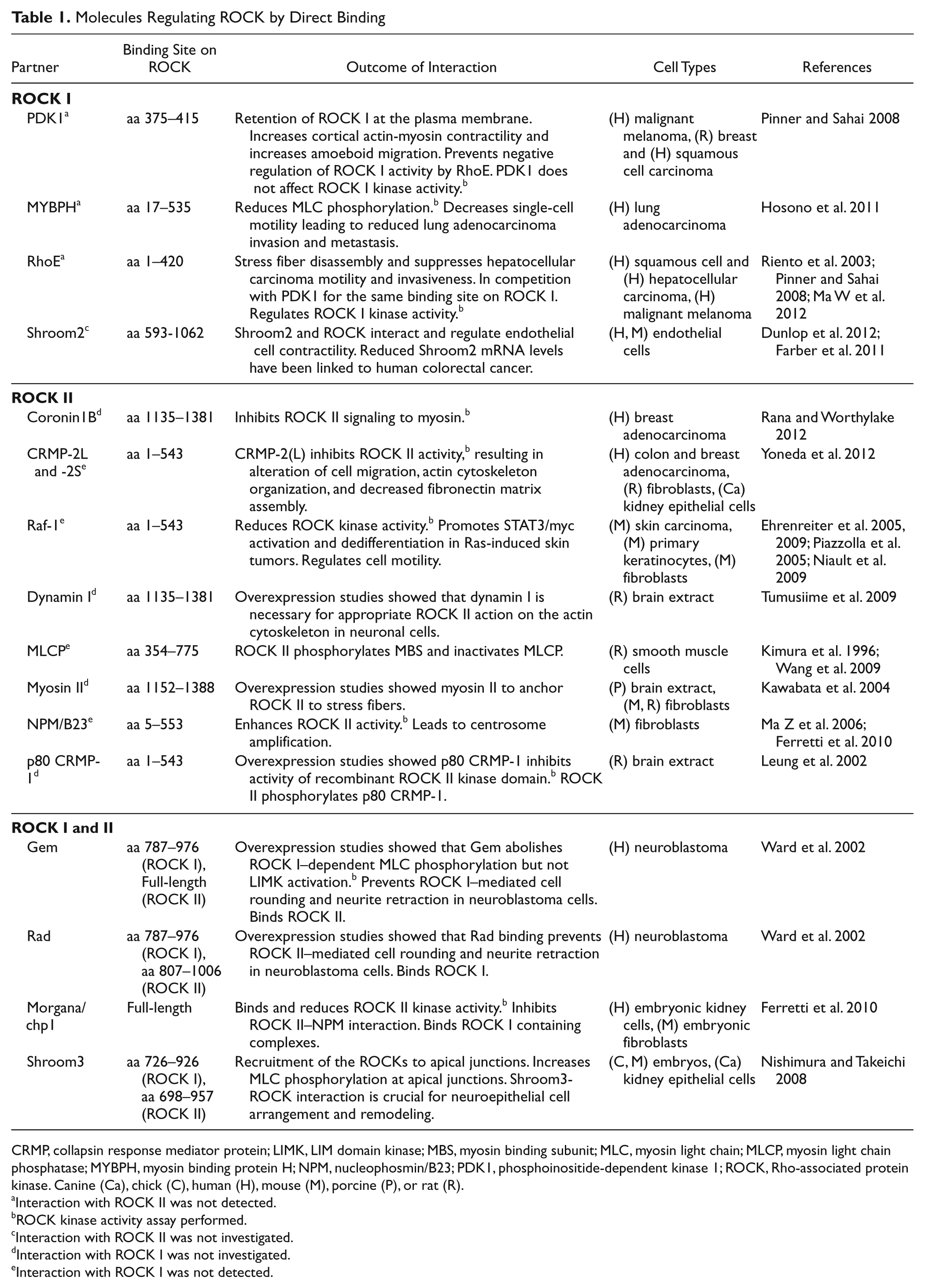
CRMP, collapsin response mediator protein; LIMK, LIM domain kinase; MBS, myosin binding subunit; MLC, myosin light chain; MLCP, myosin light chain phosphatase; MYBPH, myosin binding protein H; NPM, nucleophosmin/B23; PDK1, phosphoinositide-dependent kinase 1; ROCK, Rho-associated protein kinase. Canine (Ca), chick (C), human (H), mouse (M), porcine (P), or rat (R).
Interaction with ROCK II was not detected.
ROCK kinase activity assay performed.
Interaction with ROCK II was not investigated.
Interaction with ROCK I was not investigated.
Interaction with ROCK I was not detected.
Enhanced actin filament bundling and increased cellular contractility can drive the formation of stress fibers in a myosin II–dependent manner. Myosin II activation requires phosphorylation of MLC, which is regulated by several kinases, including ROCK. ROCK modulates phosphorylation states of MLC by inhibition of MLCP activity through phosphorylation, as well as direct phosphorylation of MLC (Amano et al. 1996; Burridge and Chrzanowska-Wodnicka 1996; Kimura et al. 1996; Kawabata et al. 2004; Wang et al. 2009). Stress fibers terminate at the plasma membrane in specialized sites known as focal adhesions (Wehrle-Haller 2012). Focal adhesions consist of large dynamic protein complexes, which, in addition to being the anchoring point for the actin cytoskeleton, are also sites of cell adhesion to the ECM. Hence, they become sites for the bidirectional transmission of signals and force between the cell and the ECM.
Anchoring of actin filaments to integral proteins of the plasma membrane can be driven by ROCK phosphorylation of ERM protein family members (Matsui et al. 1998). ERM proteins contain both a C-terminal actin-binding subunit and an N-terminal 4.1/ezrin/radixin/moesin (FERM) domain, which allows interactions with plasma membrane proteins such as CD44 (Tsukita et al. 1994). The actin cytoskeleton is further stabilized by the activation of the LIMK1/2 through phosphorylation by ROCK (Ohashi et al. 2000; Amano et al. 2001). Active LIMK1/2 phosphorylates cofilin, an actin-depolymerization factor, leading to inactivation of cofilin and reduced depolymerization of actin filaments (Arber et al. 1998). Rho-ROCK signaling is also implicated in cell cycle regulation. For example, polyploidization naturally occurs in megakaryocytes due to an incomplete mitosis, which is related to a partial defect in Rho-ROCK activation, and leads to an abnormal contractile ring lacking myosin IIA (Lordier et al. 2008). Moreover, Rho-ROCK signaling increases cyclin D1 and Cip1 protein levels, stimulating G1/S cell cycle progression (Croft and Olson 2006).
Despite a high degree of homology between the two ROCKs, as well as the fact that they share several common substrates, studies have clearly shown that the two ROCK isoforms also have distinct and non-redundant functions. By knockdown studies using small interfering RNA, we showed that, in fibroblasts, ROCK I is essential for the formation of stress fibers and focal adhesions, whereas ROCK II is required for myosin II–dependent phagocytosis (Yoneda et al. 2005). This difference is partially due to localization of the two ROCKs to distinct subcellular compartments; ROCK II (but not ROCK I) is localized to the cell periphery in both fibroblasts and breast cancer cells (Fig. 2). Moreover, we have recently identified a molecular mechanism of ROCK II–specific activity regulation as discussed below (Yoneda et al. 2012). Mice with targeted deletion of ROCK I or ROCK II exhibit some similar phenotypes—that is, they have open eyelids and an open body wall at birth due to disorganization of actin filaments in the epithelial cells (Shimizu Y et al. 2005; Thumkeo et al. 2005). However, differences in function between the two isoforms have been observed in ROCK I and II knockout studies. ROCK II knockout mice show intrauterine growth retardation caused by deregulation of the placenta, limited axonal growth after trauma to the central nervous system, and enhanced adipogenesis (Thumkeo et al. 2003; Noguchi et al. 2007; Duffy et al. 2009), whereas studies of ROCK I knockout mice have suggested that the protein is involved in cardiac fibrosis development, cardiomyocyte apoptosis, insulin resistance, and acute inflammation (Zhang YM et al. 2006; Noma et al. 2008; Lee et al. 2009; Vemula et al. 2010).
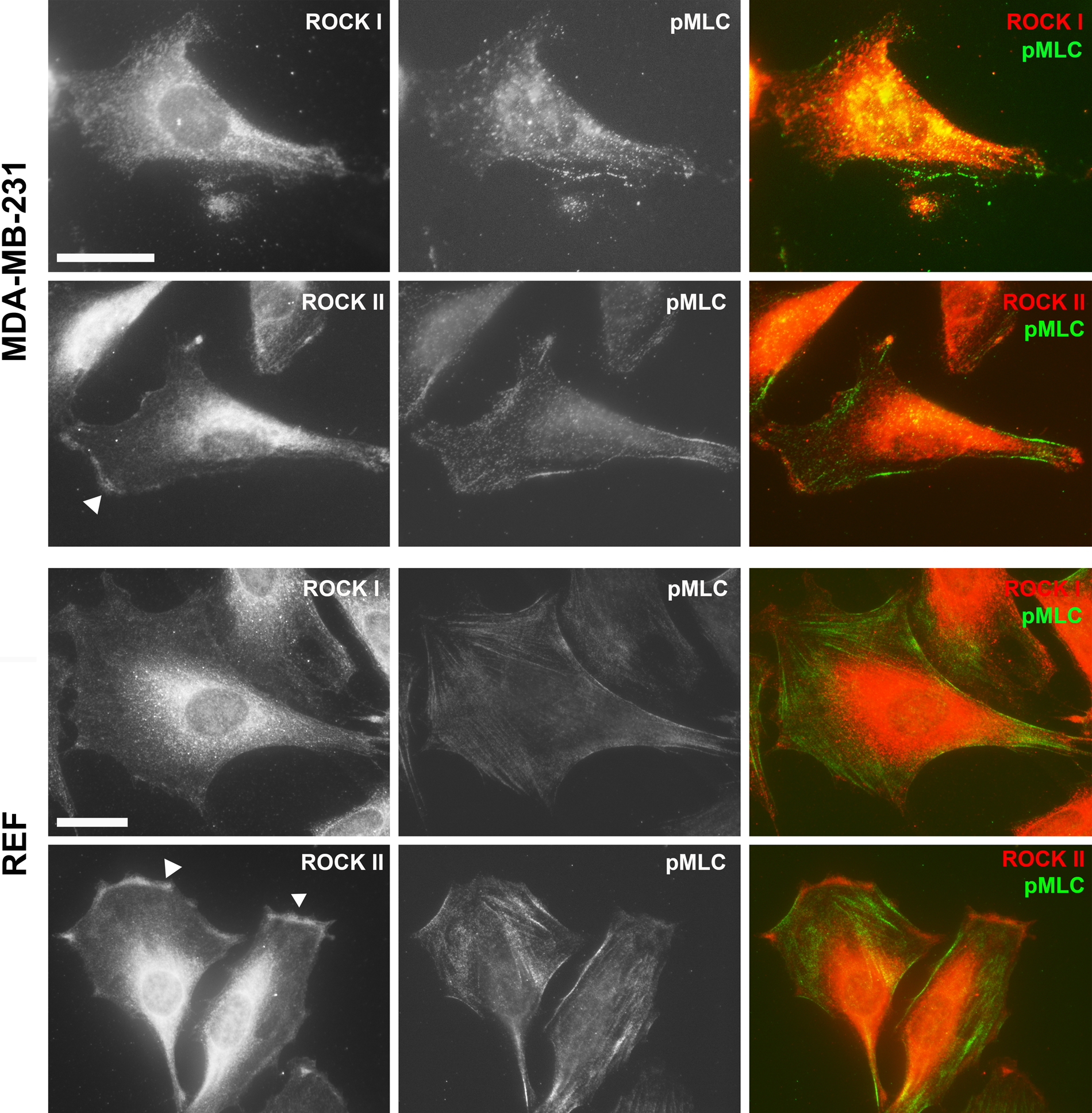
Cellular localization of Rho-associated protein kinase (ROCK) I and II. Immunofluorescence staining of ROCK I and II proteins together with phosphorylated myosin light chain (MLC) in human breast carcinoma cells (MDA-MB-231) and rat embryo fibroblasts (REF) with goat anti-ROCK I or ROCK II and rabbit anti-pMLC2 (Ser19) antibodies as described previously (Yoneda et al. 2005). ROCK II (arrowheads) but not ROCK I was detected at the cell periphery. Bar = 25 µm.
Expression of ROCKs in Human Cancers
Total activity of ROCKs reflects both their expression and activation status. In this section, the expression of ROCKs in cancer will be discussed.
Somatic mutations in both ROCK genes, some of which encode the constitutively active form, have been identified in human cancer genomes originating from cancer cell lines and human primary tumors (Greenman et al. 2007; Forbes et al. 2008, 2010). Three of the identified mutations in the ROCK 1 gene have been analyzed: two leading to premature termination of translation at Y405 and S1126 in primary human breast cancers and one leading to a substitution of proline 1193 with a serine in a non–small cell lung carcinoma cell line (Fig. 1A) (Greenman et al. 2007; Lochhead et al. 2010). All three mutations cause increased kinase activity due to the absence of autoinhibition (Lochhead et al. 2010). Mutations have also been identified in the ROCK 2 gene in primary stomach carcinoma and malignant melanoma cell lines (Greenman et al. 2007), two of which (Y1174 and S1194P) are equivalent to the mutations identified in ROCK 1 in similar positions (Fig. 1A). This suggests that these mutations in ROCK 2 cause increased kinase activity. A third mutation in ROCK 2 leads to premature termination of translation at W138 (Fig. 1A), producing a protein that retains a little of the kinase domain, thereby making its ability to exhibit kinase activity doubtful (Fig. 1A) (Lochhead et al. 2010).
Elevated protein levels of ROCKs have been described in several human cancers, but it should be noted that increased protein expression may not necessarily correlate with an increase in total activity of ROCK (discussed in detail below). Nevertheless, protein levels of both ROCKs were elevated in human breast cancer, and high ROCK I expression has been reported to correlate with increased tumor grade as well as poor overall survival (Lane et al. 2008). In addition, high levels of ROCK I protein expression in osteosarcoma correlate with poor overall survival (Liu X et al. 2011). High expression of ROCK II protein has been found to be associated with more aggressive behavior in hepatocellular carcinomas (Wong et al. 2009). Elevated ROCK II protein expression levels have also been reported in colon and bladder cancers and are associated with shorter disease-free survival in patients with bladder cancer (Kamai, Tsujii, et al. 2003; Vishnubhotla et al. 2007). Increased protein expression of the two ROCK isoforms is associated with different types of cancer, but whether ROCK I and II expression promotes disease progression or is a consequence of disease progression remains to be established (Hahmann and Schroeter 2010).
Regulation of ROCK Activity in Cancer
Regulation of ROCK activity occurs in several ways: through regulation of the activation processes, via alteration of the subcellular localization of ROCKs, and by interaction with regulatory molecules (Table 1). As key activator of ROCK, the level of the GTP-bound form of Rho greatly influences ROCK activation. Somatic mutations in RHO genes (RHOA, RHOB, and RHOC) have been found in several different cancers, including breast, lung, ovary, and intestine (Forbes et al. 2008, 2010), but the role and impact of these remain unclear. Rho, however, is overexpressed (mRNA and protein levels) and hyperactivated (due to altered expression and activity of Rho regulatory molecules guanine nucleotide exchange factor [GEF], GTPase activating proteins [GAPs], and Rho GDP-dissociation inhibitors [RhoGDIs]) in several different types of cancer, including breast, colon, and lung cancer, as well as metastatic melanoma (Fritz et al. 1999, 2002; Clark et al. 2000; Burbelo et al. 2004; Harding and Theodorescu 2010; Vigil et al. 2010). RhoA, RhoB, and RhoC, forming a subfamily, exhibit a high degree of homology in amino acid sequence, with, for example, only six non-conservative amino acid substitutions between RhoA and RhoC (Clark et al. 2000). However, the different Rho isoforms, once activated by binding of GTP, show different binding affinity for downstream effector molecules, such as Formin-like 2 and ROCK. Formin-like 2 interacts with RhoC but not RhoA or B (Kitzing et al. 2010). Similarly, the ROCK-binding ability of RhoC is higher than that of RhoA (Sahai and Marshall 2002). Overexpression or increased activation of RhoA protein is associated with advanced stages of human cancer, including invasion and metastasis of testicular germ cell, urinary tract, and cervical cancers (Kamai et al. 2001; Kamai, Kawakami, et al. 2003; He et al. 2010). Depletion of the RhoC gene in mice leads to dramatic inhibition of lung metastasis of malignant melanoma cells (due to a decrease in cell motility and survival) but does not affect tumor initiation and development (Hakem et al. 2005). Enhanced expression of RhoC mRNA and protein has been reported to correlate with a motile and invasive phenotype of breast cancer cells in human clinical samples as well as in a human cancer xenograft model, suggesting significant roles for RhoC in the progression, but not initiation, of cancer (van Golen et al. 1999; Clark et al. 2000; Kleer et al. 2002). Contrary to the significance of the RhoA- and RhoC-ROCK pathways in cancer progression, RhoB protein expression has been shown to have inhibitory effects on migration, invasion, and metastasis of human carcinoma cells via suppression of the Ras/PI3 kinase/Akt pathway (Jiang et al. 2004). Quite interestingly, RhoB protein expression was significantly reduced in tissue from both primary tumor and lymph node metastasis in patients with bladder cancer, in whom protein levels of RhoA and RhoC, as well as both ROCKs, were elevated (Kamai, Tsujii, et al. 2003).
Downstream of active GTP-bound Rho, ROCK activity-dependent cellular contractility can be altered in response to physical properties of the tumor microenvironment such as stiffness (Kraning-Rush et al. 2012). In addition, enhanced ROCK activity can also contribute to increased ECM stiffness, which is often associated with cancer (Jaalouk and Lammerding 2009). Both force-driven deformation and protease cleavage of the ECM are often required for cancer cell migration within high-density ECM (Wyckoff et al. 2006; Kraning-Rush et al. 2012). The deformation of the ECM is executed both by cancer-associated fibroblasts (CAFs) and by cancer cells (Gaggioli et al. 2007). Increased ROCK-dependent myosin II–mediated contractility in CAFs leads to creation of so-called tracks in which the cancer cells can migrate, following the leading fibroblasts (Gaggioli et al. 2007). This type of migration seems to be especially important in collective cancer cell migration (Gaggioli et al. 2007). The molecular background has not been completely elucidated, but migration appears to be initiated by proinflammatory cytokines often observed in the tumor environment and dependent on signaling regulating cellular contractility (Gaggioli et al. 2007; Sanz-Moreno et al. 2011). In a mouse skin tumor model, activation of ROCK II leads to elevated tissue stiffness, via increased collagen deposition, as well as increased tumor number and size, illustrating a relationship between cellular tension, tissue stiffness, and tumor progression (Samuel et al. 2011). In breast cancer, ROCK I appears to be activated in response to increased matrix density (Raviraj et al. 2012). These findings could indicate both cellular and temporal differences in activation of the two kinases in disease progression.
Subcellular Localization
In order for carcinoma cells to migrate and invade the surrounding stroma, they often undergo epithelial-mesenchymal transition. Here, altered cell-cell and cell-ECM interactions lead to release of cells from the surrounding tissue, a common step in both single-cell migration and collective migration of cancer cells (Chaffer and Weinberg 2011). Despite the majority of the ROCK pool being diffusely located in the cytoplasm, a fraction is localized to intracellular sites of cell-cell contacts and regulates the assembly of tight and adherens junctions (Walsh et al. 2001; Sahai and Marshall 2002). In epithelial and neuroepithelial cells, the Apx/Shrm domain 2 (ASD2) of Shroom3, an actin-binding protein, directly binds to the coiled-coil domain of ROCK and drives the relocalization of ROCK to cell-cell contacts, as well as the subsequent construction of apical junctions due to phosphorylation of MLC (Nishimura and Takeichi 2008). In colon carcinoma cells, however, ROCK signaling downstream of RhoC disrupts cell-cell contacts and blocks the formation of adherens junctions, thereby increasing the cellular migratory/invasive potential (Sahai and Marshall 2002). Similarly, in endothelial cells, another member of the Shroom family, Shroom2, binds ROCK via its ASD2 domain and drives cellular contractility. Depletion of Shroom2 from these cells leads to increased angiogenesis due to disorganization of the actin cytoskeleton (Farber et al. 2011). Reduced mRNA expression of Shroom2 has been linked to human colorectal cancer (Dunlop et al. 2012), possibly due to altered ROCK signaling and increased angiogenesis as observed in endothelial cells.
In cancer, metabolism of lipids (e.g., fatty acid and phospholipids) is often altered, leading to changes in lipid composition and availability. This can change lipid-dependent cell signaling, including Rho-ROCK signaling (Sawada et al. 2002; Santos and Schulze 2012). Binding of lipids, as well as proteins, to the PH domain can relocalize ROCK II to subcellular compartments, such as specific membrane areas or stress fibers (Feng et al. 1999; Kawabata et al. 2004; Yoneda et al. 2005; Tumusiime et al. 2009), whereas proteins binding to the kinase domain all seem, not surprisingly, to alter the kinase activity (Table 1). Considering this, one could speculate that binding of lipids or proteins to the PH domain of ROCK II controls its subcellular localization and thereby ultimately the signaling pathway.
Regulatory Molecules
Rho-ROCK signaling is part of a network where crosstalk between different small GTPases and their downstream molecules has a great impact on cell behavior. Interplay between Rho and Rac signaling is a key factor in determining the migratory phenotype of melanoma cells (Sanz-Moreno et al. 2008). Here Rho-ROCK signaling enhances myosin II–mediated contractility and drives amoeboid migration, which is associated with certain types of carcinoma, lymphomas, and leukemia. At the same time, Rho-ROCK signaling inhibits Rac1-dependent mesenchymal migration downstream of β3 integrin and Src (Sanz-Moreno et al. 2008; Ahn et al. 2012). In contrast, activation of Rac inhibits Rho-ROCK signaling via Wiskott-Aldrich syndrome protein family member 2 (WAVE-2) and drives mesenchymal migration, similar to migration of fibroblasts (Sanz-Moreno et al. 2008). In Ras-driven tumorigenesis, crosstalk between the Rho and Ras pathways promotes epithelial dedifferentiation. Here Raf1, a serine/threonine kinase acting downstream of the small GTPase Ras (Moodie et al. 1993) and an endogenous ROCK II inhibitor, regulates ROCK II kinase activity by direct binding of the kinase domain (Table 1) (Ehrenreiter et al. 2005; Piazzolla et al. 2005; Niault et al. 2009). The significance of this interaction was demonstrated in a chemical model of skin carcinogenesis using Raf-1 knockout mice (Ehrenreiter et al. 2009). In neuroblastoma cells, Gem and Rad, members of the Ras small GTPase family, interrupt ROCK effects by direct binding to the coiled-coil domain: Gem prevents ROCK I–induced cell rounding and neurite retraction, whereas Rad has the similar effect on ROCK II. This suggests a role for Gem and Rad binding in regulation of ROCK isoform-specific functions. Gem interaction further abolished ROCK I–mediated phosphorylation of MLC but not of LIMK1/2 (Table 1), indicating that Gem binding could guide ROCK I to a specific pathway or subcellular location (Ward et al. 2002).
With an increasing number of isoform-specific interaction partners identified, a new level of ROCK kinase activity regulation is emerging. Most of these interaction partners, even though ubiquitously expressed (Uhlén et al. 2005, 2010), have been identified in, or connected to, cancer. Interaction of ROCK I and phosphoinositide-dependent kinase 1 (PDK1) at the cell periphery in malignant melanoma has been suggested to play a role in amoeboid cancer cell migration, by increasing contraction of actin-myosin filaments lining the cell periphery (Table 1) (Wyckoff et al. 2006; Pinner and Sahai 2008). ROCK I-PDK1 interaction is antagonized by RhoE (Table 1), which leads to disassembly of stress fibers. This suggests an inhibitory effect of RhoE on ROCK I activity (Riento et al. 2003; Pinner and Sahai 2008). Both liver and prostate cancer have elevated PDK1 and ROCK I mRNA expression and decreased RhoE mRNA expression (Ma W et al. 2012). This could indicate increased ROCK I activity at the plasma membrane, which has been reported to be associated with cortical myosin II–dependent contractility and a high migratory potential (Rhodes et al. 2004, 2007; Pinner and Sahai 2008). Reduced mRNA levels of RhoE further correlate with disease progression in breast and prostate cancer, possibly due to increased ROCK I activity (Pinner and Sahai 2008; Belgiovine et al. 2010). Similarly, morgana/chp-1 and nucleophosmin/B23 (NPM/B23) compete for binding on ROCK II, thereby regulating ROCK II activity, centrosome duplication, and neoplastic transformation (Ma Z et al. 2006; Ferretti et al. 2010).
Downregulation of endogenous ROCK inhibitors in cancer could potentiate more aggressive migratory and invasive behavior. Our laboratory recently described a splice variant of collapsin response mediator protein–2 (CRMP-2L) to be an important regulator of ROCK II–dependent actin cytoskeleton reorganization and migratory behavior in cancer cells (Yoneda et al. 2012). CRMP-2L binds to the ROCK II kinase domain (Table 1), and reduction in total CRMP-2 protein levels in a colon carcinoma cell line that endogenously expresses both CRMP-2S and -2L leads to increased cell migration and increased activity of ROCK II but not of ROCK I. Exogenous expression of CRMP-2L in a colon cancer cell line, established from lymph node metastases, reduced ROCK-dependent cell migration significantly. A similar effect was observed with myosin binding protein H (MYBPH) (Table 1). Downregulation of MYBPH, an inhibitor of ROCK I, led to increased single-cell motility, cancer cell invasion, and metastasis in lung adenocarcinoma (Hosono et al. 2011).
Many of the regulatory molecules discussed in this review have altered expression profiles and levels in cancer as compared with normal tissue and cell lines (Uhlén et al. 2005, 2010; Yoneda et al. 2012). Studies are required to understand the role of these molecules in the regulation of ROCK in normal cells to understand how this is altered in cancer. With the identification of the natural inhibitors, it is becoming increasingly clear that protein expression levels of ROCK and Rho do not necessarily reflect the ROCK activity status, as ROCK can be inhibited by naturally occurring regulatory molecules (Table 1). Moreover, studies of these regulatory molecules demonstrate that important differences exist between the signaling that occurs through ROCK I and ROCK II. These differences affect cancer cell behavior as well as tumorigenesis and metastasis. Further investigation is needed to more completely understand both the molecular background and the impact of the differences in ROCK I and ROCK II signaling.
ROCKs: In Vivo Cancer Models
ROCK isoform-specific contributions to tumor cell behavior have been investigated using mouse and rat models. Following injection of hepatic carcinoma cells into the peritoneal cavity of rats, expression of ROCK I kinase domain or constitutively active Rho resulted in peritoneal dissemination (Itoh et al. 1999). Moreover, the metastatic rate of hepatocellular carcinoma cells in vivo was significantly reduced by expression of a dominant negative form of ROCK I (Genda et al. 1999). Whether ROCK I plays a similar role in humans is not resolved; however, ROCK II rather than ROCK I is overexpressed in human hepatocellular carcinoma (Wong et al. 2009). Using a different approach, the ROCK II kinase domain was coupled to the hormone-binding domain of the estrogen receptor and expressed in colon carcinoma cells grown as tumors in nude mice (Croft et al. 2004). Here activation of the ROCK II kinase domain/estrogen receptor fusion protein by the addition of tamoxifen led to the aggressive dissemination of tumor cells into the surrounding stroma and to the production of more vascularized tumors. Whether ROCK II plays a unique role in vascularization needs further investigation. Taking into account the different phenotypes of ROCK I and ROCK II knockout mice, it seems fair to speculate that, over time, additional in vivo studies will identify different functions in relation to cancer as well.
ROCKs: Potential Targets for Cancer Treatment?
ROCK inhibitors are in use or in clinical trials for the treatment of several clinical conditions. Fasudil, a potent adenosine triphosphate (ATP) competitor for ROCK binding, which is currently in use in Japan for treatment of cerebral vasospasm after subarachnoid hemorrhage, is leading the way for treatment of human diseases by ROCK inhibitors (Olson 2008). Clinical trials to assess the potential effect of fasudil have also been conducted for several cardiovascular conditions, including hypertension, atherosclerosis, and aortic stiffness (Olson 2008). Other ROCK inhibitors have been through phase I and II trials for glaucoma and spinal cord injury (Hahmann and Schroeter 2010).
Currently, no ROCK inhibitor is in use or in clinical trials for treatment of human cancers, but several of the ROCK inhibitors have shown effects both in cancer cell lines and in rodent cancer models, supporting the overall importance of ROCK signaling in the development and progression of cancer. Fasudil, Wf-536, Y-27632, and, most recently, RKI-1447 have all been shown to reduce tumor progression of hepatocellular and lung carcinomas, myeloma, and breast cancers in mice (Itoh et al. 1999; Takamura et al. 2001; Nakajima et al. 2003; Ying et al. 2006; Patel et al. 2012). Fasudil and Wf-536 also attenuate cancer-associated angiogenesis in vivo and in vitro lung carcinoma models (Nakajima et al. 2003; Zhang Z et al. 2012). Administration of Slx-2119, a potent selective ROCK II inhibitor, in a human tumor xenograft model resulted in a significant dose-dependent delay in tumor growth with acceptable toxicity (Shifrin et al. 2005; Boerma et al. 2008). Together, these data suggest a potential application for ROCK inhibitors not only for reducing metastasis but also for reducing primary tumor growth. As ROCK inhibitors also target other kinases, and the ROCK knockout mice have not been used in cancer-associated studies, the role of ROCK in cancer initiation and progression remains unclear.
Due to the role of ROCK in regulating vascular tone, the administration of ROCK inhibitors can lead to a lowering of blood pressure. However, induction of pressure overload cardiac hypertrophy in mice leads to elevated ROCK I, but not ROCK II, expression (Hahmann and Schroeter 2010). From this, one could speculate that targeting ROCK II would have less toxicity than inhibitors targeting either both isoforms or ROCK I specifically. Attempts to produce more specific and clinically suitable ROCK inhibitors are ongoing, with increased focus on isoform-specific regulation and inhibition (Hahmann and Schroeter 2010).
Concluding Remarks
ROCK activity and signaling are key elements in invasive and metastatic cancer cell behavior, as well as in tumor growth and cancer-associated alterations of ECM. As discussed in this review, it is becoming increasingly clear that ROCK signaling depends on several factors, including cell type, subcellular location, and downstream interaction partners, and that signaling by the two ROCKs is partially non-redundant. Regulation of ROCK isoform (ROCK I and II) activities appears to be separated both spatially and temporally.
ROCK kinase activity is greatly influenced by the expression of inhibitory proteins such as CRMP-2L and MYBPH. As a consequence, protein expression levels of Rho and ROCK do not necessarily correlate with ROCK activity level. Moreover, as natural regulators of ROCK, such as Gem, only inhibit phosphorylation of some downstream targets, the need for tools able to detect the true active form of the ROCK is obvious. Such tools will help elucidate the regulation of ROCK activation, the subcellular localization of active/inactive pools of the kinases, and the conditions in which ROCK I and II become activated.
Both in vivo and in vitro studies implicate ROCK as a potential target for cancer treatment, especially to prevent metastasis. The knowledge obtained from mouse models on the specific roles of the two ROCKs might be limited but implicates ROCK I and ROCK II in cancer cell dissemination, formation of metastasis, and tumor vascularization. Further studies are needed to elucidate the unique roles of ROCK I and ROCK II in the development and progression of different human cancers. Tissue-specific conditional knockout mice for both ROCKs would be of great use for this purpose.
With increasing knowledge of the regulation and localization of ROCK in different cellular settings, the prospect of producing anticancer drugs targeting ROCK is increasing. The goal is to reduce tumor growth, decrease metastasis formation, and improve disease outcome.
Footnotes
Acknowledgements
We thank Professor John R. Couchman for helpful discussions and Linda Raab for editorial assistance. We apologize to our colleagues whose work was not cited due to space limitations.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully acknowledge support from the Faculty of Health Sciences at the University of Copenhagen (MM-F), The Danish Cancer Society and Arvid Nilsson’s Fond (UMW), and The Novo Nordisk Foundation, The Danish Cancer Research Foundation, and The Danish Medical Research Council (AY).