
Abstract
The widespread use of whole genome analysis based on array comparative genomic hybridization in diagnostics and research has led to a continuously growing number of microdeletion and microduplication syndromes (MMSs) connected to certain phenotypes. These MMSs also include increasing instances in which the critical region can be reciprocally deleted or duplicated. This review catalogues the currently known MMSs and the corresponding critical regions including phenotypic consequences. Besides the pathogenic pathways leading to such rearrangements, the different detection methods and their limitations are discussed. Finally, the databases available for distinguishing between reported benign or pathogenic copy number alterations are highlighted. Overall, a review of MMSs that previously were also denoted “genomic disorders” or “contiguous gene syndromes” is given.
Keywords
Intellectual disability in humans is characterized by significantly impaired cognitive functioning in skills such as communicating, taking care of oneself, and social interactions. Factors such as maternal drug abuse during pregnancy, perinatal oxygen distress, or postnatal infections can be reasons for intellectual disability; however, causative genetic alterations can often be identified. These genetic reasons for intellectual disability in human can be studied by different approaches, related to the assumed underlying genetic defect. Nowadays, banding cytogenetics is still the most widely used initial test in routine diagnostics. If a normal karyotype is observed, further tests may include molecular cytogenetics, to exclude cryptic rearrangements, or molecular genetics. During the past few years an increasing number of so-called contiguous gene syndromes (CGSs) have been identified, mainly in patients with intellectual disability along with a limited number of other syndromes or diseases. CGSs are caused by an aberrant copy number (gain or loss) of a specific subchromosomal region. Originally, CGSs were considered to have critical regions of two or more genes, located in close proximity to each other. Meanwhile, it is known for some of these syndromes that many genes may be involved in the usually duplicated or deleted region, but only one of them is gene-dosage sensitive and causative for the specific clinical signs. Thus, the denomination CGS has been replaced by the designation microdeletion or microduplication syndromes (MMSs).
Emerging Numbers of MMSs
One model system for MMSs is the Charcot–Marie–Tooth syndrome type 1A (CMT1A) caused by a duplication of the PMP22 gene and its reciprocal microdeletion syndrome hereditary neuropathy with liability to pressure palsies (HNPPs) (Lupski, 1999). Although before the year 2000 only some dozens of MMSs were known, more and more MMSs have been identified with the availability of new technologies for high-resolution analyses of entire genomes. In particular, the array comparative genomic hybridization (aCGH) technique, which entered the human genome research and diagnostic fields in the end of the last century (Pinkel et al. 1998), was groundbreaking. The hallmark of aCGH is the identification of submicroscopic gains or losses of euchromatic material, recently called benign or pathogenic copy number variations (CNVs). Although for a pathogenic CNV, correlation with a certain syndrome/phenotype is possible, no such relation is known (yet) for benign CNVs. A pathogenic effect of a CNV is suggested if it is de novo (e.g., Alesi et al. 2011) or if a specific CNV cannot be found in numerous unrelated healthy individuals studied in parallel (e.g., Willat et al. 2005).
Meanwhile, the common use of aCGH in research and diagnostics has led to an increase of known benign as well as disease-causing pathogenic CNVs, which is especially reflected in the still-growing numbers of publications. From 1990 to 2011, 200 papers concerning new MMSs were published (Fig. 1). Keeping in mind that a common cause for recurrent microdeletions and microduplications is the structure of the human genome, which has countless repeats that can result in non-allelic homologue recombination (NAHR), the expected total number of reciprocal MMSs should be much higher (Gu et al. 2008). Theoretically, for every microdeletion syndrome there should be a reciprocal microduplication syndrome. However, there are at present 211 microdeletion syndromes versus only 79 microduplication syndromes reported (Table 1, Suppl. Table 1, Figure 2). This is a 2.5:1 ratio for a total of 267 different genomic loci with MMSs. Only for 56 of these, loci are reported as reciprocal/colocalizing MMSs, that is, 21%. This is due to several reasons; for instance, meiotic errors leading to duplication and deletion should occur at equal frequencies, but recent studies indicate that early selection during gametogenesis favors either one or the other (Turner et al. 2008). In fact, it is a general observation that microduplications appear to result in a milder or no clinical phenotype compared with the reciprocal microdeletion. This is even the case on the chromosomal level: trisomies of whole chromosomes or supernumerary marker chromosomes are better tolerated (Liehr et al. 2011) than are autosomal monosomies.
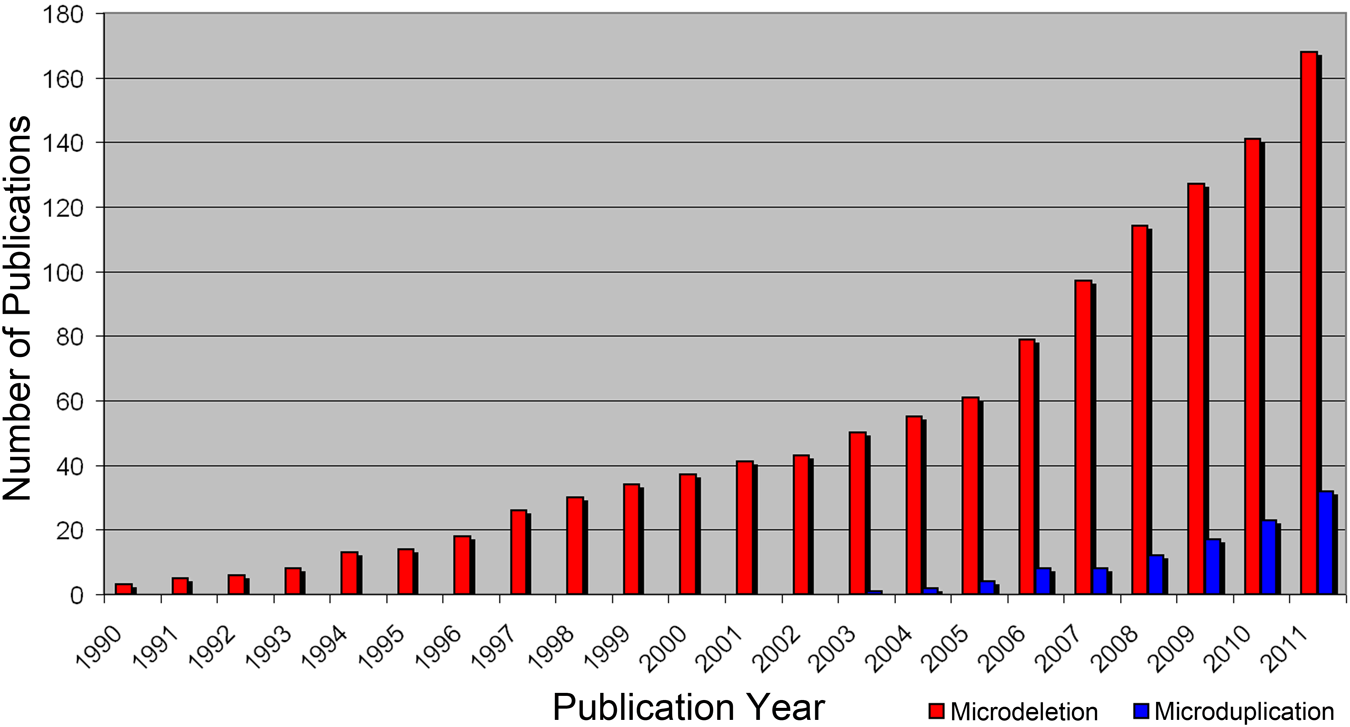
Growing numbers of publication per year found by searching for the terms microdeletion syndrome new and microduplication syndrome new in Pub Med (http://www.ncbi.nlm.nih.gov/pubmed).
All Known Microdeletions and/or Reciprocal Microduplications up to January 2012 by Chromosomes from pter to qter That Are Reported at Least in Two Different Studies or in More Than One Individual

Reported genes in certain MMSs are given in italics in brackets; abbreviations that were also used in Fig. 2 are given in capital letters after the slash. If known, the OMIM number is reported as well as the cytogenetic localization and start and end positions in kilobasepairs (kb), human genome version 18 [hg18]. Additional information on locus specific bacterial artificial chromosomes for every region, as well as the reference are given in a supplementary table. Dash/empty stands for no known reciprocal MMS up to Janurary 2012.

Schematic overview of all genomic microdeletion and microduplication regions reported at least twice. Red arrows indicate reported microdeletions, blue arrows microduplications, and mixed red/blue arrows reciprocal microduplication and microdeletion regions. For details on each indicated region and abbreviations, refer to Table 1.
Causes for Recurrence of MMSs
The basis of recurrent genomic rearrangements such as deletions, duplications, insertions, inversions, and translocations is the architecture of the primate/human genome. Innumerable repetitive elements serve as substrates for illegitimate intra- or interchromosomal/chromatide recombination during meiosis as well as in mitosis. This human specific genomic instability is not only causative, for example, for MMSs, but also has a great impact on genome evolution and flexibility in terms of gaining new gene functions or direct gene dosage effects by euchromatic copy number alterations (Marques-Bonet and Eichler, 2009; Gazave et al. 2011). Most recurrent genomic rearrangements are mediated by sequences such as segmental duplications (Bailey and Eichler, 2006) and low copy repeats flanking a certain region and allow NAHR leading to a high number of same-size de novo rearrangements (Stankiewicz and Lupski 2002) or SINEs/LINEs/LTRs (Korbel et al. 2007; Kidd et al. 2008). The second, rarer cause of genomic rearrangements is non-homologous end joining (NHEJ) following double stranded breaks, which is a cell repair mechanism and not directly mediated by specific sequence features (Lieber 2008). NHEJ can affect the same region but not the exact breakpoint in a group of patients covering a dosage-sensitive gene in the shortest region of overlap that all patients share. Also, NHEJ appears to be common in instable genome regions such as fragile sites, where some of the microdeletions and duplications are located (Schwartz et al. 2005; Bena et al. 2010; Mrasek et al. 2010). The third proposed model is DNA replication–based fork stalling and template switching, which accounts for complex rearrangements and is at least facilitated by one recurrent breakpoint through specific elements such as palindrome or cruciform DNA sequences (Lee C et al. 2007).
Moreover, recurrent MMSs can arise from an inversion polymorphism in one of the parental genomes (e.g., Gimelli et al. 2003; Koolen et al. 2006) or from a balanced translocation (Shaffer 2001). These inversion polymorphisms occur between inverted homologous sequences and are predisposed for NAHR in meiosis through inversion loop formation leading to recombinant products with deletions and duplications (Bhatt et al. 2009).
Methods to Detect and Analyze MMSs
Genomic Microarrays
Genomic microarrays refer to the principle of chromosome-based comparative genomic hybridization (CGH; Kallioniemi et al. 1992), where test DNA and control DNA are differentially labeled, mixed in a 1:1 ratio, and hybridized together with Cot 1 DNA to metaphase spreads of a normal donor. This special fluorescence in situ hybridization (FISH) procedure results in a yellow staining of regions unaffected by CNV. Green and red colors along the chromosomes indicate genetic loss or gain in the DNA derived from the patient. Currently, hybridization is no longer done on metaphase spreads but on slides with spots of defined genomic sequences. This leads to a higher and better resolution compared with CGH depending on the number and the size of the probes used (Pinkel et al. 1998; Lee C et al. 2007). Besides copy number alterations, single nucleotide polymorphism (SNP)–based aCGH provides additional information on loss of heterozygosity, indicating a deletion or uniparental isodisomy.
Depending on the resolution, target size of the different platforms [bacterial artificial chromosomes (BACs), fosmids, oligonucleotides, SNPs], and the analysis criteria, MMSs as well as benign CNVs can be detected with varying accuracy. Thus, especially for benign CNVs, problems may arise when data from different platforms are compared by annotating them, for example, in the “database of genomic variants” (DGV). The exact sizes of the corresponding benign CNV might be available after the currently ongoing “1000 genomes project” (Sudmant et al. 2010) is finished.
In most cases, aCGH is used as a genome-wide screening method. Therefore, after all technical problems are solved, the main challenge is in the interpretation of all the genomic data. This step, together with the verification of an aberration (see below), is the most labor intensive. The most critical decision is whether a CNV is considered benign or disease causative, along with final interpretation.
Fluorescence In Situ Hybridization
Molecular cytogenetics, especially the FISH technique, is used for direct analysis of a certain suspected MMS critical region by applying locus-specific DNA probes. FISH is used when a physician suspects a specific MMS or for verification of aCGH data. The advantage of the FISH approach is that single-cell information in the context of metaphase chromosomes becomes available. Besides the visualization of a balanced chromosomal aberration in a parent (leading to unbalanced situations in the index patient), mosaicism and complex rearrangements in a patient can also be detected (Mkrtchyan et al. 2010; Fig. 3).
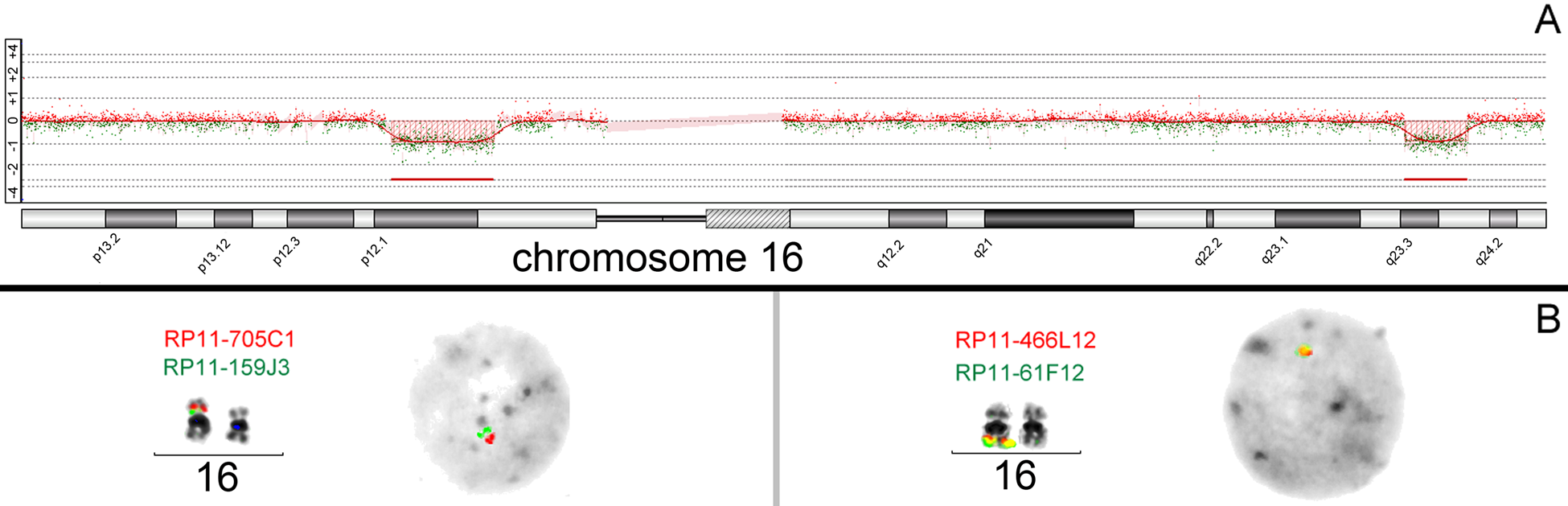
(A) Example of microdeletion findings in array comparative genomic hybridization (aCGH) (Agilent Human Genome CGH Microarray 180k) in a female with karyotype 46,XX. The patient showed two de novo microdeletions (hg18): arr 16p12.1p11.2(22.665685–28.536945)x1/del(16)(p12.1p11.2)(5.871269–6.106456 Mb) arr 16q23.3q24.1(80.662135–84.286121)x1/del(16)(q23.3q24.1)(3.623986–3.648342 Mb) (B) The aCGH result was confirmed by fluorescence in situ hybridization with bacterial artificial chromosome clones located in the deletion regions. The microdeletions were confirmed on metaphase chromosomes and interphase nuclei.
One important limitation of the FISH approach is that the size of locus-specific probes should not be less than 10 kb (Liehr et al. 1997). Furthermore, duplications are harder to verify by FISH than deletions; however, this drawback can be overcome by analysis of interphase nuclei in addition to metaphase spreads and/or the use of software signal intensity and size measurements (Weise et al. 2008).
For classic, that is, well-known, MMSs, a panel of commercial available probes can be used. For all other genomic regions to be tested for CNVs, BACs and fosmids generated by the human genome project with annotated sequence/location/size information are good sources for directed microduplication and deletion testing (Weise et al. 2009). A list with suggested BAC probes for the currently known MMSs regions is given in Suppl. Table 1.
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction (qPCR) was initially established as a method for quantifying different levels of gene expression. However, under the need to verify aCGH results, qPCR is now also routinely applied to detect CNVs (Weksberg et al. 2005). The qPCR technique provides a quantitative measurement of DNA CNVs of (nearly) any region of interest. The main limitations or problems are to find accurate primer pairs, to optimize the PCR conditions, and to detect mosaicism. Additionally, when using the parents of an index patient for CNV verification, balanced rearrangements with a higher recurrence risk in offspring are not recognizable.
Multiplex Ligation-Dependent Probe Amplification
Multiplex ligation-dependent probe amplification (MLPA) is a directed multiplex PCR-based method. Only those primers that hybridize to the target sequences are amplified, and the resulting products can be analyzed by capillary electrophoresis. MLPA allows the detection of abnormal copy numbers of up to 50 different genomic sequences at the same time. Comparing the peak pattern obtained to that of reference samples indicates which sequences show aberrant copy numbers. The technique is commercially available for several sets of MMSs and therefore can serve as a time- and cost-reduced first or second screening step method (Jehee et al. 2011). Again, the limitations are the same as in aCGH and qPCR: no proper detection of mosaicism and no verification of balanced origin in one parent. In addition, MLPA has the disadvantage that it covers only a limited number of loci. Furthermore, for newly described MMSs, a specific MLPA test has to be designed.
Databases for MMSs
To facilitate the analysis, especially of aCGH data, several databases are available summarizing genomic imbalances under different aspects. Before using these databases, one should check which version of the human genome sequence is used in one’s own aCGH platform and the database, as the coordinates move in different versions. The currently used version is hg19 or Build 37.3, but not all databases listed below refer to this version and need to be updated or converted by the user.
The first step for analysis and interpretation of aCGH results is the classification of a detected CNV as benign or disease causative. This question can be answered best by the use of genome browsers showing both reported benign and pathological CNVs, such as the genome browser from the University of California, Santa Cruz (UCSC; http://genome.ucsc.edu/cgi-bin/hgGateway), the U.S. National Institutes of Health genome browser (NCBI; http://www.ncbi.nlm.nih.gov), or the Ensemble genome browser (http://www.ensembl.org/Homo_sapiens). These genome browsers also provide information on BAC or fosmid clones that can be used for FISH verification of the CNV. Furthermore, the direct sequence can be downloaded or BLASTed for qPCR and MLPA design. Some of the genome browsers’ subdatabases are also directly available.
Copy Number Variation and Polymorphism
Database of genomic variants (DGV): http://projects.tcag.ca/variation
Catalogue of structural and copy number variation in the human genome
Chromosome Anomaly Collection: http://www.ngrl.org.uk/Wessex/collection
Collection of Unbalanced Chromosome Abnormalities (UBCAs) and Euchromatic Variants (EVs) visible by light microscope but without any phenotypic effect
Small supernumerary marker chromosomes. http://www.med.uni-jena.de/fish/sSMC/00START.htm
Collection of all available case reports on small supernumerary marker chromosomes (sSMCs) with definition of critical regions for partial trisomies due to the presence of sSMCs
Catalogues of Pathological Imbalances
Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER): https://decipher.sanger.ac.uk/syndromes
An interactive Web-based database that incorporates a suite of tools designed to aid the interpretation of submicroscopic chromosomal imbalance
European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA): http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp
Database that collects and provides cytogenetic and clinical information on rare chromosomal disorders, including microdeletions and microduplications
The Chromosome Microdeletion/duplication Collection: http://www.ngrl.org.uk/wessex/microdel_collection.htm
Collection of MMS with the aim to interpret results of array CGH analysis
Literature and Educational Resources
Chromosomal Variation in Man: http://www.wiley.com/legacy/products/subject/life/borgaonkar/
Interactive database and searching tool by chromosomes and regions on reviewed literature on all common and rare chromosomal alterations and abnormalities
Online Mendelian inheritance in man (OMIM): http://www.ncbi.nlm.nih.gov/omim
Comprehensive, authoritative, and timely compendium of human genes and genetic phenotypes with several search options
GeneReviews: http://www.ncbi.nlm.nih.gov/books/NBK1116
Expert-authored, peer-reviewed disease descriptions that apply genetic testing to the diagnosis, management, and genetic counseling of patients and families with specific inherited conditions
Orpha net: http://www.orpha.net
Reference portal for information on rare diseases and orphan drugs for helping to improve the diagnosis, care, and treatment of patients with rare diseases
Conclusion and Outlook
Submicroscopic microdeletions and microduplications are only one aspect of variations taking place in the human genome. As most MMSs patients are unable to reproduce due to the severity of their intellectual disability, these kinds of diseases are more an expression of the structure of the human genome than inherited disorders. Due to recent progress in current diagnostic panels, MMSs can now be discovered during conventional diagnostics. Keeping in mind the limitations of every technique per se, clinicians should analyze patients with intellectual disability using careful stepwise analysis. Detection of whole chromosome trisomies and monosomies as well as gross chromosomal rearrangements should be studied via banding and molecular cytogenetic techniques. Such aberrations, especially when present in low mosaic levels, might be easily missed when only performing targeted molecular techniques such as qPCR or MLPA alone. Targeted FISH, qPCR or MLPA, and aCGH could be the next steps. It is noteworthy that rare MMSs will not be identified by any method other than array CGH due to the small size of the anomaly.
New techniques such as high throughput and fast next-generation sequencing (NGS) of whole genomes will produce much more data and will detect variations that will be difficult to interpret, which could be the highest limitation. Also, NGS will have limitations such as detection of copy numbers, low resolution in repetitive regions, and findings that need to be verified in the patient and also in the parent samples by aCGH, FISH, qPCR, MLPA, and even conventional cytogenetics. Overall, the older approaches like the aforementioned should not be neglected, as they have at least two important advantages: (1) in most cases results are relatively easy to interpret due to long-standing experience, and (2) large financial resources are not needed for their implementation.
Supplemental Material
sj-pdf-1-jhc-10.1369_0022155412440001 – Supplemental material for Microdeletion and Microduplication Syndromes
Supplemental material, sj-pdf-1-jhc-10.1369_0022155412440001 for Microdeletion and Microduplication Syndromes by Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena, David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova and Thomas Liehr in Journal of Histochemistry & Cytochemistry
Supplemental Material
sj-xls-2-jhc-10.1369_0022155412440001 – Supplemental material for Microdeletion and Microduplication Syndromes
Supplemental material, sj-xls-2-jhc-10.1369_0022155412440001 for Microdeletion and Microduplication Syndromes by Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena, David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova and Thomas Liehr in Journal of Histochemistry & Cytochemistry
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported in parts by the BMBF/DLR BRA 09/020 and BLR 10/006, and the Else Kröner-Fresenius-Stiftung 2011_A42.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.